
Identification of SNPs and Candidate Genes Associated with Monocyte/Lymphocyte Ratio and Neutrophil/Lymphocyte Ratio in Duroc × Erhualian F2 Population

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. MLR and NLR of Duroc × Erhualian F2 Resource Population Exhibited High Heritability
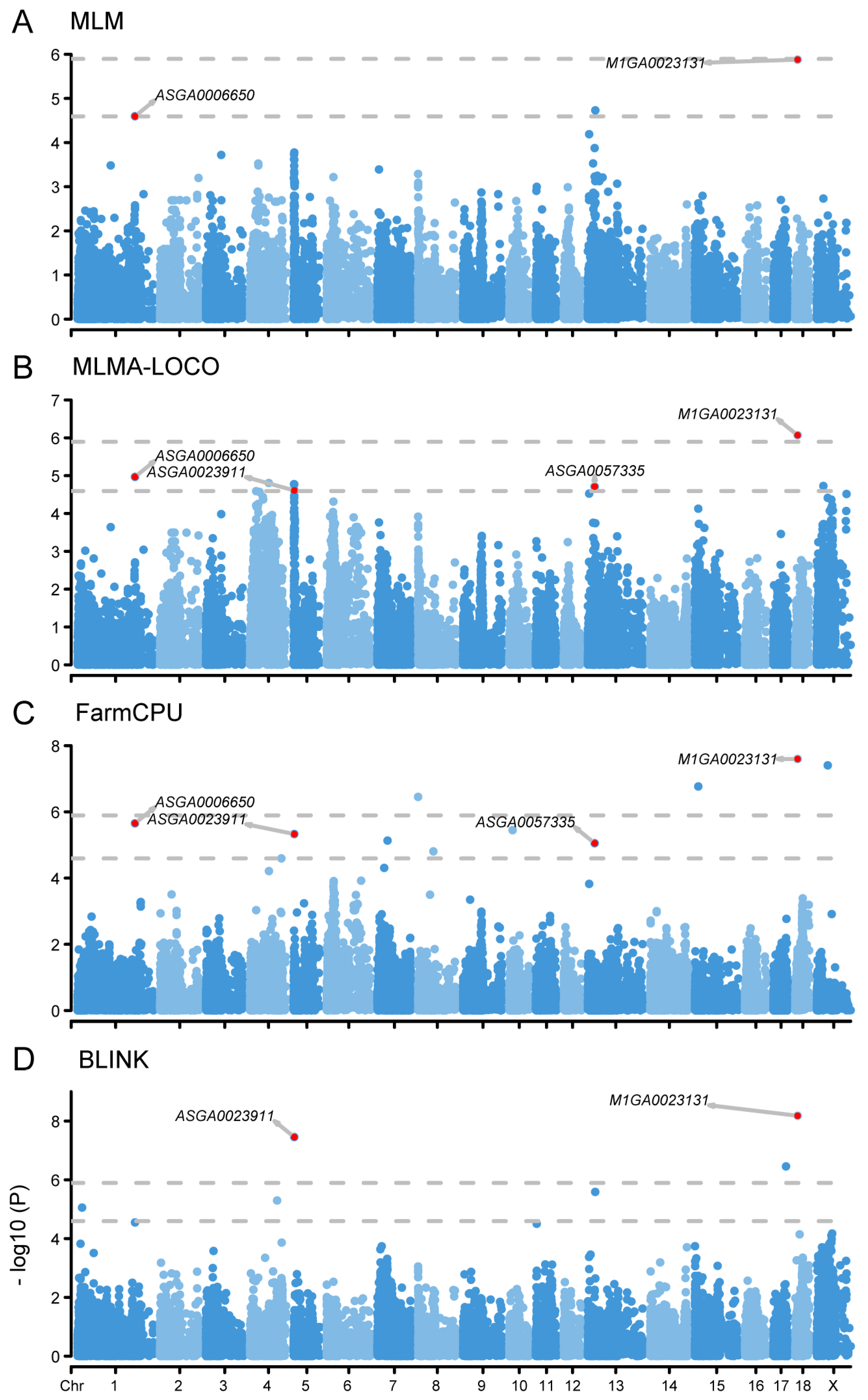
2.2. GWAS Analysis Identified Five SNPs Associated with MLR Trait
2.3. GWAS Analysis Identified Four SNPs Associated with NLR Trait
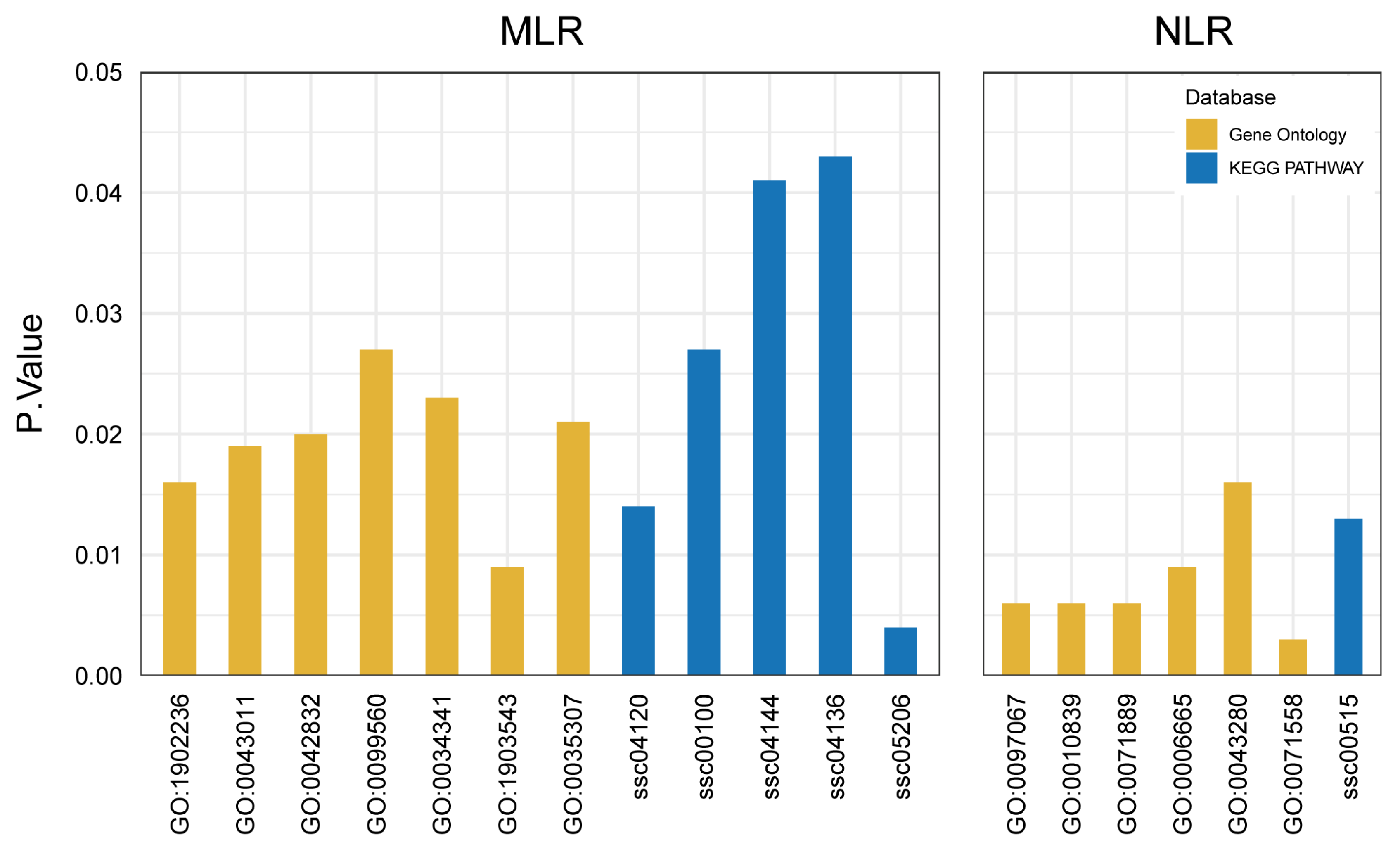
2.4. Functional Enrichment Analysis Identified Multiple Candidate Genes for MLR and NLR
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Phenotype
4.3. Genotype
4.4. Estimation of Genetic Variance and Heritability
4.5. Genome-Wide Association Study
4.6. Identification and Functional Analysis of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. A brief outline of the immune system. Methods Mol. Biol. 2014, 1184, 3–12. [Google Scholar] [PubMed]
- Margraf, A.; Perretti, M. Immune Cell Plasticity in Inflammation: Insights into Description and Regulation of Immune Cell Phenotypes. Cells 2022, 11, 1824. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fan, Z.-s. Research progress on immune cell plasticity and immune pathogenesis. Chin. J. Immunol. 2018, 12, 641–646. [Google Scholar]
- Bogdanos, D.P.; Sakkas, L.I. Enterococcus gallinarum as a component of the Autoinfectome: The gut-liver-autoimmune rheumatic disease axis is alive and kicking. Mediterr. J. Rheumatol. 2018, 29, 187–189. [Google Scholar] [CrossRef]
- Baynes, R.E.; Dedonder, K.; Kissell, L.; Mzyk, D.; Marmulak, T.; Smith, G.; Tell, L.; Gehring, R.; Davis, J.; Riviere, J.E. Health concerns and management of select veterinary drug residues. Food Chem. Toxicol. 2016, 88, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Centner, T.J.; Alvey, J.C.; Stelzleni, A.M. Beta agonists in livestock feed: Status, health concerns, and international trade. J. Anim. Sci. 2014, 92, 4234–4240. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.H.; Liu, X.D.; Wang, H.Y.; Liu, X.L.; Li, X.Y.; Wu, Z.F.; Zhu, M.J.; Zhao, S.H. Genomewide association studies for hematological traits and T lymphocyte subpopulations in a Duroc × Erhualian F2 resource population. J. Anim. Sci. 2016, 94, 5028–5041. [Google Scholar] [CrossRef]
- Pabst, R. The pig as a model for immunology research. Cell Tissue Res. 2020, 380, 287–304. [Google Scholar] [CrossRef]
- Zahorec, R. Ratio of neutrophil to lymphocyte counts--rapid and simple parameter of systemic inflammation and stress in critically ill. Bratisl. Lek. Listy 2001, 102, 5–14. [Google Scholar]
