
A Korean Family Presenting with Renal Cysts and Maturity-Onset Diabetes of the Young Caused by a Novel In-Frame Deletion of HNF1B

Abstract
:1. Introduction
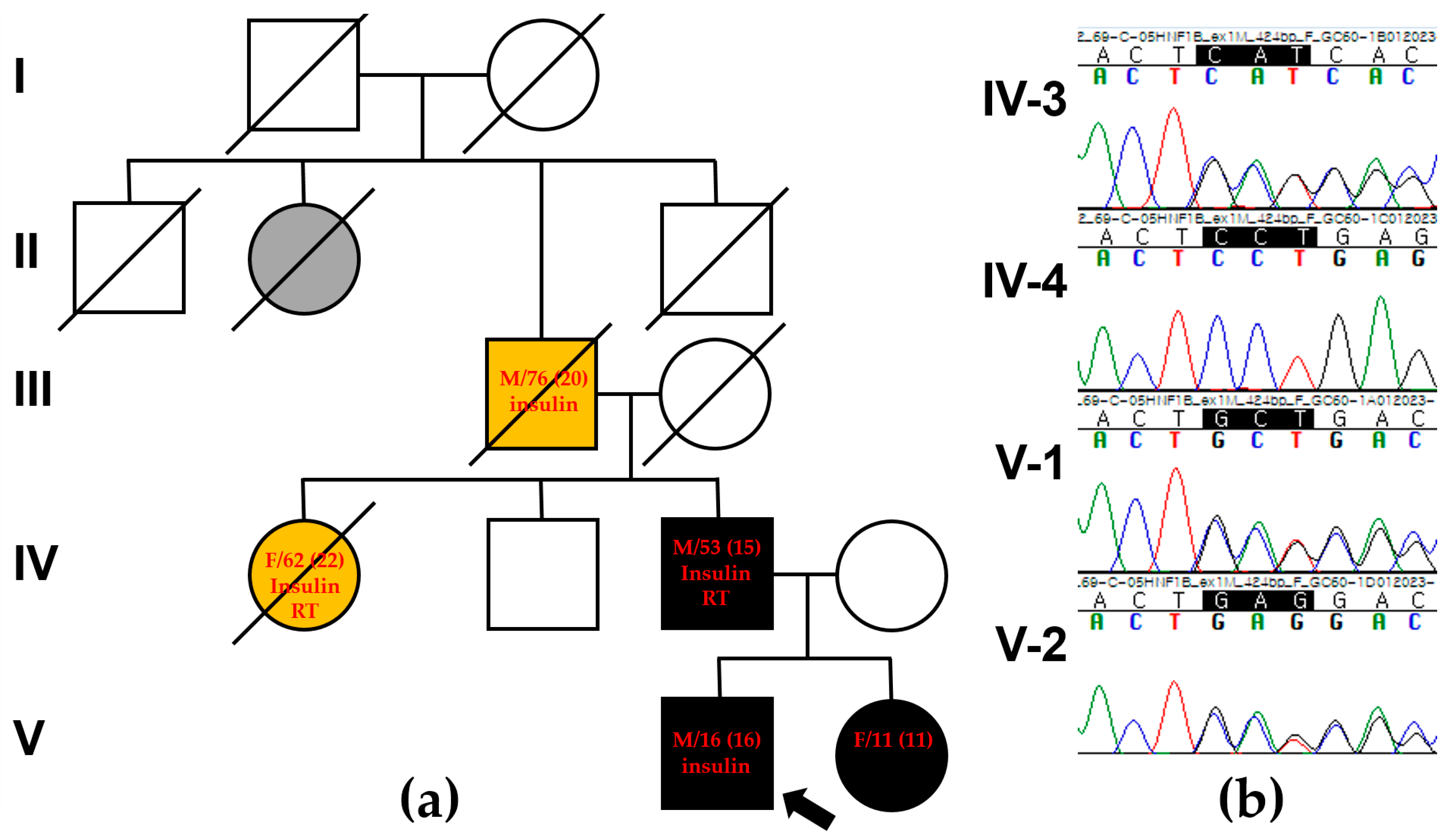
2. Case Presentation
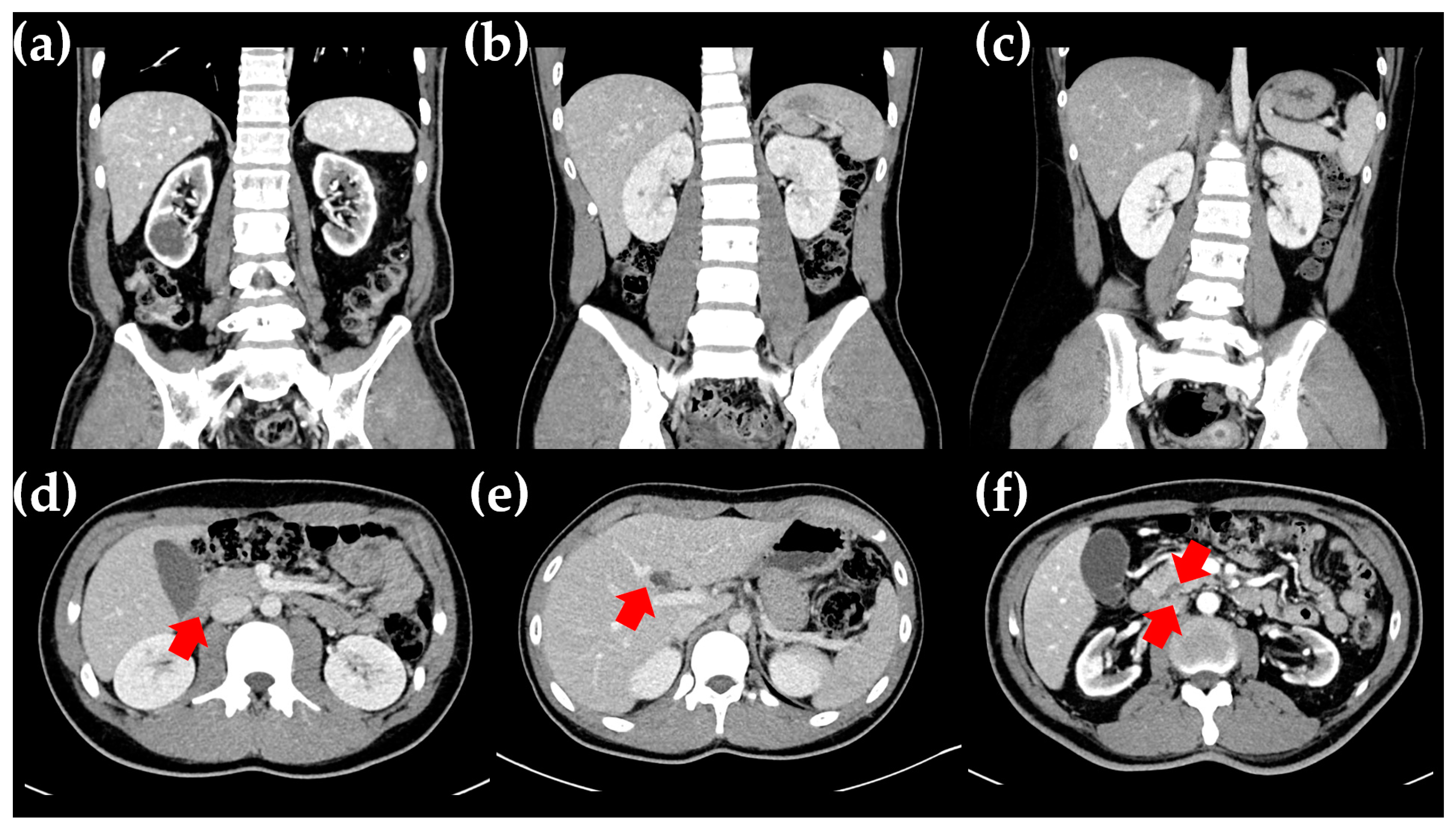
2.1. The Proband
2.2. The Proband’s Father
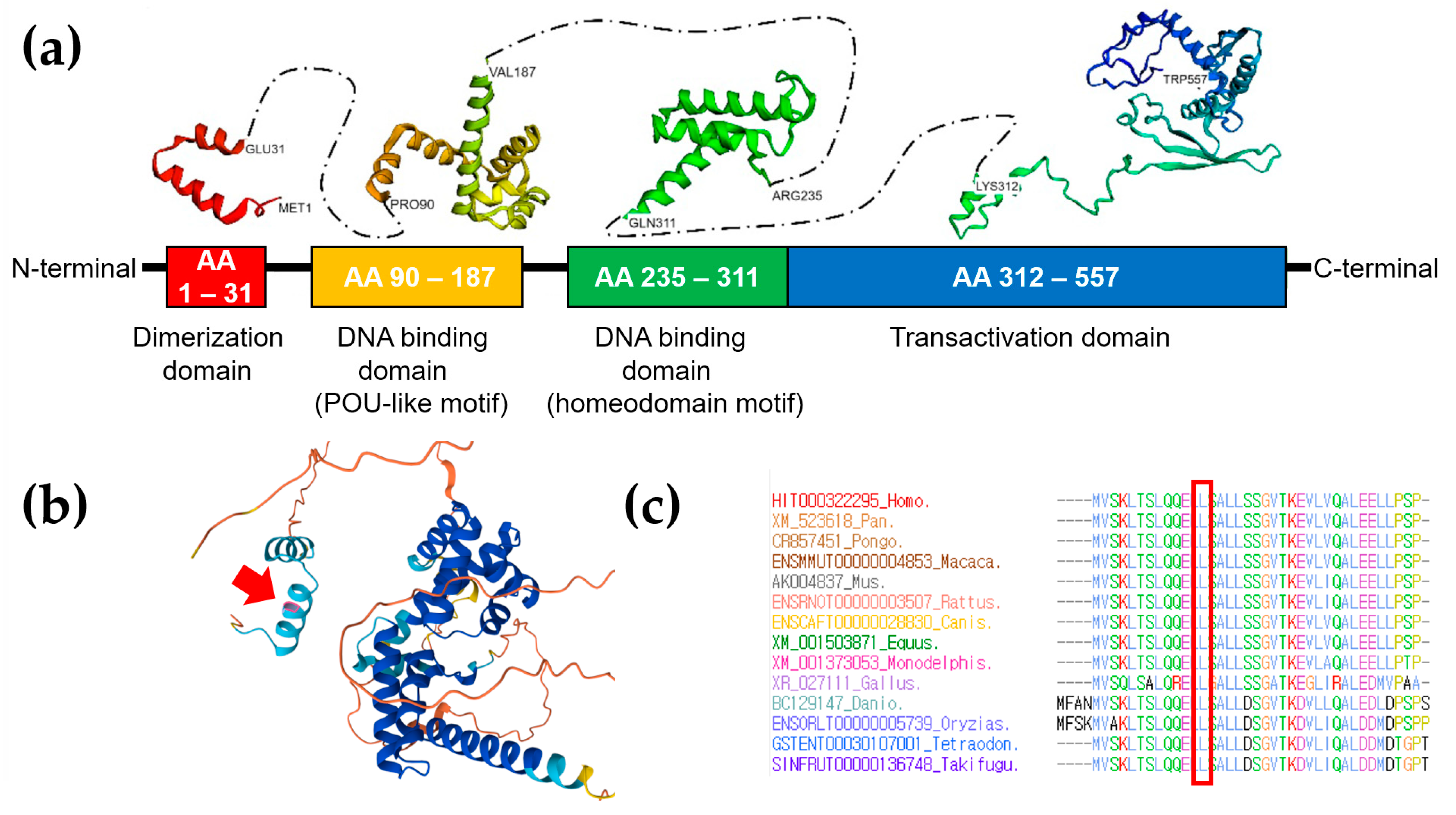
3. Genetic Testing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gardner, D.S.; Tai, E.S. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab. Syndr. Obes. Targets Ther. 2012, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Colclough, K.; Ellard, S.; Hattersley, A.; Patel, K. Syndromic monogenic diabetes genes should be tested in patients with a clinical suspicion of maturity-onset diabetes of the young. Diabetes 2022, 71, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Iwasaki, N.; Hara, M.; Furuta, H.; Hinokio, Y.; Cockburn, B.N.; Lindner, T.; Yamagata, K.; Ogata, M.; Tomonaga, O.; et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat. Genet. 1997, 17, 384–385. [Google Scholar] [CrossRef] [PubMed]
- Clissold, R.L.; Hamilton, A.J.; Hattersley, A.T.; Ellard, S.; Bingham, C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 2015, 11, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). In Clinical Diabetes and Endocrinology; Springer: Cham, Switzerland, 2020; Volume 6, pp. 1–10. [Google Scholar]
- Ulinski, T.; Lescure, S.; Beaufils, S.; Guigonis, V.; Decramer, S.; Morin, D.; Clauin, S.; Desche, G.; Bensman, A.; Bellanné-Chantelot, C. Renal phenotypes related to hepatocyte nuclear factor-1β (TCF2) mutations in a pediatric cohort. J. Am. Soc. Nephrol. 2006, 17, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Faguer, S.; Chassaing, N.; Bandin, F.; Prouheze, C.; Garnier, A.; Casemayou, A.; Huart, A.; Schanstra, J.P.; Calvas, P.; Decramer, S.; et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 2014, 86, 1007–1015. [Google Scholar] [CrossRef]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel). Hum. Mutat. 2016, 37, 28–35. [Google Scholar] [CrossRef]
