
Replicative Endothelial Cell Senescence May Lead to Endothelial Dysfunction by Increasing the BH2/BH4 Ratio Induced by Oxidative Stress, Reducing BH4 Availability, and Decreasing the Expression of eNOS

 ,
,  , , , ,
, , , ,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
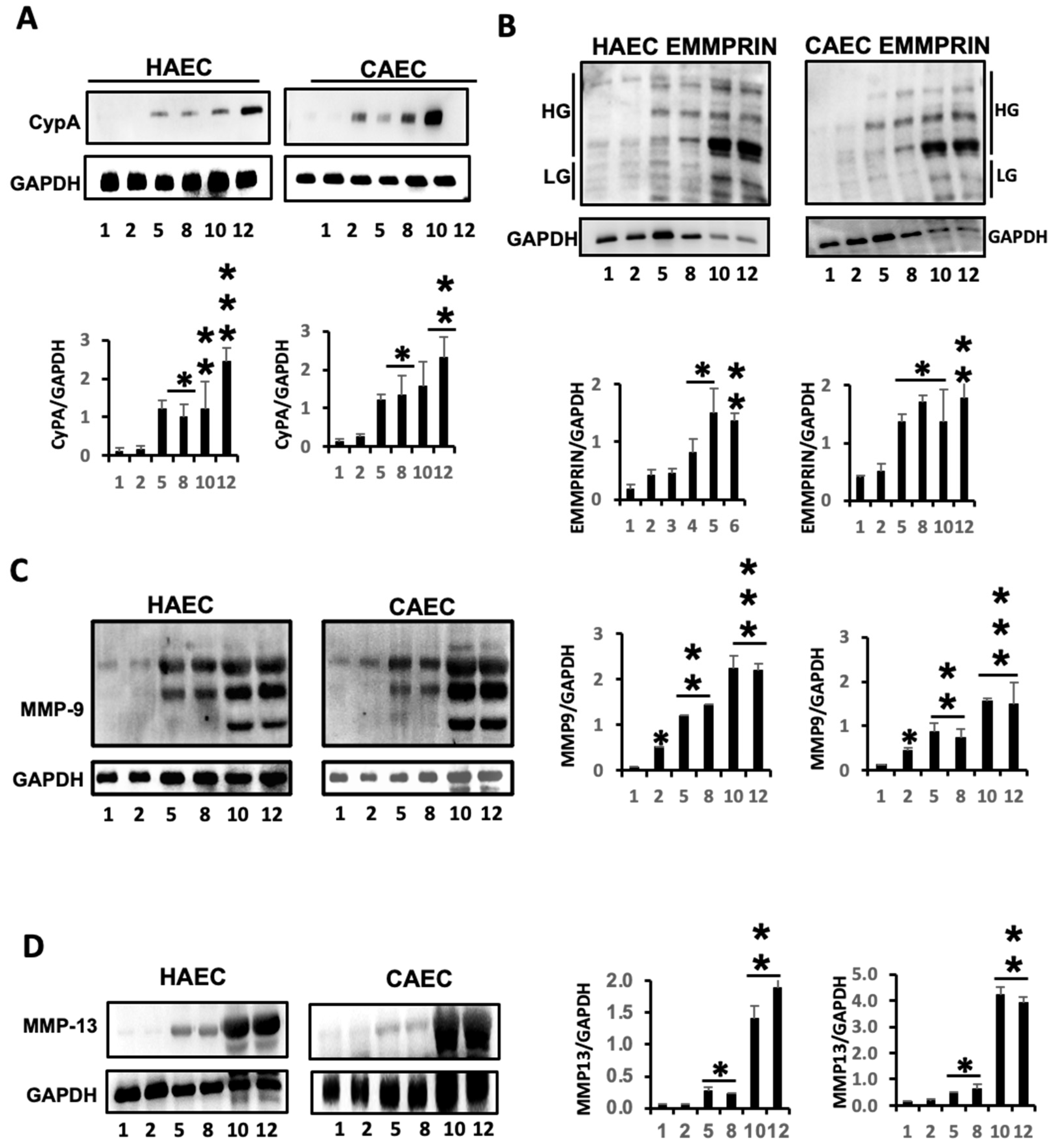
2.1. ES Induced the Expression of Inflammatory Markers in Endothelial Cells
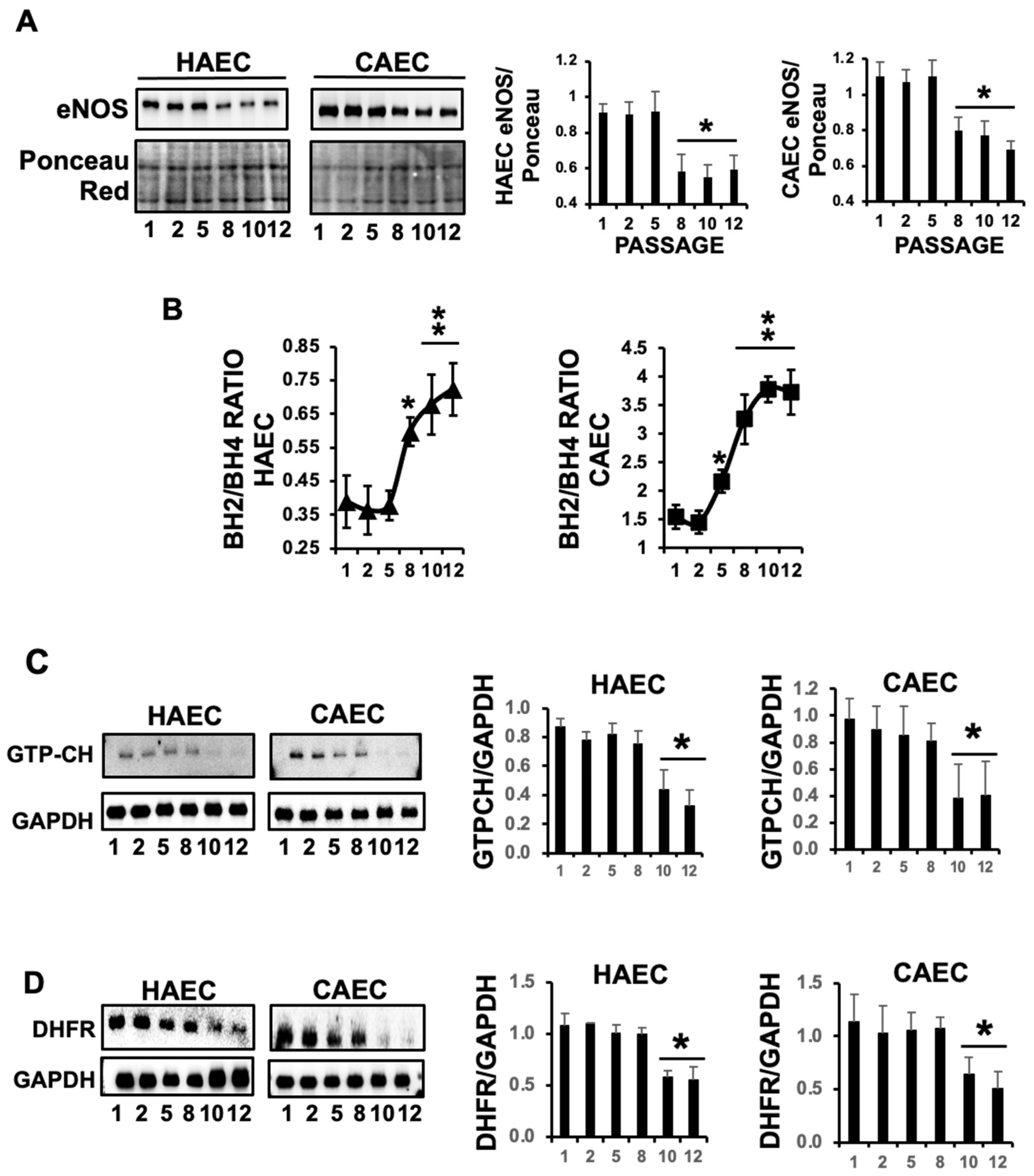
2.2. The Levels of eNOS Were Markedly Reduced in Response to Replicative ES, and Promoted eNOS Cofactor BH4 Deprivation
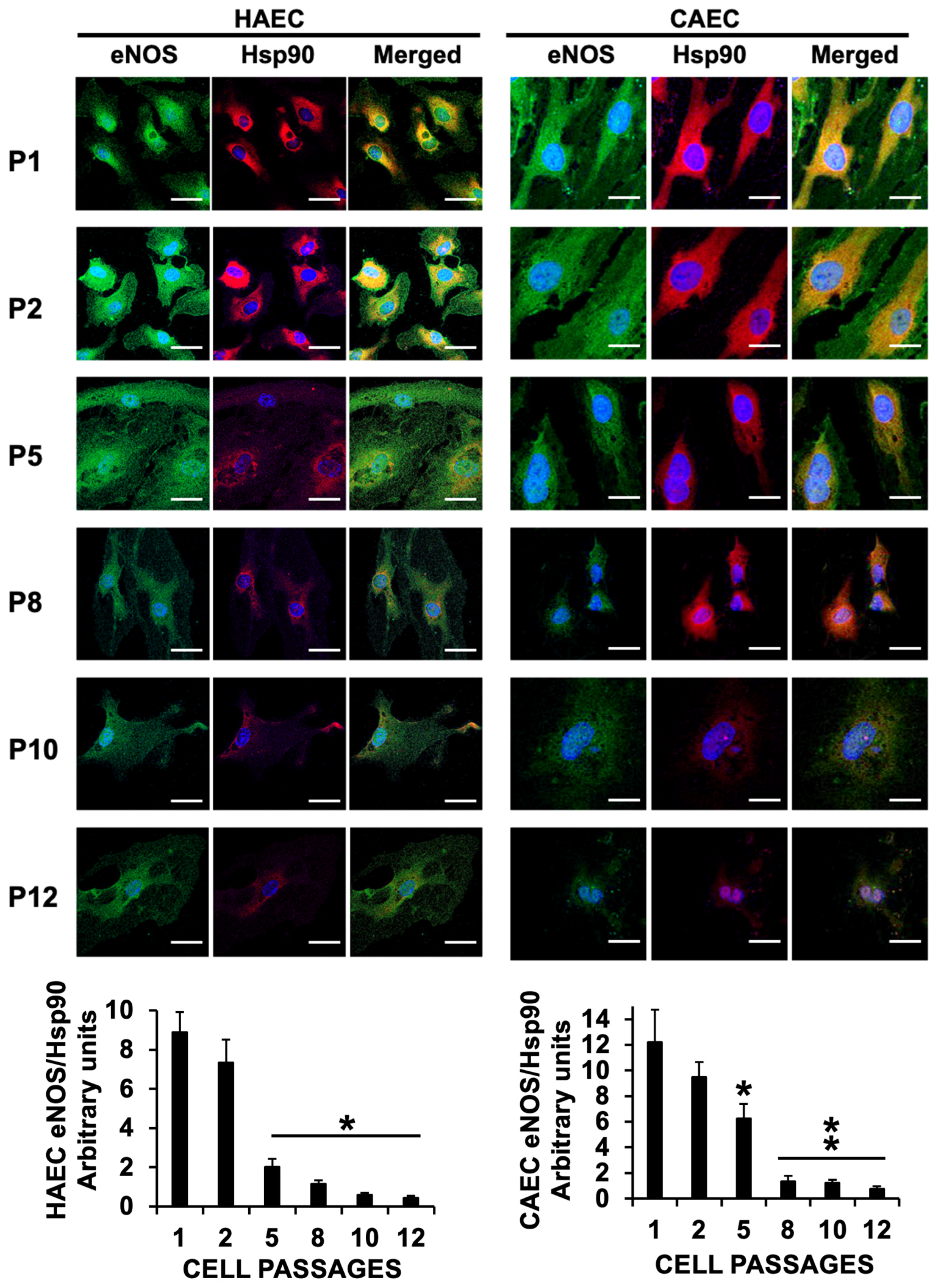
