
SIRT1 Regulates Mitochondrial Damage in N2a Cells Treated with the Prion Protein Fragment 106–126 via PGC-1α-TFAM-Mediated Mitochondrial Biogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
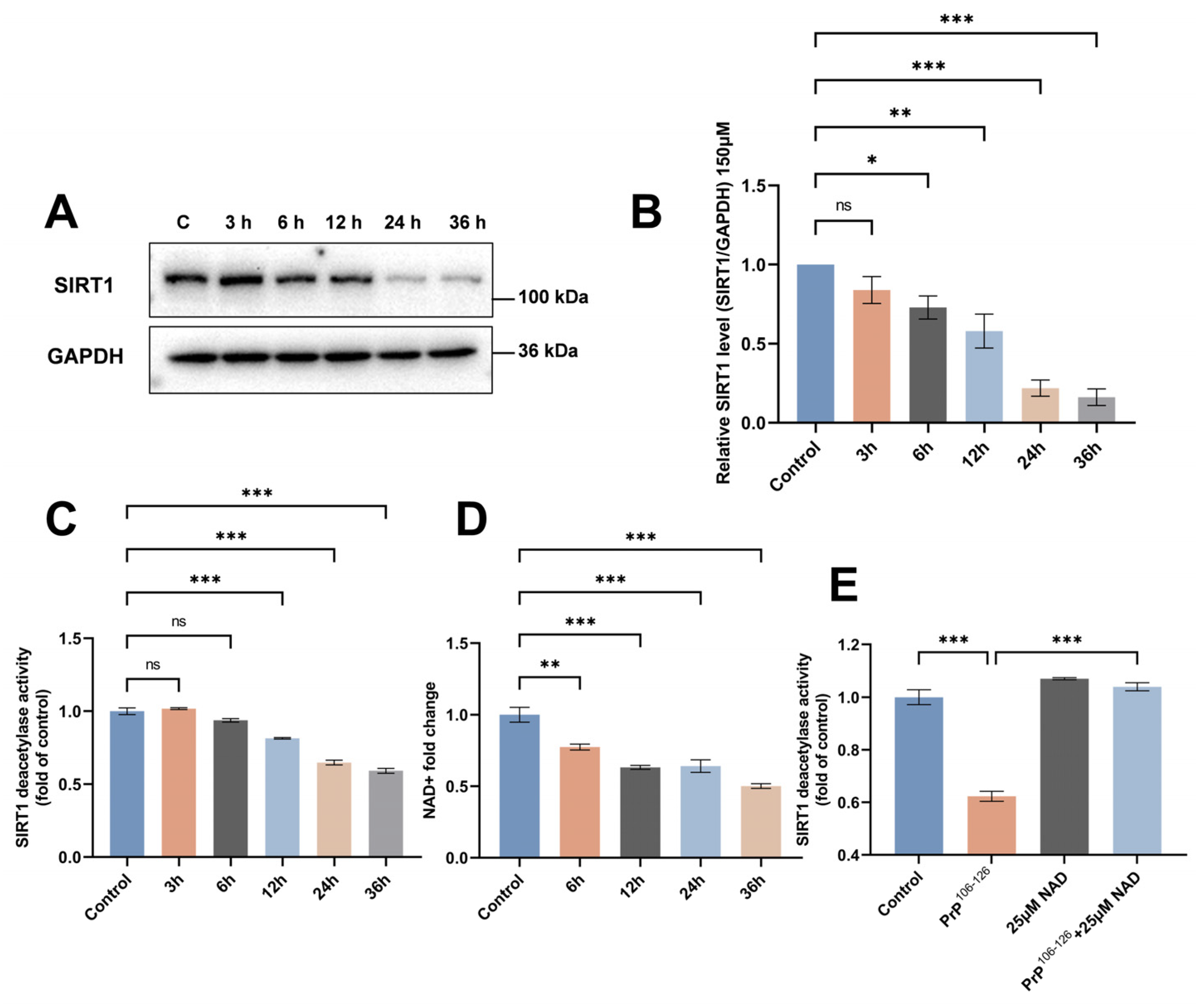
2.1. SIRT1 Expression and Deacetylase Activity Are Downregulated in PrP106–126-Exposed N2a Cells
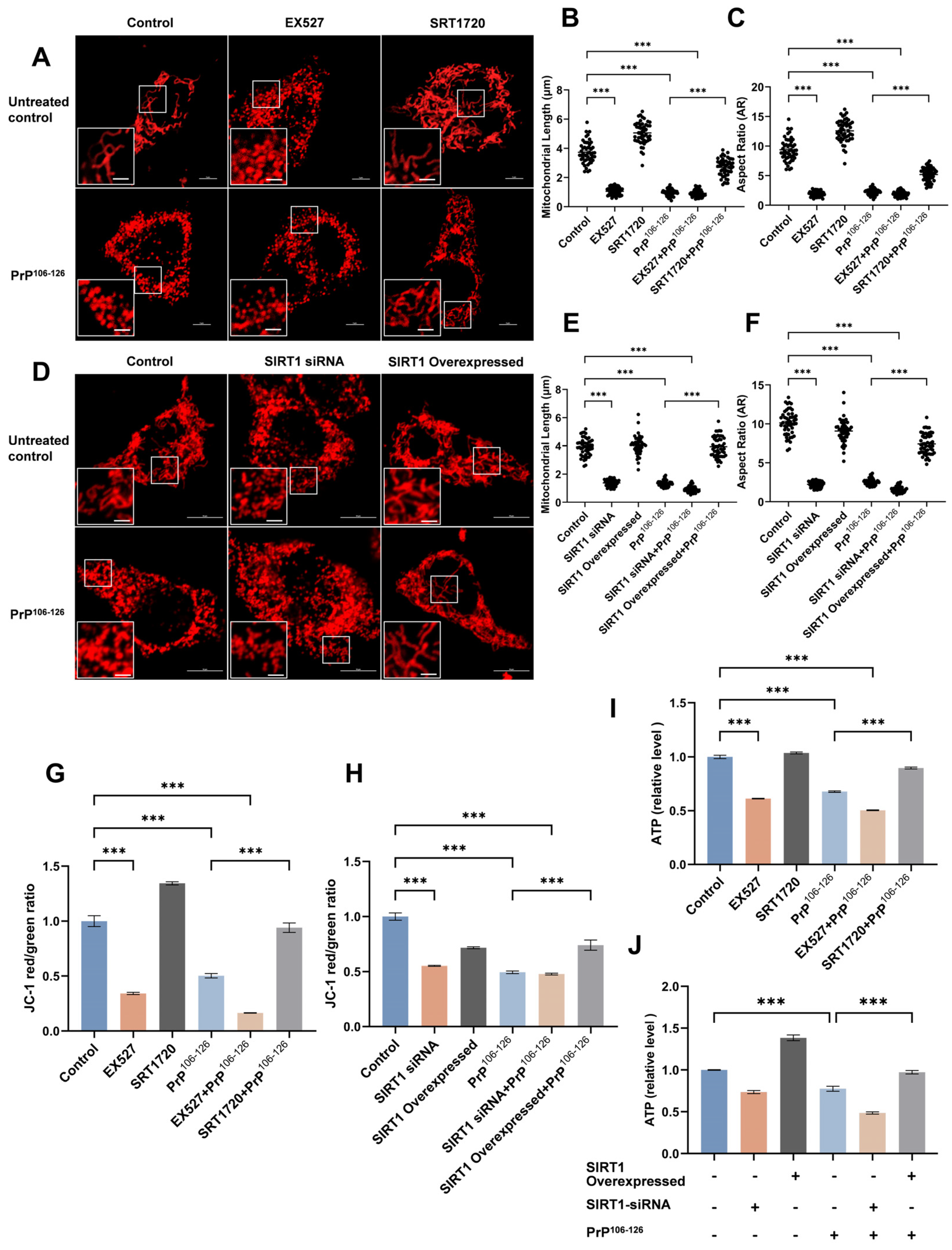
2.2. SIRT1 Participates in PrP106–126-Induced Mitochondrial Morphological Damage and Dysfunction
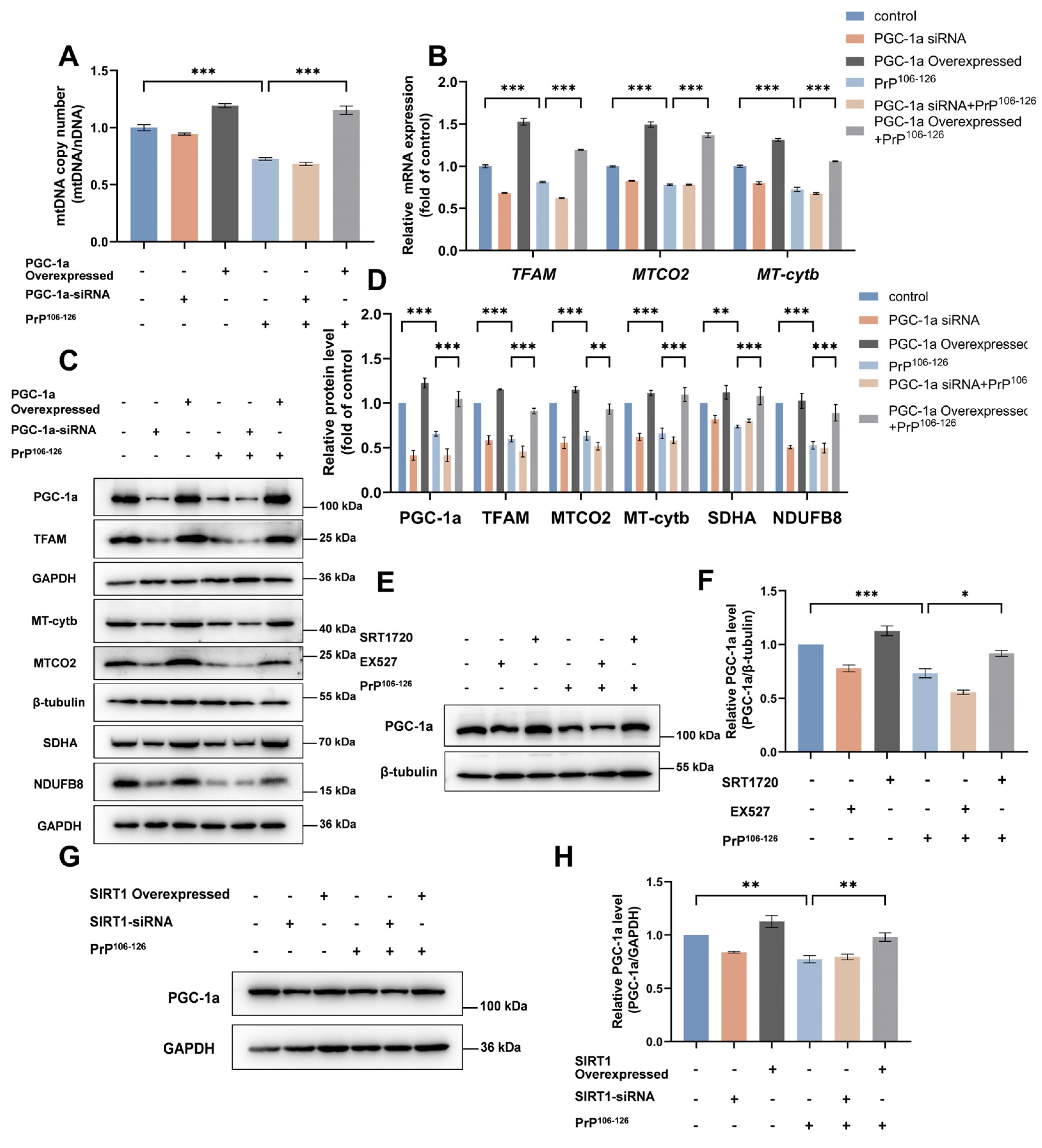
2.3. SIRT1 Overexpression and Activation Alleviate PrP106–126-Induced Impairment of Mitochondrial Biogenesis
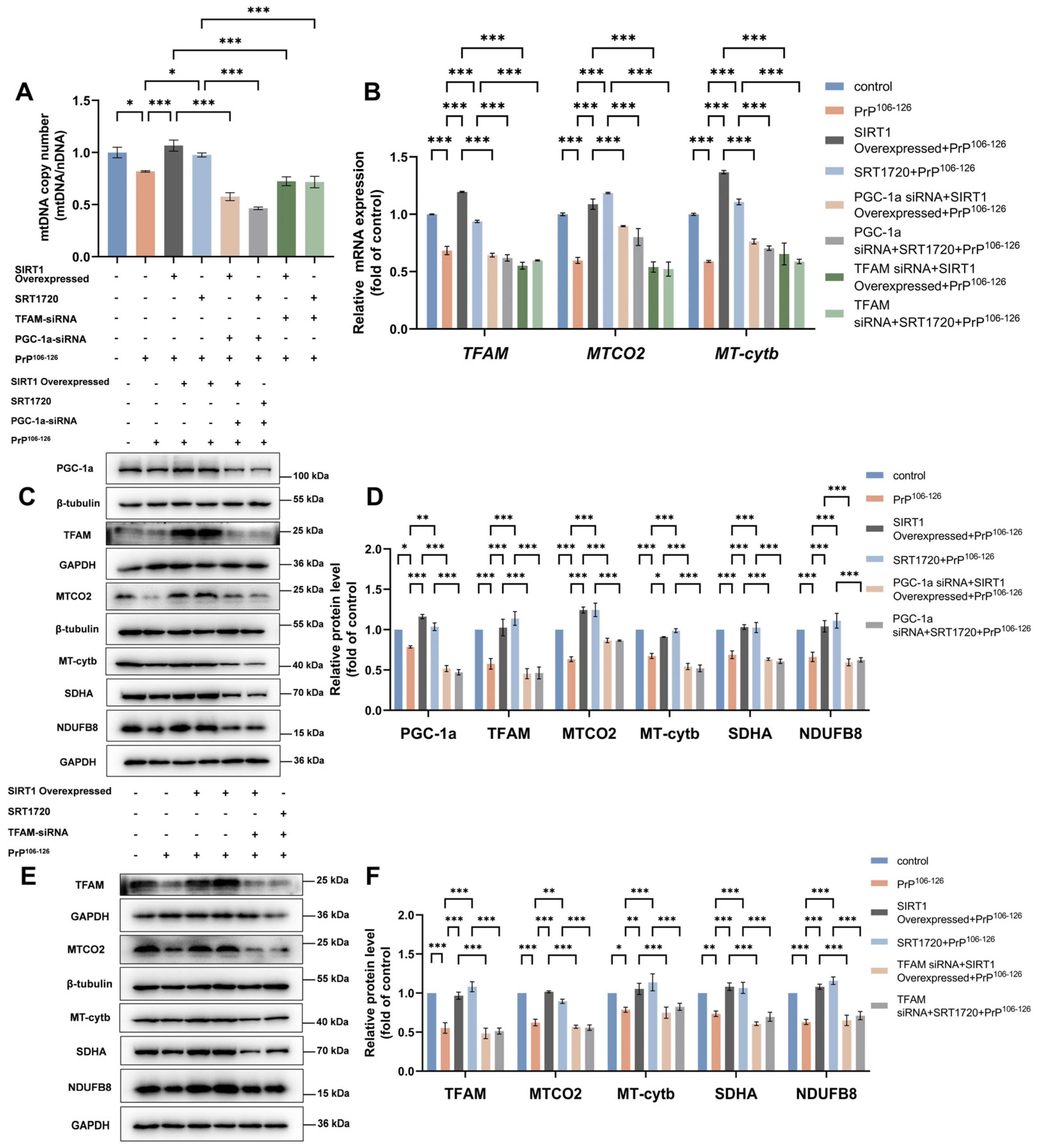
2.4. TFAM Is Required for SIRT1-Mediated Mitochondrial Biogenesis in PrP106–126-Treated N2a Cells
2.5. SIRT1 Regulates Mitochondrial Biogenesis through the PGC-1α-TFAM Signaling Pathway in a PrP106–126-Induced Prion Model
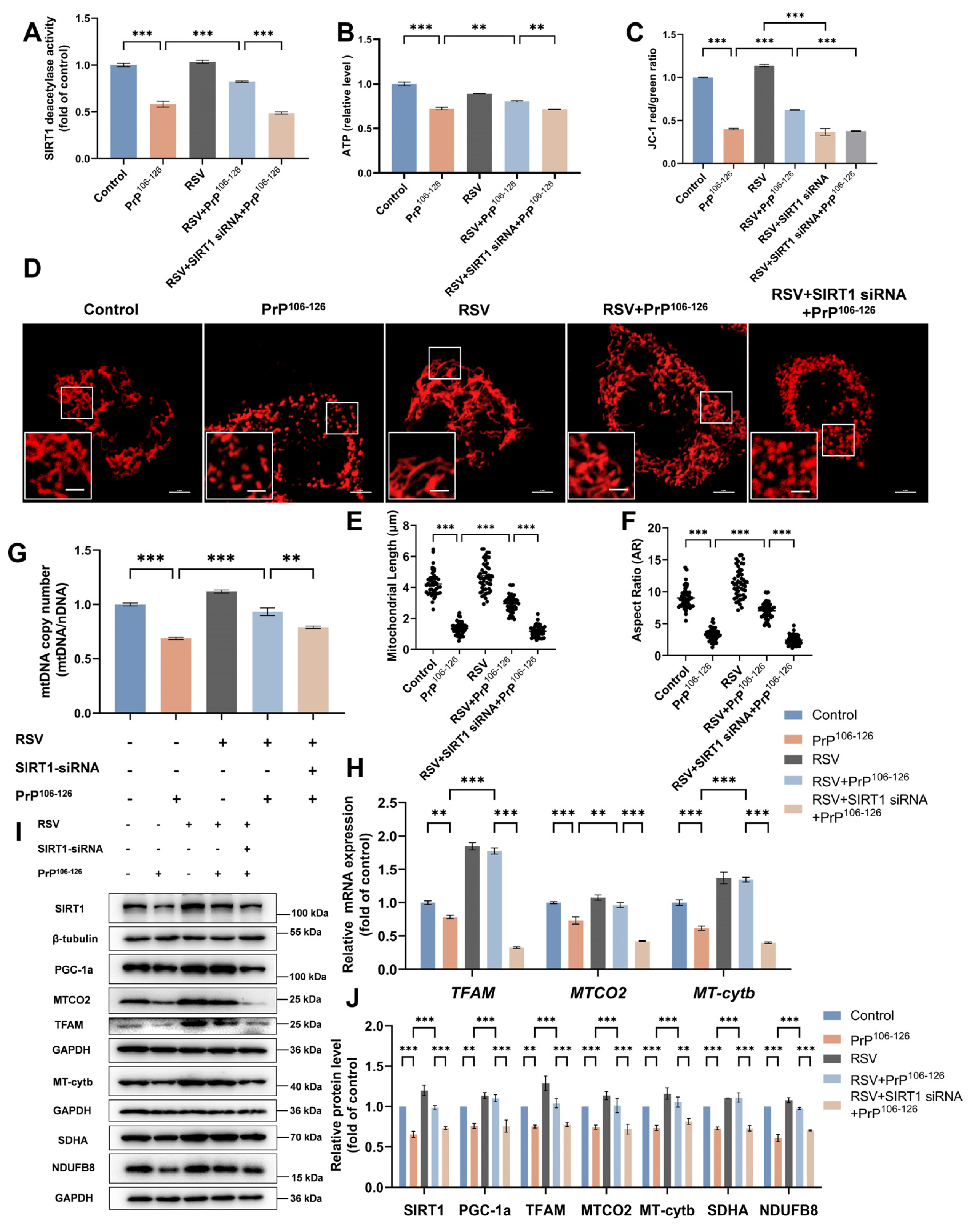
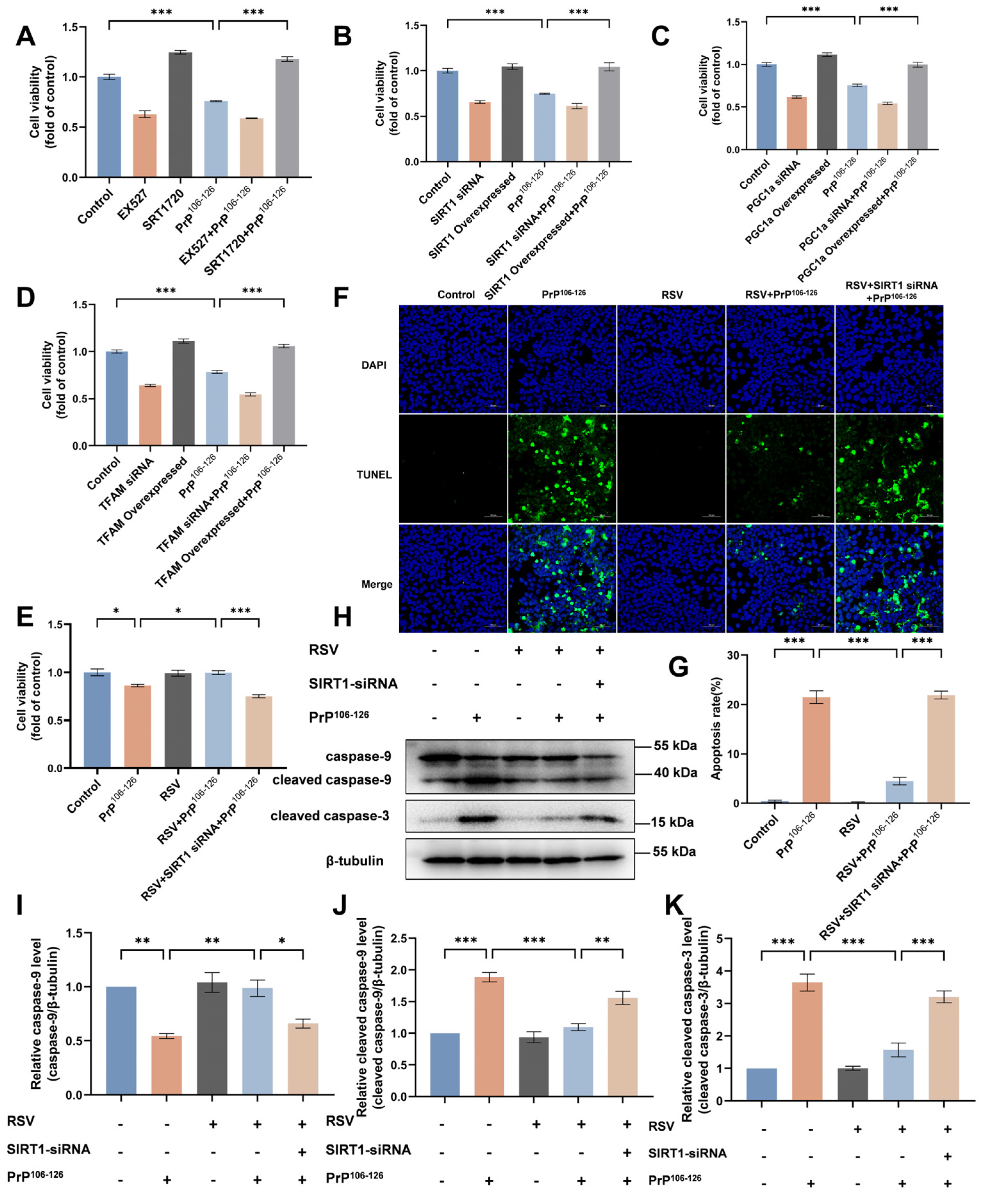
2.6. Resveratrol (RSV) Ameliorates PrP106–126-Induced Mitochondrial Dysfunction by Activating SIRT1-Dependent Mitochondrial Biogenesis in N2a Cells
2.7. RSV Supplementation Attenuates PrP106–126-Induced N2a Cell Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Small Interfering RNAs, Plasmids, and Transfections
4.3. Western Blot Assays
4.4. Measurement of SIRT1 Deacetylase Activity
4.5. Measurement of NAD+ Levels
4.6. Detection of ATP
4.7. Measurement of the Mitochondrial Membrane Potential
4.8. Immunofluorescence Microscopy
4.9. Reverse Transcription-Quantitative Real-Time PCR (RT-qPCR)
4.10. Assessment of the Mitochondrial DNA (mtDNA) Copy Numbers
4.11. Cell Viability Assays
4.12. TUNEL Assays
4.13. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Orge, L.; Lima, C.; Machado, C.; Tavares, P.; Mendonça, P.; Carvalho, P.; Silva, J.; Pinto, M.D.L.; Bastos, E.; Pereira, J.C.; et al. Neuropathology of Animal Prion Diseases. Biomolecules 2021, 11, 466. [Google Scholar] [CrossRef]
- Pritzkow, S.; Gorski, D.; Ramirez, F.; Soto, C. Prion Dissemination through the Environment and Medical Practices: Facts and Risks for Human Health. Clin. Microbiol. Rev. 2021, 34, e00059-19. [Google Scholar] [CrossRef] [PubMed]