- Bohn, S.; Thomas, H.; Turan, G.; Ellard, S.; Bingham, C.; Hattersley, A.T.; Ryffel, G.U. Distinct molecular and morphogenetic properties of mutations in the human HNF1β gene that lead to defective kidney development. J. Am. Soc. Nephrol. 2003, 14, 2033–2041. [Google Scholar] [CrossRef]
- Igarashi, P.; Shao, X.; Mcnally, B.T.; Hiesberger, T. Roles of HNF-1β in kidney development and congenital cystic diseases. Kidney Int. 2005, 68, 1944–1947. [Google Scholar] [CrossRef]
- Ferrè, S.; Igarashi, P. New insights into the role of HNF-1β in kidney (patho)physiology. Pediatr. Nephrol. 2019, 34, 1325–1335. [Google Scholar] [CrossRef]
- Çubuk, H.; Yalçın Çapan, Ö. A Review of Functional Characterization of Single Amino Acid Change Mutations in HNF Transcription Factors in MODY Pathogenesis. Protein J. 2021, 40, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Bellanné-Chantelot, C.; Chauveau, D.; Gautier, J.-F.; Dubois-Laforgue, D.; Clauin, S.; Beaufils, S.; Wilhelm, J.-M.; Boitard, C.; Noël, L.-H.; Velho, G. Clinical spectrum associated with hepatocyte nuclear factor-1β mutations. Ann. Intern. Med. 2004, 140, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Edghill, E.L.; Bingham, C.; Ellard, S.; Hattersley, A.T. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J. Med. Genet. 2006, 43, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, G.; Hoyer, J.; Thiel, C.T.; Schaefer, J.; Zapke, M.; Krumbiegel, M.; Kraus, C.; Zweier, M.; Uebe, S.; Ekici, A.B.; et al. Prenatal diagnosis of HNF1B-associated renal cysts: Is there a need to differentiate intragenic variants from 17q12 microdeletion syndrome? Prenat. Diagn. 2019, 39, 1136–1147. [Google Scholar] [CrossRef]
- Barbacci, E.; Reber, M.; Ott, M.-O.; Breillat, C.; Huetz, F.; Cereghini, S. Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development 1999, 126, 4795–4805. [Google Scholar] [CrossRef]
- Cereghini, S.; Ott, M.-O.; Power, S.; Maury, M. Expression patterns of vHNF1 and HNF1 homeoproteins in early postimplantation embryos suggest distinct and sequential developmental roles. Development 1992, 116, 783–797. [Google Scholar] [CrossRef]
