
Alzheimer’s Disease as a Membrane Dysfunction Tauopathy? New Insights into the Amyloid Cascade Hypothesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
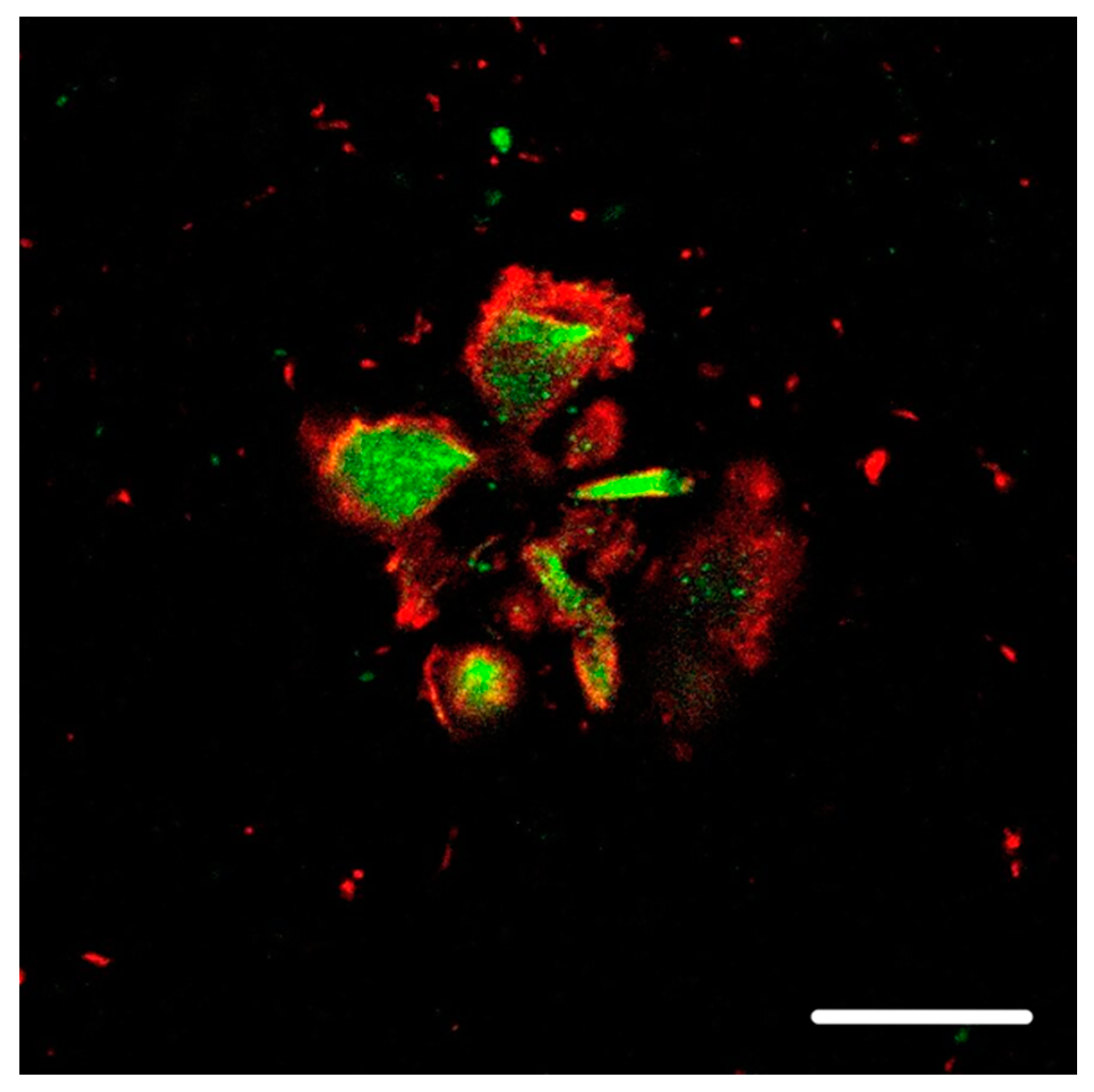
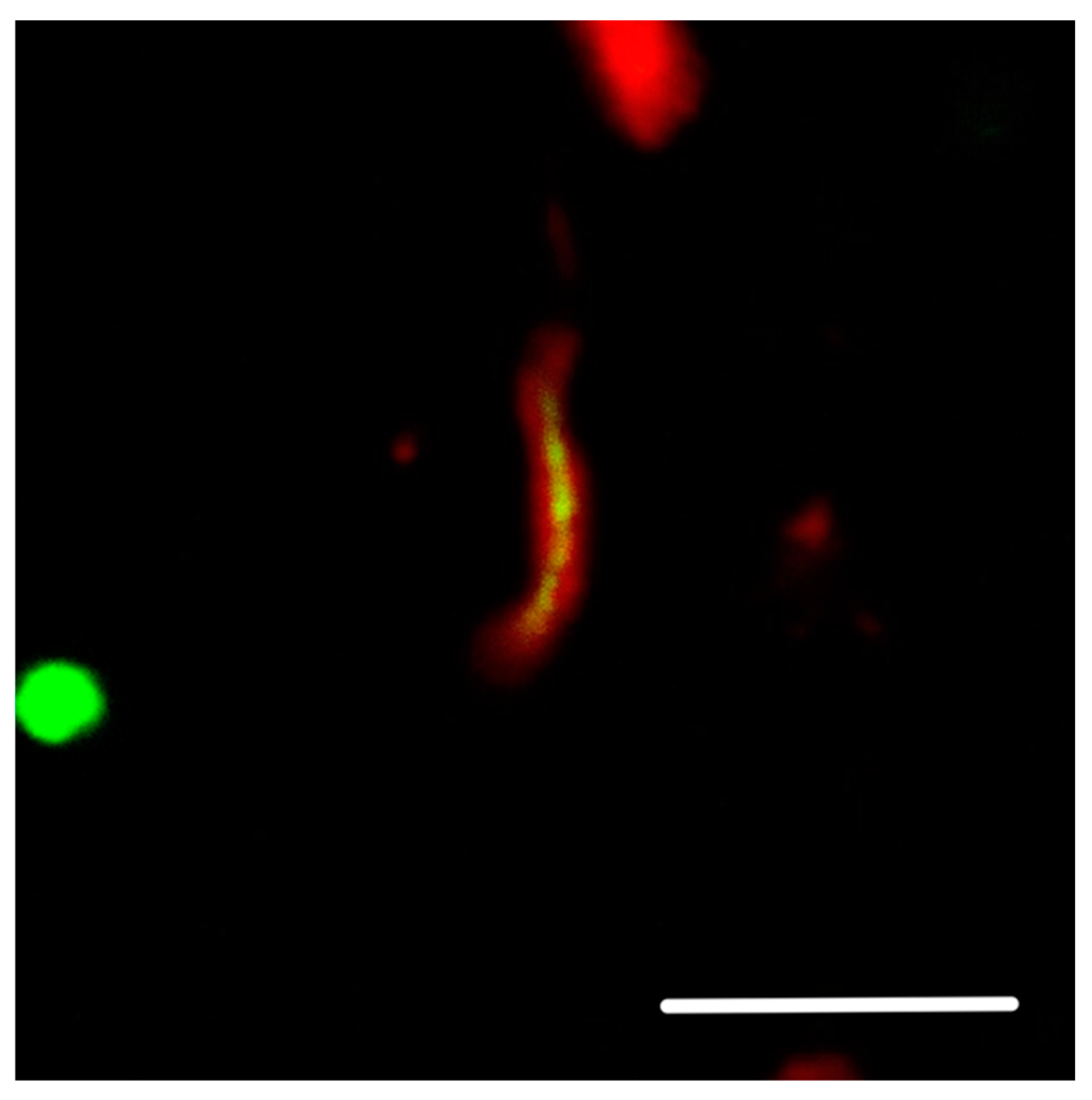
2. Preliminary Study: Hyperphosphorylated Protein Tau Aggregates in the Vicinity of the Neuron Membrane
3. Hypothesis
4. Supporting Circumstances
- (1)
- As mentioned before, the distribution of neurofibrillary tangles in the brain correlates with the clinical development of AD and characterizes the stages of disease on neuropathological investigation much more than Aβ plaques [7].
- (2)
- (3)
- Aβ plaques, both neuritic or diffuse, are located in the cortex, where neuronal bodies and axonal proximal–initial segments of cortical neurites are not myelinated [10,11]. In these locations, there is enough space either for the production of neuritic plaques or subsequently for inhibiting the internalization of LRP1-Aβ42 anchored to Aβ42 fibers.
- (4)
- Aβ is not accumulated inside neurons to a significant extent in AD.