- Ji, H.; Li, Y.; Fan, Z.; Zuo, B.; Jian, X.; Li, L.; Liu, T. Monocyte/lymphocyte ratio predicts the severity of coronary artery disease: A syntax score assessment. BMC Cardiovasc. Disord. 2017, 17, 90. [Google Scholar] [CrossRef]
- Balta, S.; Demirkol, S.; Unlu, M.; Arslan, Z.; Celik, T. Neutrophil to lymphocyte ratio may be predict of mortality in all conditions. Br. J. Cancer 2013, 109, 3125–3126. [Google Scholar] [CrossRef]
- Gomes, L.T.; Morato-Conceicao, Y.T.; Gambati, A.V.M.; Maciel-Pereira, C.M.; Fontes, C.J.F. Diagnostic value of neutrophil-to-lymphocyte ratio in patients with leprosy reactions. Heliyon 2020, 6, e03369. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Fu, Z.; Huang, W.; Huang, K. Prognostic value of neutrophil-to-lymphocyte ratio in sepsis: A meta-analysis. Am. J. Emerg. Med. 2020, 38, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Kanetsky, P.A.; Egan, K.M. Exploring the prognostic value of the neutrophil-to-lymphocyte ratio in cancer. Sci. Rep. 2019, 9, 19673. [Google Scholar] [CrossRef] [PubMed]
- Raffetti, E.; Donato, F.; Casari, S.; Castelnuovo, F.; Sighinolfi, L.; Bandera, A.; Maggiolo, F.; Ladisa, N.; di Pietro, M.; Fornabaio, C.; et al. Systemic inflammation-based scores and mortality for all causes in HIV-infected patients: A MASTER cohort study. BMC Infect. Dis. 2017, 17, 193. [Google Scholar] [CrossRef]
- Hua, Y.; Sun, J.Y.; Lou, Y.X.; Sun, W.; Kong, X.Q. Monocyte-to-lymphocyte ratio predicts mortality and cardiovascular mortality in the general population. Int. J. Cardiol. 2023, 379, 118–126. [Google Scholar] [CrossRef]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on Neutrophil Function in Severe Inflammation. Front Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Marti, G.E.; Fleisher, T.A. Application of lymphocyte immunophenotyping in selected diseases. Pathol. Immunopathol. Res. 1988, 7, 319–337. [Google Scholar] [CrossRef]
- Cantor, R.M.; Lange, K.; Sinsheimer, J.S. Prioritizing GWAS Results: A Review of Statistical Methods and Recommendations for Their Application. Am. J. Hum. Genet. 2010, 86, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Zhang, X.; Johnson, A.D.; Hendricks, A.E.; Hwang, S.J.; Tanriverdi, K.; Ganesh, S.K.; Smith, N.L.; Peyser, P.A.; Freedman, J.E.; O’Donnell, C.J. Genetic associations with expression for genes implicated in GWAS studies for atherosclerotic cardiovascular disease and blood phenotypes. Hum. Mol. Genet. 2014, 23, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Andiappan, A.K.; Melchiotti, R.; Poh, T.Y.; Nah, M.; Puan, K.J.; Vigano, E.; Haase, D.; Yusof, N.; San Luis, B.; Lum, J.; et al. Genome-wide analysis of the genetic regulation of gene expression in human neutrophils. Nat. Commun. 2015, 6, 7971. [Google Scholar] [CrossRef]