- Amed, S.; Oram, R. Maturity-onset diabetes of the young (MODY): Making the right diagnosis to optimize treatment. Can. J. Diabetes 2016, 40, 449–454. [Google Scholar] [CrossRef]
- Tshivhase, A.; Matsha, T.; Raghubeer, S. Diagnosis and treatment of MODY: An updated mini review. Appl. Sci. 2021, 11, 9436. [Google Scholar] [CrossRef]
- Mateus, J.C.; Rivera, C.; O’Meara, M.; Valenzuela, A.; Lizcano, F. Maturity-onset diabetes of the young type 5 a MULTISYSTEMIC disease: A CASE report of a novel mutation in the HNF1B gene and literature review. Clin. Diabetes Endocrinol. 2020, 6, 16. [Google Scholar] [CrossRef]
- Ge, S.; Yang, M.; Cui, Y.; Wu, J.; Xu, L.; Dong, J.; Liao, L. The Clinical Characteristics and Gene Mutations of Maturity-Onset Diabetes of the Young Type 5 in Sixty-One Patients. Front. Endocrinol. 2022, 13, 911526. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Gao, Q.; Zhao, X.-Z.; Chen, Y.-Z.; Craig, L.B.; Xiong, X.-S.; Mei, C.-L.; Shi, Y.-Q.; Chen, X.-M. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin. Med. J. 2010, 123, 3326–3333. [Google Scholar] [PubMed]
- Bellanné-Chantelot, C.; Clauin, S.; Chauveau, D.; Collin, P.; Daumont, M.; Douillard, C.; Dubois-Laforgue, D.; Dusselier, L.; Gautier, J.-F.; Jadoul, M. Large genomic rearrangements in the hepatocyte nuclear factor-1β (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 2005, 54, 3126–3132. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Laforgue, D.; Cornu, E.; Saint-Martin, C.; Coste, J.; Bellanné-Chantelot, C.; Timsit, J. Diabetes, Associated Clinical Spectrum, Long-term Prognosis, and Genotype/Phenotype Correlations in 201 Adult Patients with Hepatocyte Nuclear Factor 1B (HNF1B) Molecular Defects. Diabetes Care 2017, 40, 1436–1443. [Google Scholar] [CrossRef]
- Decramer, S.; Parant, O.; Beaufils, S.; Clauin, S.; Guillou, C.; Kessler, S.; Aziza, J.; Bandin, F.; Schanstra, J.P.; Bellanné-Chantelot, C. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J. Am. Soc. Nephrol. 