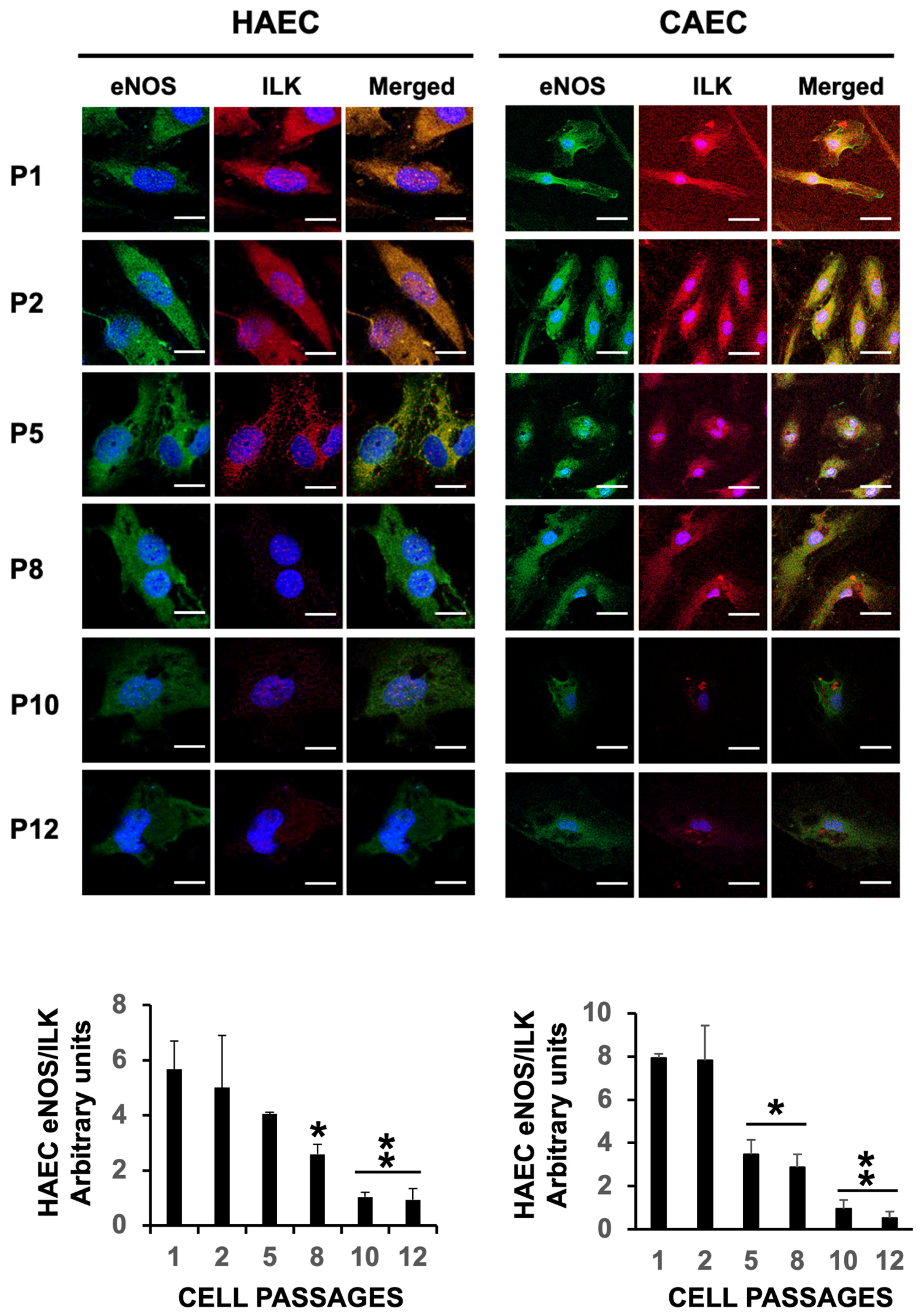
2.3. ES Induced Uncoupling of eNOS in HAEC and CAEC
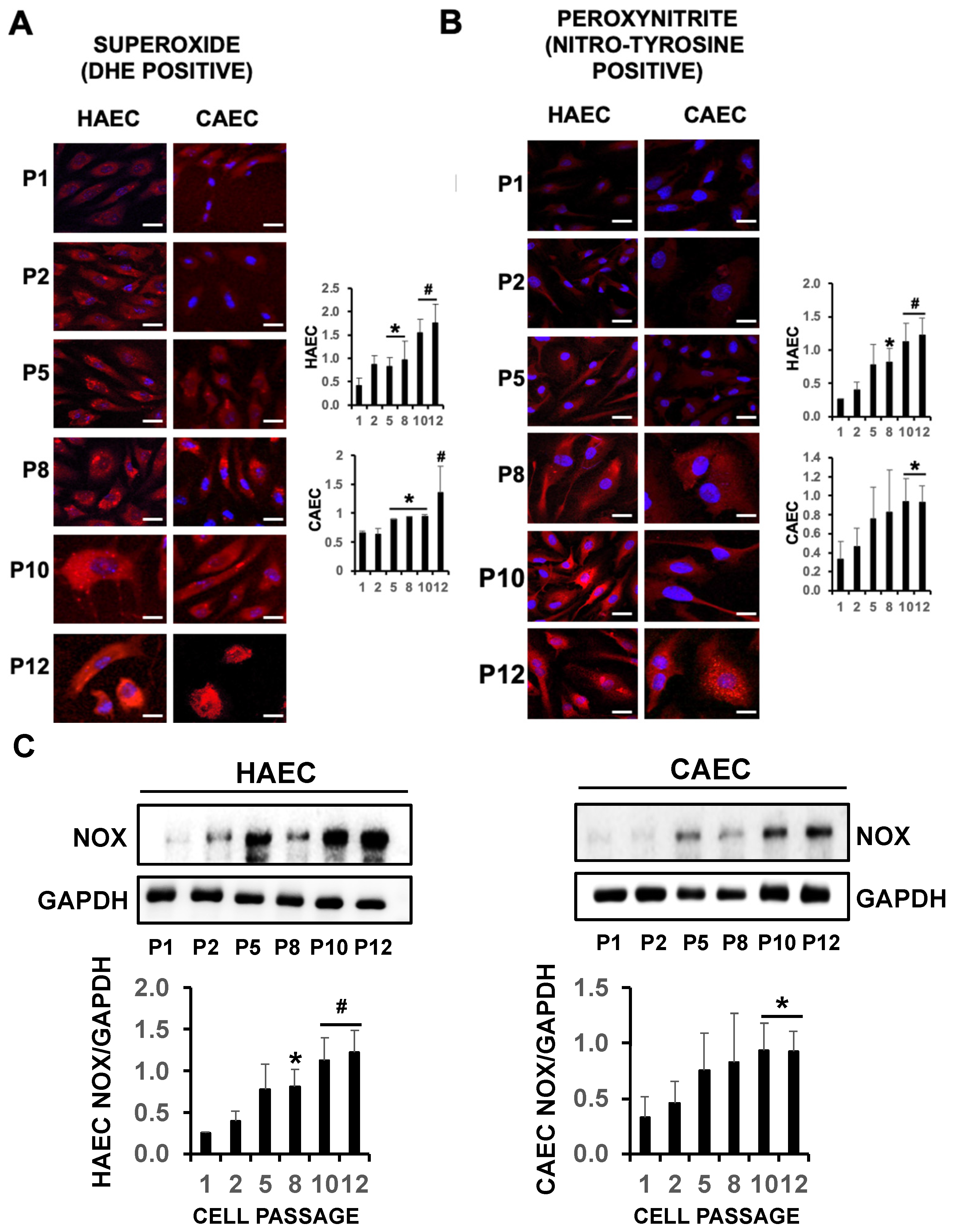
2.4. eNOS Uncoupling Led to Oxidative Stress in Senescent Endothelial Cells
3. Discussion
- -
- Antioxidants like vitamin C, vitamin E, N-acetylcysteine (NAC), or the polyphenol resveratrol can help reduce oxidative stress, improve eNOS function, and increase NO bioavailability [44]. Moreover, ascorbic acid prevents eNOS uncoupling by promoting Ser1177 phosphorylation while preventing Thr455 phosphorylation, which induces the uncoupling of eNOS [45].
- -
- -