- Lin, B.D.; Carnero-Montoro, E.; Bell, J.T.; Boomsma, D.I.; de Geus, E.J.; Jansen, R.; Kluft, C.; Mangino, M.; Penninx, B.; Spector, T.D.; et al. 2SNP heritability and effects of genetic variants for neutrophil-to-lymphocyte and platelet-to-lymphocyte ratio. J. Hum. Genet. 2017, 62, 979–988. [Google Scholar] [CrossRef]
- Roth, K.; Proll-Cornelissen, M.J.; Henne, H.; Appel, A.K.; Schellander, K.; Tholen, E.; Grosse-Brinkhaus, C. Multivariate genome-wide associations for immune traits in two maternal pig lines. BMC Genom. 2023, 24, 492. [Google Scholar] [CrossRef]
- Dauben, C.M.; Proll-Cornelissen, M.J.; Heuss, E.M.; Appel, A.K.; Henne, H.; Roth, K.; Schellander, K.; Tholen, E.; Grosse-Brinkhaus, C. Genome-wide associations for immune traits in two maternal pig lines. BMC Genom. 2021, 22, 717. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Yang, J.A.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Liu, X.L.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z.W. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Huang, M.; Liu, X.L.; Zhou, Y.; Summers, R.M.; Zhang, Z.W. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 2019, 8, giy154. [Google Scholar] [CrossRef]
- Sun, H.; Cheng, Y.; He, Y.; Cheng, C.; Zhao, H.; Yang, S.; Wei, M.; Yang, J.; Liang, S.; Bai, C.; et al. Genome-wide association studies for the number of piglets born alive and dead in Dongliao black pigs. Anim. Genet. 2024, 55, 282–285. [Google Scholar] [CrossRef]
- Zeng, M.; Wang, B.H.; Liu, L.; Yang, Y.L.; Tang, Z.L. Genome-wide association study identifies 12 new genetic loci associated with growth traits in pigs. J. Integr. Agric. 2024, 23, 217–227. [Google Scholar] [CrossRef]
- Wang, J.; Peng, W.; Chen, L.; Kangzhu, Y.; Zhong, J. Assessment of Genomic Prediction Strategies after Animal Genome-Wide Association Study. 2022. [Google Scholar] [CrossRef]
- Adhikari, M.; Kantar, M.B.; Longman, R.J.; Lee, C.N.; Oshiro, M.; Caires, K.; He, Y. Genome-wide association study for carcass weight in pasture-finished beef cattle in Hawai’i. Front. Genet. 2023, 14, 1168150. [Google Scholar] [CrossRef]
- Bai, X.; Yang, T.; Putz, A.M.; Wang, Z.; Li, C.; Fortin, F.; Harding, J.C.S.; Dyck, M.K.; PigGen, C.; Dekkers, J.C.M.; et al. Investigating the genetic architecture of disease resilience in pigs by genome-wide association studies of complete blood count traits collected from a natural disease challenge model. BMC Genom. 2021, 22, 535. [Google Scholar] [CrossRef]