2007, 18, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Gambella, A.; Kalantari, S.; Cadamuro, M.; Quaglia, M.; Delvecchio, M.; Fabris, L.; Pinon, M. The Landscape of HNF1B Deficiency: A Syndrome Not Yet Fully Explored. Cells 2023, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Türkvatan, A.; Erden, A.; Türkoğlu, M.A.; Yener, Ö. Congenital variants and anomalies of the pancreas and pancreatic duct: Imaging by magnetic resonance cholangiopancreaticography and multidetector computed tomography. Korean J. Radiol. 2013, 14, 905–913. [Google Scholar] [CrossRef]
- Bockenhauer, D.; Jaureguiberry, G. HNF1B-associated clinical phenotypes: The kidney and beyond. Pediatr. Nephrol. 2016, 31, 707–714. [Google Scholar] [CrossRef]
- Sagen, J.V.; Bostad, L.; Njølstad, P.R.; Søvik, O. Enlarged nephrons and severe nondiabetic nephropathy in hepatocyte nuclear factor-1beta (HNF-1beta) mutation carriers. Kidney Int. 2003, 64, 793–800. [Google Scholar] [CrossRef]
- Faguer, S.; Decramer, S.; Chassaing, N.; Bellanné-Chantelot, C.; Calvas, P.; Beaufils, S.; Bessenay, L.; Lengelé, J.P.; Dahan, K.; Ronco, P.; et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011, 80, 768–776. [Google Scholar] [CrossRef]
- Kotalova, R.; Dusatkova, P.; Cinek, O.; Dusatkova, L.; Dedic, T.; Seeman, T.; Lebl, J.; Pruhova, S. Hepatic phenotypes of HNF1B gene mutations: A case of neonatal cholestasis requiring portoenterostomy and literature review. World J. Gastroenterol. 2015, 21, 2550–2557. [Google Scholar] [CrossRef]
- Okorn, C.; Goertz, A.; Vester, U.; Beck, B.B.; Bergmann, C.; Habbig, S.; König, J.; Konrad, M.; Müller, D.; Oh, J. HNF1B nephropathy has a slow-progressive phenotype in childhood—With the exception of very early onset cases: Results of the German Multicenter HNF1B Childhood Registry. Pediatr. Nephrol. 2019, 34, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Bingham, C.; Bulman, M.P.; Ellard, S.; Allen, L.I.; Lipkin, G.W.; van’t Hoff, W.G.; Woolf, A.S.; Rizzoni, G.; Novelli, G.; Nicholls, A.J. Mutations in the hepatocyte nuclear factor-1β gene are associated with familial hypoplastic glomerulocystic kidney disease. Am. J. Hum. Genet. 