- Tagliavini, F.; Prelli, F.; Verga, L.; Giaccone, G.; Sarma, R.; Gorevic, P.; Ghetti, B.; Passerini, F.; Ghibaudi, E.; Forloni, G.; et al. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 9678–9682. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Fujioka, H.; Mishra, R.S.; Li, R.; Singh, N. Prion Peptide 106–126 Modulates the Aggregation of Cellular Prion Protein and Induces the Synthesis of Potentially Neurotoxic Transmembrane PrP. J. Biol. Chem. 2002, 277, 2275–2286. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, B.; Zhao, C.; Wang, Y.; He, L.; Cui, M.; Zhu, X.; Du, W. Inhibition of human prion neuropeptide PrP106–126 aggregation by hexacoordinated ruthenium complexes. J. Inorg. Biochem. 2013, 128, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Chiesa, R.; Bugiani, O.; Salmona, M.; Tagliavini, F. Review: PrP 106–126-25 years after. Neuropathol. Appl. Neurobiol. 2019, 45, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hung, W.; Lee, M.; Huang, H.W. Membrane-mediated amyloid formation of PrP 106–126: A kinetic study. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 2422–2429. [Google Scholar] [CrossRef]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of Prion Protein Aggregation in Neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648–8669. [Google Scholar] [CrossRef]
- Moon, J.; Park, S. Prion peptide-mediated calcium level alteration governs neuronal cell damage through AMPK-autophagy flux. Cell Commun. Signal. 2020, 18, 109. [Google Scholar] [CrossRef]
- Huo, Y.; Huang, X.; Wang, Y.; Zhao, C.; Zheng, T.; Du, W. Inhibitory effects of sesquiterpene lactones on the aggregation and cytotoxicity of prion neuropeptide. Biochimie 2023, 211, 131–140. [Google Scholar] [CrossRef]
- Jeong, J.K.; Moon, M.H.; Seol, J.W.; Seo, J.S.; Lee, Y.J.; Park, S.Y. Prion peptide-mediated cellular prion protein overexpression and neuronal cell death can be blocked by aspirin treatment. Int. J. Mol. Med. 2011, 27, 689–693. [Google Scholar] [CrossRef]