- Dervishi, E.; Bai, X.; Dyck, M.K.; Harding, J.C.S.; Fortin, F.; Dekkers, J.C.M.; Plastow, G. GWAS and genetic and phenotypic correlations of plasma metabolites with complete blood count traits in healthy young pigs reveal implications for pig immune response. Front. Mol. Biosci. 2023, 10, 1140375. [Google Scholar] [CrossRef] [PubMed]
- Uemoto, Y.; Ichinoseki, K.; Matsumoto, T.; Oka, N.; Takamori, H.; Kadowaki, H.; Kojima-Shibata, C.; Suzuki, E.; Okamura, T.; Aso, H.; et al. Genome-wide association studies for production, respiratory disease, and immune-related traits in Landrace pigs. Sci. Rep. 2021, 11, 15823. [Google Scholar] [CrossRef]
- Lu, X.; Fu, W.X.; Luo, Y.R.; Ding, X.D.; Zhou, J.P.; Liu, Y.; Liu, J.F.; Zhang, Q. Genome-wide association study for T lymphocyte subpopulations in swine. BMC Genom. 2012, 13, 488. [Google Scholar] [CrossRef] [PubMed]
- Ballester, M.; Jove-Junca, T.; Pascual, A.; Lopez-Serrano, S.; Crespo-Piazuelo, D.; Hernandez-Banque, C.; Gonzalez-Rodriguez, O.; Ramayo-Caldas, Y.; Quintanilla, R. Genetic architecture of innate and adaptive immune cells in pigs. Front. Immunol. 2023, 14, 1058346. [Google Scholar] [CrossRef] [PubMed]
- Flori, L.; Gao, Y.; Laloë, D.; Lemonnier, G.; Leplat, J.J.; Teillaud, A.; Cossalter, A.M.; Laffitte, J.; Pinton, P.; de Vaureix, C.; et al. Immunity Traits in Pigs: Substantial Genetic Variation and Limited Covariation. PLoS ONE 2011, 6, e22717. [Google Scholar] [CrossRef] [PubMed]
- Clapperton, M.; Diack, A.B.; Matika, O.; Glass, E.J.; Gladney, C.D.; Mellencamp, M.A.; Hoste, A.; Bishop, S.C. Traits associated with innate and adaptive immunity in pigs: Heritability and associations with performance under different health status conditions. Genet. Sel. Evol. 2009, 41, 54. [Google Scholar] [CrossRef] [PubMed]
- Elcheva, I.A.; Gowda, C.P.; Bogush, D.; Gornostaeva, S.; Fakhardo, A.; Sheth, N.; Kokolus, K.M.; Sharma, A.; Dovat, S.; Uzun, Y.; et al. IGF2BP family of RNA-binding proteins regulate innate and adaptive immune responses in cancer cells and tumor microenvironment. Front. Immunol. 2023, 14, 1224516. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, C.H.; Lee, S.W. Exosomal Transmission of MicroRNA from HCV Replicating Cells Stimulates Transdifferentiation in Hepatic Stellate Cells. Mol. Ther. Nucleic Acids 2019, 14, 483–497. [Google Scholar] [CrossRef]
- Chen, J.; Jin, L.; Yan, M.; Yang, Z.; Wang, H.; Geng, S.; Gong, Z.; Liu, G. Serum Exosomes from Newborn Piglets Restrict Porcine Epidemic Diarrhea Virus Infection. J. Proteome Res. 2019, 18, 1939–1947. [Google Scholar] [CrossRef]