- Renin–angiotensin–aldosterone inhibitors. Angiotensin II activates NADPH oxidases through AT1 receptor stimulation. Therefore, drugs interfering with the renin–angiotensin–aldosterone system reduce vascular oxidative stress and improve NO bioactivity. Renin inhibitors like Aliskiren prevent NADPH oxidase and increase the levels of BH4, eNOS, and eNOS activity by the induction of eNOS Ser1177 phosphorylation [48,49]. In this regard, ACE inhibitors also prevent eNOS uncoupling by increasing BH4 levels and decreasing nitrotyrosine. Similarly, aldosterone inhibitors like Eplenorone also reduce NADPH oxidase-mediated BH4 oxidation, thus preventing the uncoupling of eNOS [50].
- -
- -
- Beta blockers. Nebivolol prevents eNOS uncoupling by reducing oxidative stress through NAPH oxidase inhibition [53]. Interestingly, Nebivolol, but no other betablockers, exerts this effect, and hence, further research is required to fully elucidate the precise mechanism of action.
- -
- Beta3-adrenergic agonists like CL316243 is shown to increase eNOS recoupling by reducing oxidative stress and eNOS glutathionylation, a main source of eNOS uncoupling, in pulmonary arterial hypertension [54].
- -
- PDE5 inhibitors including Sildenafil, may prevent eNOS uncoupling by preventing the breakdown of cyclic GMP, which might help in maintaining eNOS function [55].
- -
- Metformin and GLP1 receptor agonists. Pharmacological treatment of diabetes mellitus includes the administration of Metformin and, more recently, GLP1 analogs with significant results. Metformin recoupled eNOS by upregulating GTPCH1 and BH4 levels and attenuated the upregulation of p47-phox [56], while the GLP1 analog liraglutide has shown a strong capacity of eNOS uncoupling prevention by reducing oxidative stress and S-glutathionylation and increasing NO bioavailability [57].
- -
- BH4 and Sepiapterin are effective treatments for cardiovascular conditions, including hypertension, atherosclerosis [58], coronary artery disease [59], diabetes [60], and aging. However, in some cases, the beneficial effect goes beyond NO synthesis [61]. A key limitation of BH4 treatment is the rapid oxidation in the bloodstream, which significantly reduces its effectiveness as a therapeutic tool. To overcome this limitation, others have recently encapsulated BH4 into nanoparticles as a promising tool, showing the efficacy after delivery in NO bioavailability and restoring endothelial function in diabetic rats [62].