- (5)
5. Animal Models
6. Discussion
7. Conclusions
8. Material and Methods
8.1. Patients
8.2. Tissue Samples
8.3. Immunofluorescence and Immunohistochemistry
8.3.1. Primary Antibodies
8.3.2. Secondary Antibodies
8.4. Confocal Microscopy
8.4.1. Figure 1
8.4.2. Figure 2 and Figure 3
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jankovska, N.; Olejar, T.; Matej, R. Extracellular Amyloid Deposits in Alzheimer’s and Creutzfeldt-Jakob Disease: Similar Behavior of Different Proteins? Int. J. Mol. Sci. 2020, 22, 7. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jankovska, N.; Olejar, T.; Kukal, J.; Matej, R. Different Morphology of Neuritic Plaques in the Archicortex of Alzheimer’s Disease with Comorbid Synucleinopathy: A Pilot Study. Curr. Alzheimer’s Res. 2020, 17, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006, 66, 1837–1844. [Google Scholar] [CrossRef]
- Bennett, D.A.; Schneider, J.A.; Aggarwal, N.T.; Arvanitakis, Z.; Shah, R.C.; Kelly, J.F.; Fox, J.H.; Cochran, E.J.; Arends, D.; Treinkman, A.D.; et al. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006, 27, 169–176. [Google Scholar] [CrossRef]
- Jones, S.L.; Svitkina, T.M. Axon Initial Segment Cytoskeleton: Architecture, Development, and Role in Neuron Polarity. Neural Plast. 2016, 2016, 6808293. [Google Scholar] [CrossRef]
- Yamada, R.; Kuba, H. Structural and Functional Plasticity at the Axon Initial Segment. Front. Cell Neurosci. 2016, 10, 250. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Claeysen, S.; Giannoni, P.; Ismeurt, C. The 5xFAD Mouse Model of Alzheimer’s Disease. The Neuroscience of Dementia 1. In Diagnosis and Management in Dementia; Academic Press: Cambridge, MA, USA, 2020; Chapter 13; pp. 207–221. [Google Scholar] [CrossRef]
- Youmans, K.L.; Tai, L.M.; Kanekiyo, T.; Stine, W.B., Jr.; Michon, S.-C.; Nwabuisi-Heath, E.; Manelli, A.M.; Fu, Y.; Riordan, S.; Roher, A.E.; et al. Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol. Neurodegener. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, C.L.; Kokjohn, T.A.; Whiteside, C.M.; Macias, M.P.; Kalback, W.M.; Sabbagh, M.N.; Beach, T.G.; Vassar, R.; Roher, A.E. Molecular Differences and Similarities Between Alzheimer’s Disease and the 5XFAD Transgenic Mouse Model of Amyloidosis. Biochem. Insights 2013, 6, BCI-S13025. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Park, S.; Lee, H.; Kim, Y. Thioflavin-positive tau aggregates complicating quantification of amyloid plaques in the brain of 5XFAD transgenic mouse model. Sci. Rep. 2021, 11, 1617. [Google Scholar] [CrossRef]
- Tohda, C.; Urano, T.; Umezaki, M.; Nemere, I.; Kuboyama, T. Diosgenin is an exogenous activator of 1,25D3-MARRS/Pdia3/ERp57 and improves Alzheimer’s disease pathologies in 5XFAD mice. Sci. Rep. 2012, 2, 535. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Cirrito, J.R.; Liu, C.C.; Shinohara, M.; Li, J.; Schuler, D.R.; Shinohara, M.; Holtzman, D.M.; Bu, G. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J. Neurosci. 2013, 33, 19276–19283. [Google Scholar] [CrossRef]
- Citron, M.; Diehl, T.S.; Gordon, G.; Biere, A.L.; Seubert, P.; Selkoe, D.J. Evidence that the 42- and 40-amino acid forms of amyloid beta protein are generated from the beta-amyloid precursor protein by different protease activities. Proc. Natl. Acad. Sci. USA 1996, 93, 13170–13175. [Google Scholar] [CrossRef]
- Bayer, T.A. N-Truncated Aβ Starting at Position Four-Biochemical Features, Preclinical Models, and Potential as Drug Target in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 710579. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Zampar, S.; Weggen, S. N-Terminally Truncated Aβ Peptide Variants in Alzheimer’s Disease. In Alzheimer’s Disease [Internet]; Wisniewski, T., Ed.; Codon Publications: Brisbane, Australia, 2019; Chapter 7. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Dickson, D.W. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2011; pp. 62–68. ISBN 978140519632. [Google Scholar]
- Bernstein, S.L.; Dupuis, N.F.; Lazo, N.D.; Wyttenbach, T.; Condron, M.M.; Bitan, G.; Teplow, D.B.; Shea, J.-E.; Ruotolo, T.; Robinson, C.V.; et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef]