- Pan, B.; Yang, L.; Wang, J.; Wang, Y.; Wang, J.; Zhou, X.; Yin, X.; Zhang, Z.; Zhao, D. c-Abl Tyrosine Kinase Mediates Neurotoxic Prion Peptide-Induced Neuronal Apoptosis via Regulating Mitochondrial Homeostasis. Mol. Neurobiol. 2014, 49, 1102–1116. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.H.; Wang, Y.C.; Zhang, L.M.; Duan, S.R. Protective effect of edaravone against PrP106-126-induced PC12 cell death. J. Biochem. Mol. Toxicol. 2010, 24, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Zhao, D.; Shah, S.Z.A.; Hassan, M.F.; Zhu, T.; Song, Z.; Zhou, X.; Yang, L. Parkin Overexpression Ameliorates PrP106-126-Induced Neurotoxicity via Enhanced Autophagy in N2a Cells. Cell. Mol. Neurobiol. 2017, 37, 717–728. [Google Scholar] [CrossRef]
- Thellung, S.; Villa, V.; Corsaro, A.; Arena, S.; Millo, E.; Damonte, G.; Benatti, U.; Tagliavini, F.; Florio, T.; Schettini, G. p38 MAP Kinase Mediates the Cell Death Induced by PrP106-126 in the SH-SY5Y Neuroblastoma Cells. Neurobiol. Dis. 2002, 9, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, M.; Zhang, X.; Li, Z.; Yang, D.; Zhao, M.; Wang, D.; Sun, Z.; Ehsan, S.; Li, W.; et al. PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease. Cell Death Dis. 2022, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, D.; Wu, W.; Yang, W.; Ali Shah, S.Z.; Zhao, Y.; Duan, Y.; Wang, L.; Zhou, X.; Zhao, D.; et al. DLP1-dependent mitochondrial fragmentation and redistribution mediate prion-associated mitochondrial dysfunction and neuronal death. Aging Cell 2018, 17, e12693. [Google Scholar] [CrossRef]
- Wu, W.; Zhao, D.; Shah, S.Z.A.; Zhang, X.; Lai, M.; Yang, D.; Wu, X.; Guan, Z.; Li, J.; Zhao, H.; et al. OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis. 2019, 10, 710. [Google Scholar] [CrossRef]
- Xu, D.-C.; Chen, Y.; Xu, Y.; ShenTu, C.-Y.; Peng, L.-H. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef]
- Rehman, M.U.; Sehar, N.; Dar, N.J.; Khan, A.; Arafah, A.; Rashid, S.; Rashid, S.M.; Ganaie, M.A. Mitochondrial dysfunctions, oxidative stress and neuroinflammation as therapeutic targets for neurodegenerative diseases: An update on current advances and impediments. Neurosci. Biobehav. Rev. 2023, 144, 104961. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Liu, Z.; Qi, X. Mitochondrial Quality Control Strategies: Potential Therapeutic Targets for Neurodegenerative Diseases? Front. Neurosci. 2021, 15, 746873. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Liu, H.; Xu, S.; Wang, C.; Deng, Y.; Xu, B.; Yang, T.; Sun, J.; Liu, W. The Beneficial Role of Sirtuin 1 in Preventive or Therapeutic Options of Neurodegenerative Diseases. Neuroscience 2022, 504, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, D.; Liu, W.; Li, S.; Chen, J.; Shen, Y.; Wang, F.; Hu, L.; Liu, C. Disruption of the Circadian Clock Alters Antioxidative Defense via the SIRT1-BMAL1 Pathway in 6-OHDA-Induced Models of Parkinson’s Disease. Oxidative Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, Y.; Yang, Y.; Huang, X.; Sun, C. SIRT1 Protects Dopaminergic Neurons in Parkinson’s Disease Models via PGC-1α-Mediated Mitochondrial Biogenesis. Neurotox. Res. 2021, 39, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Yang, Y.; Chang, W.; Fang, W.; Huang, S.; Chou, S.; Lo, Y. Hyperbaric Oxygen Therapy Improves Parkinson’s Disease by Promoting Mitochondrial Biogenesis via the SIRT-1/PGC-1α Pathway. Biomolecules 2022, 12, 661. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Wu, A. The Protective Mechanism of SIRT1 in the Regulation of Mitochondrial Biogenesis and Mitochondrial Autophagy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ottenberg, G.; Sferrazza, G.F.; Hubbs, C.; Fallahi, M.; Rumbaugh, G.; Brantley, A.F.; Lasmezas, C.I. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 2015, 138, 992–1008. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Moon, M.; Jeong, J.; Seol, J.; Lee, Y.; Park, B.; Park, S. SIRT1, a histone deacetylase, regulates prion protein-induced neuronal cell death. Neurobiol. Aging 2012, 33, 1110–1120. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Shi, Q.; Zhang, B.; Chen, C.; Chen, L.; Sun, J.; Wang, H.; Xiao, K.; Dong, X. Scrapie Infection in Experimental Rodents and SMB-S15 Cells Decreased the Brain Endogenous Levels and Activities of Sirt1. J. Mol. Neurosci. 2015, 55, 1022–1030. [Google Scholar] [CrossRef]
- Singulani, M.P.; Pereira, C.P.M.; Ferreira, A.F.F.; Garcia, P.C.; Ferrari, G.D.; Alberici, L.C.; Britto, L.R. Impairment of PGC-1α-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp. Gerontol. 2020, 133, 110882. [Google Scholar] [CrossRef]