2001, 68, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Nozu, K.; Goto, Y.; Kamei, K.; Ito, S.; Sato, H.; Emi, M.; Nakanishi, K.; Tsuchiya, S.; Iijima, K. HNF1B alterations associated with congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol. 2010, 25, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Sanna-Cherchi, S.; Warady, B.A.; Furth, S.L.; Kaskel, F.J.; Gharavi, A.G. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr. Nephrol. 2011, 26, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Edghill, E.L.; Stals, K.; Oram, R.A.; Shepherd, M.H.; Hattersley, A.T.; Ellard, S. HNF1B deletions in patients with young-onset diabetes but no known renal disease. Diabet. Med. 2013, 30, 114–117. [Google Scholar] [CrossRef]
- Kolatsi-Joannou, M.; Bingham, C.; Ellard, S.; Bulman, M.P.; Allen, L.I.S.; Hattersley, A.T.; Woolf, A.S. Hepatocyte nuclear factor-1beta: A new kindred with renal cysts and diabetes and gene expression in normal human development. J. Am. Soc. Nephrol. 2001, 12, 2175–2180. [Google Scholar] [CrossRef]
- Verdeguer, F.; Le Corre, S.; Fischer, E.; Callens, C.; Garbay, S.; Doyen, A.; Igarashi, P.; Terzi, F.; Pontoglio, M. A mitotic transcriptional switch in polycystic kidney disease. Nat. Med. 2010, 16, 106–110. [Google Scholar] [CrossRef]
- Hiesberger, T.; Bai, Y.; Shao, X.; McNally, B.T.; Sinclair, A.M.; Tian, X.; Somlo, S.; Igarashi, P. Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J. Clin. Investig. 2004, 113, 814–825. [Google Scholar] [CrossRef]
- Heidet, L.; Decramer, S.; Pawtowski, A.; Moriniere, V.; Bandin, F.; Knebelmann, B.; Lebre, A.-S.; Faguer, S.; Guigonis, V.; Antignac, C. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 2010, 5, 1079. [Google Scholar] [CrossRef]
- Madariaga, L.; Moriniere, V.; Jeanpierre, C.; Bouvier, R.; Loget, P.; Martinovic, J.; Dechelotte, P.; Leporrier, N.; Thauvin-Robinet, C.; Jensen, U.B. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin. J. Am. Soc. Nephrol. 2013, 8, 1179. [Google Scholar] [CrossRef]