- Gunther, E.C.; Strittmatter, S.M. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J. Mol. Med. 2009, 88, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Yokota, O.; Terada, S.; Ishizu, H.; Ujike, H.; Ishihara, T.; Nakashima, H.; Yasuda, M.; Kitamura, Y.; Uéda, K.; Checler, F.; et al. NACP/alpha-synuclein, NAC, and beta-amyloid pathology of familial Alzheimer’s disease with the E184D presenilin-1 mutation: A clinicopathological study of two autopsy cases. Acta Neuropathol. 2002, 104, 637–648. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Misasi, R. Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. Int. J. Mol. Sci. 2021, 22, 6016. [Google Scholar] [CrossRef]
- Sallaberry, C.A.; Voss, B.J.; Majewski, J.; Biernat, J.; Mandelkow, E.; Chi, E.Y.; Vander Zanden, C.M. Tau and Membranes: Interactions That Promote Folding and Condensation. Front. Cell Dev. Biol. 2021, 9, 725241. [Google Scholar] [CrossRef]
- Narita, M.; Holtzman, D.M.; Schwartz, A.L.; Bu, G. Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J. Neurochem. 1997, 69, 1904–1911. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Arélin, K.; Kinoshita, A.; Whelan, C.M.; Irizarry, M.C.; Rebeck, G.W.; Strickland, D.K.; Hyman, B.T. LRP and senile plaques in Alzheimer’s disease: Colocalization with apolipoprotein E and with activated astrocytes. Brain Res. Mol. Brain Res. 2002, 15, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.; Schmeidler, J.; Katsel, P.; Hof, P.R.; Haroutunian, V. Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiol. Aging 2012, 33, 628.e1–628.e14. [Google Scholar] [CrossRef]
- Kang, D.E.; Pietrzik, C.U.; Baum, L.; Chevallier, N.; Merriam, D.E.; Kounnas, M.Z.; Wagner, S.L.; Troncoso, J.C.; Kawas, C.H.; Katzman, R.; et al. Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Investig. 2000, 106, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Haggarty, S.J. Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies. Int. J. Mol. Sci. 2020, 21, 8948. [Google Scholar] [CrossRef]
- Hori, T.; Eguchi, K.; Wang, H.Y.; Miyasaka, T.; Guillaud, L.; Taoufiq, Z.; Mahapatra, S.; Yamada, H.; Takei, K.; Takahashi, T. Microtubule assembly by tau impairs endocytosis and neurotransmission via dynamin sequestration in Alzheimer’s disease synapse model. eLife 2022, 11, e73542. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Jankovska, N.; Matej, R.; Olejar, T. Extracellular Prion Protein Aggregates in Nine Gerstmann-Sträussler-Scheinker Syndrome Subjects with Mutation P102L: A Micromorphological Study and Comparison with Literature Data. Int. J. Mol Sci. 2021, 22, 13303. [Google Scholar] [CrossRef]
- Risacher, S.L.; Farlow, M.R.; Bateman, D.R.; Epperson, F.; Tallman, E.F.; Richardson, R.; Murrell, J.R.; Unverzagt, F.W.; Apostolova, L.G.; Bonnin, J.M.; et al. Detection of tau in Gerstmann-Sträussler-Scheinker disease (PRNP F198S) by [18F]Flortaucipir PET. Acta Neuropathol. Commun. 2018, 6, 114. [Google Scholar] [CrossRef]
- Mabrouk, R.; Gotkiewicz, M.; Rauramaa, T.; Tanila, H. DAPI (4’.;6-diamidino-2-phenylindole) Stains Compact Amyloid Plaques. J. Alzheimer’s Dis. 2022, 88, 949–955. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olejar, T.; Jankovska, N.; Matej, R. Alzheimer’s Disease as a Membrane Dysfunction Tauopathy? New Insights into the Amyloid Cascade Hypothesis. Int. J. Mol. Sci. 2024, 25, 9689. https://doi.org/10.3390/ijms25179689
Olejar T, Jankovska N, Matej R. Alzheimer’s Disease as a Membrane Dysfunction Tauopathy? New Insights into the Amyloid Cascade Hypothesis. International Journal of Molecular Sciences. 2024; 25(17):9689. https://doi.org/10.3390/ijms25179689
Chicago/Turabian StyleOlejar, Tomas, Nikol Jankovska, and Radoslav Matej. 2024. "Alzheimer’s Disease as a Membrane Dysfunction Tauopathy? New Insights into the Amyloid Cascade Hypothesis" International Journal of Molecular Sciences 25, no. 17: 9689. https://doi.org/10.3390/ijms25179689
APA StyleOlejar, T., Jankovska, N., & Matej, R. (2024). Alzheimer’s Disease as a Membrane Dysfunction Tauopathy? New Insights into the Amyloid Cascade Hypothesis. International Journal of Molecular Sciences, 25(17), 9689. https://doi.org/10.3390/ijms25179689