- Lai, M.; Li, J.; Zhang, X.; Wu, W.; Li, Z.; Sun, Z.; Zhao, M.; Yang, D.; Wang, D.; Li, W.; et al. SARM1 participates in axonal degeneration and mitochondrial dysfunction in prion disease. Neural Regen. Res. 2022, 17, 2293. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, D.; Wu, W.; Ali Shah, S.Z.; Lai, M.; Yang, D.; Li, J.; Guan, Z.; Li, W.; Gao, H.; et al. Melatonin regulates mitochondrial dynamics and alleviates neuron damage in prion diseases. Aging 2020, 12, 11139–11151. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Huang, C.; Cheng, J.; Chiou, C.; Lee, I.; Yang, Y.; Hsu, T.; Yei, C.; Lin, P.; Chen, J.; et al. SRT1720 as an SIRT1 activator for alleviating paraquat-induced models of Parkinson’s disease. Redox Biol. 2022, 58, 102534. [Google Scholar] [CrossRef]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Nandave, M.; Acharjee, R.; Bhaduri, K.; Upadhyay, J.; Rupanagunta, G.P.; Ansari, M.N. A pharmacological review on SIRT 1 and SIRT 2 proteins, activators, and inhibitors: Call for further research. Int. J. Biol. Macromol. 2023, 242, 124581. [Google Scholar] [CrossRef]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Hillen, H.S.; Morozov, Y.I.; Sarfallah, A.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription Initiation. Cell 2017, 171, 1072–1081. [Google Scholar] [CrossRef]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol. Ther. 2021, 219, 107705. [Google Scholar] [CrossRef]
- Lv, J.; Jiang, S.; Yang, Z.; Hu, W.; Wang, Z.; Li, T.; Yang, Y. PGC-1α sparks the fire of neuroprotection against neurodegenerative disorders. Ageing Res. Rev. 2018, 44, 8–21. [Google Scholar] [CrossRef]
- Chodari, L.; Dilsiz Aytemir, M.; Vahedi, P.; Alipour, M.; Vahed, S.Z.; Khatibi, S.M.H.; Ahmadian, E.; Ardalan, M.; Eftekhari, A. Targeting Mitochondrial Biogenesis with Polyphenol Compounds. Oxida. Med. Cell. Longev. 2021, 2021, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting cAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Sawant, N.; Morton, H.; Kshirsagar, S.; Reddy, A.P.; Reddy, P.H. Mitochondrial Abnormalities and Synaptic Damage in Huntington’s Disease: A Focus on Defective Mitophagy and Mitochondria-Targeted Therapeutics. Mol. Neurobiol. 2021, 58, 6350–6377. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Klein, H.; Trumpff, C.; Yang, H.; Lee, A.J.; Picard, M.; Bennett, D.A.; De Jager, P.L. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol. Neurodegener. 2021, 16, 75. [Google Scholar] [CrossRef]