- Adalat, S.; Woolf, A.S.; Johnstone, K.A.; Wirsing, A.; Harries, L.W.; Long, D.A.; Hennekam, R.C.; Ledermann, S.E.; Rees, L.; Van’t Hoff, W. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J. Am. Soc. Nephrol. 2009, 20, 1123. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, M.; Shields, B.; Hammersley, S.; Hudson, M.; McDonald, T.J.; Colclough, K.; Oram, R.A.; Knight, B.; Hyde, C.; Cox, J. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the UK pediatric diabetes population with monogenic diabetes. Diabetes Care 2016, 39, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Hansen, L.P.; Graves, M.K.; Conley, P.B.; Crabtree, G.R. HNF-1 alpha and HNF-1 beta (vHNF-1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes. Dev. 1991, 5, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Cereghini, S. Liver-enriched transcription factors and hepatocyte differentiation. FASEB J. 1996, 10, 267–282. [Google Scholar] [CrossRef] [PubMed]
- El-Khairi, R.; Vallier, L. The role of hepatocyte nuclear factor 1β in disease and development. Diabetes Obes. Metab. 2016, 18, 23–32. [Google Scholar] [CrossRef]
- Motyka, R.; Kołbuc, M.; Wierzchołowski, W.; Beck, B.B.; Towpik, I.E.; Zaniew, M. Four cases of maturity onset diabetes of the young (MODY) type 5 associated with mutations in the hepatocyte nuclear factor 1 beta (HNF1B) gene presenting in a 13-year-old boy and in adult men aged 33, 34, and 35 years in Poland. Am. J. Case Rep. 2021, 22, e928994. [Google Scholar] [CrossRef]
- Haldorsen, I.; Vesterhus, M.; Raeder, H.; Jensen, D.; Søvik, O.; Molven, A.; Njølstad, P. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet. Med. 2008, 25, 782–787. [Google Scholar] [CrossRef]
- Pearson, E.R.; Badman, M.K.; Lockwood, C.R.; Clark, P.M.; Ellard, S.; Bingham, C.; Hattersley, A.T. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1α and-1β mutations. Diabetes Care 2004, 27, 1102–1107. [Google Scholar] [CrossRef]
- Hattersley, A.T.; Greeley, S.A.; Polak, M.; Rubio-Cabezas, O.; Njølstad, P.R.; Mlynarski, W.; Castano, L.; Carlsson, A.; Raile, K.; Chi, D.V. ISPAD Clinical Practice Consensus Guidelines 2018: The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr. Diabetes 2018, 19 (Suppl. S27), 47–63. [Google Scholar] [CrossRef]