- Chen, H.; Chen, J.; Zhao, L.; Song, W.; Xuan, Z.; Chen, J.; Li, Z.; Song, G.; Hong, L.; Song, P.; et al. CDCA5, Transcribed by E2F1, Promotes Oncogenesis by Enhancing Cell Proliferation and Inhibiting Apoptosis via the AKT Pathway in Hepatocellular Carcinoma. J. Cancer 2019, 10, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef]
- Li, H.; Zhang, M.; Zheng, E. Comprehensive miRNA expression profiles in the ilea of Lawsonia intracellularis-infected pigs. J. Vet. Med. Sci. 2017, 79, 282–289. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Morgillo, F. Early use of steroids affects immune cells and impairs immunotherapy efficacy. ESMO Open 2019, 4, e000477. [Google Scholar] [CrossRef]
- Arbour, K.C.; Mezquita, L.; Long, N.; Rizvi, H.; Auclin, E.; Ni, A.; Martinez-Bernal, G.; Ferrara, R.; Lai, W.V.; Hendriks, L.E.L.; et al. Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients With Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2872–2878. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Buxbaum, J.D.; Grice, D.E. SLITRK1 binds 14-3-3 and regulates neurite outgrowth in a phosphorylation-dependent manner. Biol. Psychiatry 2009, 66, 918–925. [Google Scholar] [CrossRef]
- Mosner, M.G.; Kinard, J.L.; Shah, J.S.; McWeeny, S.; Greene, R.K.; Lowery, S.C.; Mazefsky, C.A.; Dichter, G.S. Rates of Co-occurring Psychiatric Disorders in Autism Spectrum Disorder Using the Mini International Neuropsychiatric Interview. J. Autism. Dev. Disord. 2019, 49, 3819–3832. [Google Scholar] [CrossRef]
- Tian, Z.; Ning, C.; Fu, C.; Xu, F.; Zou, C.; Zhu, Q.; Cai, J.; Wang, Y. CALCOCO2 silencing represents a potential molecular therapeutic target for glioma. Arch. Med. Sci. 2020, 16. Available online: https://api.semanticscholar.org/CorpusID:226639886 (accessed on 7 September 2024). [CrossRef]
- Kumthip, K.; Yang, D.; Li, N.L.; Zhang, Y.; Fan, M.; Sethuraman, A.; Li, K. Pivotal role for the ESCRT-II complex subunit EAP30/SNF8 in IRF3-dependent innate antiviral defense. PLoS Pathog. 2017, 13, e1006713. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, G.; Li, L.; Chen, J.; Hao, P.; Gao, Z.; Hao, J.; Xu, Z.; Wang, M.; Li, C.; et al. A novel host restriction factor MRPS6 mediates the inhibition of PDCoV infection in HIEC-6 cells. Front. Immunol. 2024, 15, 1381026. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.Y.; Liao, M.C.; Ai, X.H.; He, G.C. Increased CDCA2 Level Was Related to Poor Prognosis in Hepatocellular Carcinoma and Associated With Up-Regulation of Immune Checkpoints. Front. Med. 2022, 8, 773724. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H.H.; Li, Q.; Wu, F.; Wang, Y.; Wang, Z.Z.; Wang, X.F.; Huang, C. CDCA2 acts as an oncogene and induces proliferation of clear cell renal cell carcinoma cells. Oncol. Lett. 2020, 19, 2466–2474. [Google Scholar] [CrossRef]