- Harerimana, N.V.; Paliwali, D.; Romero Molina, C.; Bennett, D.A.; Pa, J.; Goate, A.; Swerdlow, R.H.; Andrews, S.J. The role of mitochondrial genome abundance in Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Acevedo-Cintrón, J.; Jhaldiyal, A.; Wang, H.; Andrabi, S.A.; Eacker, S.; Karuppagounder, S.S.; Brahmachari, S.; Chen, R.; Kim, H.; et al. Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons. Stem Cell Rep. 2020, 15, 629–645. [Google Scholar] [CrossRef]
- Thomas, R.R.; Keeney, P.M.; Bennett, J.P. Impaired Complex-I Mitochondrial Biogenesis in Parkinson Disease Frontal Cortex. J. Park. Dis. 2012, 2, 67–76. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Currais, A.; Soriano-Castell, D.; Schubert, D.; Maher, P. Natural products targeting mitochondria: Emerging therapeutics for age-associated neurological disorders. Pharmacol. Ther. 2021, 221, 107749. [Google Scholar] [CrossRef]
- Yin, Z.; Gao, D.; Du, K.; Han, C.; Liu, Y.; Wang, Y.; Gao, X.; Ciobica, A. Rhein Ameliorates Cognitive Impairment in an APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease by Relieving Oxidative Stress through Activating the SIRT1/PGC-1α Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 2524832. [Google Scholar] [CrossRef]
- Zhao, Q.; Tian, Z.; Zhou, G.; Niu, Q.; Chen, J.; Li, P.; Dong, L.; Xia, T.; Zhang, S.; Wang, A. SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride. Theranostics 2020, 10, 4822–4838. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Malik, N.; Ferreira, B.I.; Hollstein, P.E.; Curtis, S.D.; Trefts, E.; Weiser Novak, S.; Yu, J.; Gilson, R.; Hellberg, K.; Fang, L.; et al. Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1. Science 2023, 380, eabj5559. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Jia, J.; Yin, J.; Zhang, Y.; Xu, G.; Wang, M.; Jiang, H.; Li, L.; Zeng, X.; Zhu, D. Thioredoxin-1 Promotes Mitochondrial Biogenesis Through Regulating AMPK/Sirt1/PGC1α Pathway in Alzheimer’s Disease. ASN Neuro 2023, 15, 166769320. [Google Scholar] [CrossRef]