- Bingham, C.; Ellard, S.; Allen, L.; Bulman, M.; Shepherd, M.; Frayling, T.; Berry, P.J.; Clark, P.M.; Lindner, T.; Bell, G.I. Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1β1: 1See Editorial by Woolf, p. 1202. Kidney Int. 2000, 57, 898–907. [Google Scholar] [CrossRef]
- Amaral, S.; Palha, A.; Bogalho, P.; Silva-Nunes, J. Maturity-onset diabetes of the young secondary to HNF1B variants (HNF1B-MODY): A series of 10 patients from a single diabetes center. Diabetol. Metab. Syndr. 2023, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Maestro, M.A.; Boj, S.F.; Luco, R.F.; Pierreux, C.E.; Cabedo, J.; Servitja, J.M.; German, M.S.; Rousseau, G.G.; Lemaigre, F.P.; Ferrer, J. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum. Mol. Genet. 2003, 12, 3307–3314. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, J.W.; Baitzel, C.; Könner, A.C.; Nicholls, H.T.; Vogt, M.C.; Herrmanns, K.; Scheja, L.; Haumaitre, C.; Wolf, A.M.; Knippschild, U.; et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of HNF1B. Nature 2013, 494, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y. Maturity-onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J. Diabetes Investig. 2018, 9, 704–712. [Google Scholar] [CrossRef]
- Oba, Y.; Sawa, N.; Mizuno, H.; Hoshino, J.; Kinowaki, K.; Ohashi, K.; Morisada, N.; Iijima, K.; Yamaguchi, Y.; Ubara, Y. Autosomal Dominant Tubulointerstitial Kidney Disease HNF1B With Maturity-Onset Diabetes of the Young: A Case Report With Kidney Biopsy. Kidney Med. 2021, 3, 278–281. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Patients | Proband’s Father (VI-3) | Proband (V-1) | Proband’s Sister (V-2) | Reference Ranges | |
|---|---|---|---|---|---|
| Sex/age (year) | M/53 | M/16 | F/11 | ||
| BMI (kg/m2) | 22.5 | 22.6 | 28.4 | 18.5–22.9 | |
| HNF1B score | 14 | 14 | 10 | <8 | |
| Renal cyst | bilateral | bilateral | bilateral | ||
| Pancreatic abnormalities | Diffuse atrophic changes | Agenesis of dorsal pancreas | none | ||
| Genitourinary tract defect | none | none | none | ||
| Laboratory findings at age | at 15 y old | at 50 y old | at 16 y old | at 11 y old | |
| FBS (mg/dL) | 398 | 228 | 301 | 115 | 1–120 |
| HbA1c (%) | 18 | 7.5 | 15.4 | 6.1 | 4.5–5.6 |