- Jin, W.H.; Zhou, A.T.; Chen, J.J.; Cen, Y. CDCA2 promotes proliferation and migration of melanoma by upregulating CCAD1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6858–6863. [Google Scholar]
- Roy, S.; Guler, R.; Parihar, S.P.; Schmeier, S.; Kaczkowski, B.; Nishimura, H.; Shin, J.W.; Negishi, Y.; Ozturk, M.; Hurdayal, R.; et al. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J. Immunol. 2015, 194, 6035–6044. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Wakarchuk, W. O-glycosylation and its role in therapeutic proteins. Biosci. Rep. 2022, 42, BSR20220094. [Google Scholar] [CrossRef]
- Pinho, S.S.; Alves, I.; Gaifem, J.; Rabinovich, G.A. Immune regulatory networks coordinated by glycans and glycan-binding proteins in autoimmunity and infection. Cell. Mol. Immunol. 2023, 20, 1101–1113. [Google Scholar] [CrossRef]
- Derks, M.F.L.; Megens, H.J.; Bosse, M.; Lopes, M.S.; Harlizius, B.; Groenen, M.A.M. A systematic survey to identify lethal recessive variation in highly managed pig populations. BMC Genom. 2017, 18, 858. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.Z.; Liu, Y.; Wang, J.; Fan, H.J.; Fang, H.; Li, S.; Yuan, L.; Liu, C.C.; Peng, Y.; Zhao, W.W.; et al. Histone demethylase KDM7A is required for stem cell maintenance and apoptosis inhibition in breast cancer. J. Cell. Physiol. 2020, 235, 932–943. [Google Scholar] [CrossRef]
- Higashijima, Y.; Matsui, Y.; Shimamura, T.; Nakaki, R.; Nagai, N.; Tsutsumi, S.; Abe, Y.; Link, V.M.; Osaka, M.; Yoshida, M.; et al. Coordinated demethylation of H3K9 and H3K27 is required for rapid inflammatory responses of endothelial cells. EMBO J. 2020, 39, e103949. [Google Scholar] [CrossRef]
- Montesinos, M.D.M.; Pellizas, C.G. Thyroid Hormone Action on Innate Immunity. Front. Endocrinol. 2019, 10, 350. [Google Scholar] [CrossRef]
- De Vito, P.; Incerpi, S.; Pedersen, J.Z.; Luly, P.; Davis, F.B.; Davis, P.J. Thyroid Hormones as Modulators of Immune Activities at the Cellular Level. Thyroid 2011, 21, 879–890. [Google Scholar] [CrossRef] [PubMed]
- van der Spek, A.H.; Fliers, E.; Boelen, A. Thyroid hormone metabolism in innate immune cells. J. Endocrinol. 2017, 232, R67–R81. [Google Scholar] [CrossRef] [PubMed]