- Chen, M.; Yan, R.; Luo, J.; Ning, J.; Zhou, R.; Ding, L. The Role of PGC-1α-Mediated Mitochondrial Biogenesis in Neurons. Neurochem. Res. 2023, 48, 2595–2606. [Google Scholar] [CrossRef]
- Fan, H.; Ding, R.; Liu, W.; Zhang, X.; Li, R.; Wei, B.; Su, S.; Jin, F.; Wei, C.; He, X.; et al. Heat shock protein 22 modulates NRF1/TFAM-dependent mitochondrial biogenesis and DRP1-sparked mitochondrial apoptosis through AMPK-PGC1α signaling pathway to alleviate the early brain injury of subarachnoid hemorrhage in rats. Redox Biol. 2021, 40, 101856. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial Biogenesis Is Impaired in Osteoarthritis Chondrocytes but Reversible via Peroxisome Proliferator-Activated Receptor γ Coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Czubryt, M.P.; McAnally, J.; Fishman, G.I.; Olson, E.N. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2003, 100, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Zirden, L.C.; Puustinen, P.; Blanchette, P.; Jeong, H.; Dejgaard, K.; Siegel, P.M.; Pause, A. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 2021, 17, 3957–3975. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Hu, M.L.; Pan, Y.R.; Yong, Y.Y.; Liu, Y.; Yu, L.; Qin, D.L.; Qiao, G.; Law, B.Y.; Wu, J.M.; Zhou, X.G.; et al. Poly (ADP-ribose) polymerase 1 and neurodegenerative diseases: Past, present, and future. Ageing Res. Rev. 2023, 91, 102078. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, H.; Luo, Y.; Cui, J.; Talukder, M.; Li, J. Lycopene mitigates DEHP-induced hepatic mitochondrial quality control disorder via regulating SIRT1/PINK1/mitophagy axis and mitochondrial unfolded protein response. Environ. Pollut. 2021, 292, 118390. [Google Scholar] [CrossRef]
- Zhao, N.; Xia, J.; Xu, B. Physical exercise may exert its therapeutic influence on Alzheimer’s disease through the reversal of mitochondrial dysfunction via SIRT1-FOXO1/3-PINK1-Parkin-mediated mitophagy. J. Sport Health Sci. 2021, 10, 1–3. [Google Scholar] [CrossRef]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef]