| BUN (mg/dL) | 15 | 28.9 | 13.9 | 13.9 | 6–20 |
| Creatinine (mg/dL) | 1 | 5.52 | 1.03 | 0.58 | 0.5–1.2 |
| eGFR (mL/min per 1.73 m2) | 115 | 10.8 | 107 | 141 | 90–120 |
| AST/ALT (IU/L) | 15/21 | 16/14 | 25/12 | 17/15 | 8–40/5–41 |
| Uric acid (mg/dL) | 2.4 | 3.9 | 2 | 6 | 2.4–7 |
| Mg (mg/dL) | 1.5 | 2.3 | 1.6 | 1.8 | 1.6–2.6 |
| Ketone body (mg/L) | 380.10 | 27.1 | 371.30 | 120 | 0–120 |
| Ketone | negative | negative | negative | negative | negative |
| Autoantibodies | negative | n/a | negative | negative | negative |
| Insulin (AC) (uU/mL) | 0.90 | n/a | 0.80 | 1.5 | 1.9–23 |
| Insulin (PC) (uU/mL) | 8 | n/a | 8.5 | 10 | 1.9–23 |
| C-peptide (AC) (ng/mL) | 0.8 | 1.65 | 0.75 | 1.4 | 1.1–4.4 |
| C-peptide (PC) (ng/mL) | 2 | 1.93 | 2.13 | 3.2 | 1.1–4.4 |
| Urine sugar (mg/100mL) | 3+ | 2+ | 3+ | negative | negative |
| Urine protein (mg/100mL) | negative | 2+ | negative | negative | negative |
| Urine albumin/Cr ratio (ug/mg) | 10 | 200 | 6 | 2 | <30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.Y.; Gwack, J.; Kim, T.Y.; Park, J. A Korean Family Presenting with Renal Cysts and Maturity-Onset Diabetes of the Young Caused by a Novel In-Frame Deletion of HNF1B. Int. J. Mol. Sci. 2024, 25, 9823. https://doi.org/10.3390/ijms25189823
Han JY, Gwack J, Kim TY, Park J. A Korean Family Presenting with Renal Cysts and Maturity-Onset Diabetes of the Young Caused by a Novel In-Frame Deletion of HNF1B. International Journal of Molecular Sciences. 2024; 25(18):9823. https://doi.org/10.3390/ijms25189823
Chicago/Turabian StyleHan, Ji Yoon, Jin Gwack, Tae Yun Kim, and Joonhong Park. 2024. "A Korean Family Presenting with Renal Cysts and Maturity-Onset Diabetes of the Young Caused by a Novel In-Frame Deletion of HNF1B" International Journal of Molecular Sciences 25, no. 18: 9823. https://doi.org/10.3390/ijms25189823
APA StyleHan, J. Y., Gwack, J., Kim, T. Y., & Park, J. (2024). A Korean Family Presenting with Renal Cysts and Maturity-Onset Diabetes of the Young Caused by a Novel In-Frame Deletion of HNF1B. International Journal of Molecular Sciences, 25(18), 9823. https://doi.org/10.3390/ijms25189823