- Köllisch, G.; Kalali, B.N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005, 114, 531–541. [Google Scholar] [CrossRef]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Rajput, K.; Ansari, M.N.; Jha, S.K.; Pani, T.; Medatwal, N.; Chattopadhyay, S.; Bajaj, A.; Dasgupta, U. Ceramide Kinase (CERK) Emerges as a Common Therapeutic Target for Triple Positive and Triple Negative Breast Cancer Cells. Cancers 2022, 14, 4496. [Google Scholar] [CrossRef]
- Qing, L.; Yuan, Z. Measurement of immune parameters in growing piglets. J. -Huazhong Agric. Univ. 2001, 20, 561–563. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Tian, X.W.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Zhou, G.L.; Xu, F.J.; Qiao, J.K.; Che, Z.X.; Xiang, T.; Liu, X.L.; Li, X.Y.; Zhao, S.H.; Zhu, M.J. E-GWAS: An ensemble-like GWAS strategy that provides effective control over false positive rates without decreasing true positives. Genet. Sel. Evol. 2023, 55, 46. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Shaffer, J.P. Multiple Hypothesis-Testing. Annu. Rev. Psychol. 1995, 46, 561–584. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Wu, J.M.; Mao, X.Z.; Cai, T.; Luo, J.C.; Wei, L.P. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Trait | 1 ± SE | 2 ± SE | 3 ± 4 SE |
|---|---|---|---|
| MLR | 0.008 ± 0.002 | 0.004 ± 0.001 | 0.649 ± 0.080 |
| NLR | 1.384 ± 0.273 | 0.627 ± 0.118 | 0.688 ± 0.076 |
| SNP | 1 Chr | Position | Model | 2 p-Value |
|---|---|---|---|---|
| INRA0058297 | 2 | 7110358 | MLMA-LOCO | 1.51 × 10−5 |
| BLINK | 2.71 × 10−7 * | |||
| ASGA0022554 | 4 | 116094650 | MLM | 9.41 × 10−6 |
| MLMA-LOCO | 6.07 × 10−6 | |||
| H3GA0032308 | 11 | 54602515 | FarmCPU | 8.25 × 10−7 * |
| BLINK | 2.27 × 10−8 * | |||
| DRGA0011794 | 12 | 25068363 | MLMA-LOCO | 2.28 × 10−5 |
| BLINK | 6.92 × 10−7 * | |||
| ALGA0075031 | 14 | 9571032 | MLMA-LOCO | 2.54 × 10−5 |
| FarmCPU | 2.98 × 10−12 * |
| SNP | 1 Chr | Position | Model | 2 p-Value |
|---|---|---|---|---|
| ASGA0006650 | 1 | 223397796 | MLM | 2.53 × 10−5 |
| MLMA-LOCO | 1.08 × 10−5 | |||
| FarmCPU | 2.21 × 10−6 | |||
| ASGA0023911 | 5 | 2488153 | MLMA-LOCO | 2.47 × 10−5 |
| FarmCPU | 4.67 × 10−6 | |||
| BLINK | 3.50 × 10−8 * | |||
| ASGA0057335 | 13 | 26494790 | MLMA-LOCO | 1.94 × 10−5 |
| FarmCPU | 8.84 × 10−6 | |||
| M1GA0023131 | 18 | 9230153 | MLM | 1.32 × 10−6 * |
| MLMA-LOCO | 8.52 × 10−7 * | |||
| FarmCPU | 2.52 × 10−8 * | |||
| BLINK | 6.60 × 10−9 * |
| Trait | Gene | 1 Chr | 2 Term | Database | ID | 3 p-Value |
|---|---|---|---|---|---|---|
| MLR | SYVN1 | 2 | Ubiquitin mediated proteolysis | KEGG PATHWAY | ssc04120 | 0.014 |
| negative regulation of endoplasmic reticulum stress-induced intrinsic apoptotic signaling pathway | Gene Ontology | GO:1902236 | 0.016 | |||
| TM7SF2 | 2 | Steroid biosynthesis | KEGG PATHWAY | ssc00100 | 0.027 | |