- Lei, M.; Cong, L.; Liu, Z.; Liu, Z.; Ma, Z.; Liu, K.; Li, J.; Deng, Y.; Liu, W.; Xu, B. Resveratrol reduces DRP1-mediated mitochondrial dysfunction via the SIRT1-PGC1α signaling pathway in manganese-induced nerve damage in mice. Environ. Toxicol. 2021, 37, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Guo, Y.; Song, L.; Wen, H.; Sun, N.; Wang, Y.; Qi, B.; Liang, Q.; Geng, J.; Liu, X.; et al. Nicotinamide riboside promotes Mfn2-mediated mitochondrial fusion in diabetic hearts through the SIRT1-PGC1α-PPARα pathway. Free Radic. Biol. Med. 2022, 183, 75–88. [Google Scholar] [CrossRef]
- Ungurianu, A.; Zanfirescu, A.; Margină, D. Sirtuins, resveratrol and the intertwining cellular pathways connecting them. Ageing Res. Rev. 2023, 88, 101936. [Google Scholar] [CrossRef]
- Shaito, A.; Al-Mansoob, M.; Ahmad, S.; Haider, M.Z.; Eid, A.H.; Posadino, A.M.; Pintus, G.; Giordo, R. Resveratrol-mediated regulation of mitochondria biogenesis-associated pathways in neurodegenerative diseases: Molecular insights and potential therapeutic applications. Curr. Neuropharmacol. 2022, 21, 1184. [Google Scholar] [CrossRef] [PubMed]
- Corpas, R.; Grinan-Ferre, C.; Rodriguez-Farre, E.; Pallas, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience Against Alzheimer Neurodegeneration Through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 902–915. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Li, J.; Li, Z.; Yang, D.; Wang, D.; Sun, Z.; Wen, P.; Gou, F.; Dai, Y.; Ji, Y.; et al. SIRT1 Regulates Mitochondrial Damage in N2a Cells Treated with the Prion Protein Fragment 106–126 via PGC-1α-TFAM-Mediated Mitochondrial Biogenesis. Int. J. Mol. Sci. 2024, 25, 9707. https://doi.org/10.3390/ijms25179707
Zhao M, Li J, Li Z, Yang D, Wang D, Sun Z, Wen P, Gou F, Dai Y, Ji Y, et al. SIRT1 Regulates Mitochondrial Damage in N2a Cells Treated with the Prion Protein Fragment 106–126 via PGC-1α-TFAM-Mediated Mitochondrial Biogenesis. International Journal of Molecular Sciences. 2024; 25(17):9707. https://doi.org/10.3390/ijms25179707
Chicago/Turabian StyleZhao, Mengyang, Jie Li, Zhiping Li, Dongming Yang, Dongdong Wang, Zhixin Sun, Pei Wen, Fengting Gou, Yuexin Dai, Yilan Ji, and et al. 2024. "SIRT1 Regulates Mitochondrial Damage in N2a Cells Treated with the Prion Protein Fragment 106–126 via PGC-1α-TFAM-Mediated Mitochondrial Biogenesis" International Journal of Molecular Sciences 25, no. 17: 9707. https://doi.org/10.3390/ijms25179707