| CDCA5 | 2 | MicroRNAs in cancer | KEGG PATHWAY | ssc05206 | 0.004 | |
| BATF2 | 2 | Endocytosis | KEGG PATHWAY | ssc04144 | 0.041 | |
| myeloid dendritic cell differentiation | Gene Ontology | GO:0043011 | 0.019 | |||
| defense response to protozoan | Gene Ontology | GO:0042832 | 0.020 | |||
| ATG2A | 2 | Autophagy—other | KEGG PATHWAY | ssc04136 | 0.043 | |
| MIR192 | 2 | MicroRNAs in cancer | KEGG PATHWAY | ssc05206 | 0.004 | |
| SLITRK1 | 11 | synaptic membrane adhesion | Gene Ontology | GO:0099560 | 0.027 | |
| CALCOCO2 | 12 | response to interferon-gamma | Gene Ontology | GO:0034341 | 0.023 | |
| SNF8 | 12 | positive regulation of exosomal secretion | Gene Ontology | GO:1903543 | 0.009 | |
| IGF2BP1 | 12 | MicroRNAs in cancer | KEGG PATHWAY | ssc05206 | 0.004 | |
| CDCA2 | 14 | positive regulation of protein dephosphorylation | Gene Ontology | GO:0035307 | 0.021 | |
| NLR | KLF9 | 1 | cellular response to thyroid hormone stimulus | Gene Ontology | GO:0097067 | 0.006 |
| negative regulation of keratinocyte proliferation | Gene Ontology | GO:0010839 | 0.006 | |||
| TBC1D22A | 5 | 14-3-3 protein binding | Gene Ontology | GO:0071889 | 0.006 | |
| CERK | 5 | sphingolipid metabolic process | Gene Ontology | GO:0006665 | 0.009 | |
| GRAMD4 | 5 | positive regulation of cysteine-type endopeptidase activity involved in apoptotic process | Gene Ontology | GO:0043280 | 0.016 | |
| POMGNT2 | 13 | Mannose type O-glycan biosynthesis | KEGG PATHWAY | ssc00515 | 0.013 | |
| KDM7A | 18 | histone demethylase activity (H3-K27 specific) | Gene Ontology | GO:0071558 | 0.003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, J.; Xu, M.; Xu, F.; Che, Z.; Han, P.; Dai, X.; Miao, N.; Zhu, M. Identification of SNPs and Candidate Genes Associated with Monocyte/Lymphocyte Ratio and Neutrophil/Lymphocyte Ratio in Duroc × Erhualian F2 Population. Int. J. Mol. Sci. 2024, 25, 9745. https://doi.org/10.3390/ijms25179745
Qiao J, Xu M, Xu F, Che Z, Han P, Dai X, Miao N, Zhu M. Identification of SNPs and Candidate Genes Associated with Monocyte/Lymphocyte Ratio and Neutrophil/Lymphocyte Ratio in Duroc × Erhualian F2 Population. International Journal of Molecular Sciences. 2024; 25(17):9745. https://doi.org/10.3390/ijms25179745
Chicago/Turabian StyleQiao, Jiakun, Minghang Xu, Fangjun Xu, Zhaoxuan Che, Pingping Han, Xiangyu Dai, Na Miao, and Mengjin Zhu. 2024. "Identification of SNPs and Candidate Genes Associated with Monocyte/Lymphocyte Ratio and Neutrophil/Lymphocyte Ratio in Duroc × Erhualian F2 Population" International Journal of Molecular Sciences 25, no. 17: 9745. https://doi.org/10.3390/ijms25179745