- -
- Folic acid has been shown to improve cardiovascular health, at least by inducing BH4-dependent eNOS activation. Therefore, folic acid supplements may be effective in preventing the uncoupling of eNOS in cardiovascular complications [63].
4. Materials and Methods
4.1. Reagents and Equipment
4.2. HAEC and CAEC Cell Culture
4.3. Replicative Senescence Assay: SA-β-Gal Staining
4.4. Confocal Microscopy
4.5. Immunoblotting
4.6. Quantification of BH4 and BH2 Levels
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: Current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 2023, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- López-Rivera, E.; Lizarbe, T.R.; Martínez-Moreno, M.; López-Novoa, J.M.; Rodríguez-Barbero, A.; Rodrigo, J.; Fernández, A.P.; Alvarez-Barrientos, A.; Lamas, S.; Zaragoza, C. Matrix metalloproteinase 13 mediates nitric oxide activation of endothelial cell migration. Proc. Natl. Acad. Sci. USA 2005, 102, 3685–3690. [Google Scholar] [CrossRef]
- Herranz, B.; Marquez, S.; Guijarro, B.; Aracil, E.; Aicart-Ramos, C.; Rodriguez-Crespo, I.; Serrano, I.; Rodríguez-Puyol, M.; Zaragoza, C.; Saura, M. Integrin-linked kinase regulates vasomotor function by preventing endothelial nitric oxide synthase uncoupling: Role in atherosclerosis. Circ. Res. 2012, 110, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- del Nogal, M.; Troyano, N.; Calleros, L.; Griera, M.; Rodriguez-Puyol, M.; Rodriguez-Puyol, D.; Ruiz-Torres, M.P. Hyperosmolarity induced by high glucose promotes senescence in human glomerular mesangial cells. Int. J. Biochem. Cell Biol. 2014, 54, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Sosa, P.; Alcalde-Estevez, E.; Plaza, P.; Troyano, N.; Alonso, C.; Martínez-Arias, L.; Evelem de Melo Aroeira, A.; Rodriguez-Puyol, D.; Olmos, G.; López-Ongil, S.; et al. Hyperphosphatemia Promotes Senescence of Myoblasts by Impairing Autophagy Through Ilk Overexpression, A Possible Mechanism Involved in Sarcopenia. Aging Dis. 2018, 9, 769–784. [Google Scholar] [CrossRef]
- Reventun, P.; Sánchez-Esteban, S.; Cook-Calvete, A.; Delgado-Marín, M.; Roza, C.; Jorquera-Ortega, S.; Hernandez, I.; Tesoro, L.; Botana, L.; Zamorano, J.L.; et al. Endothelial ILK induces cardioprotection by preventing coronary microvascular dysfunction and endothelial-to-mesenchymal transition. Basic Res. Cardiol. 2023, 118, 28. [Google Scholar] [CrossRef]
- Reventun, P.; Alique, M.; Cuadrado, I.; Márquez, S.; Toro, R.; Zaragoza, C.; Saura, M. iNOS-Derived Nitric Oxide Induces Integrin-Linked Kinase Endocytic Lysosome-Mediated Degradation in the Vascular Endothelium. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1272–1281. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: Molecular mechanisms as potential therapeutic targets. Cell. Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Bouly, M.; Bourguignon, M.P.; Roesch, S.; Rigouin, P.; Gosgnach, W.; Bossard, E.; Royere, E.; Diguet, N.; Sansilvestri-Morel, P.; Bonnin, A.; et al. Aging increases circulating BH2 without modifying BH4 levels and impairs peripheral vascular function in healthy adults. Transl. Res. 2021, 238, 36–48. [Google Scholar] [CrossRef]
- Simonet, S.; Gosgnach, W.; Billou, L.; Lucats, L.; Royere, E.; Crespo, C.; Lapret, I.; Ragonnet, L.; Moreau, K.; Vayssettes-Courchay, C.; et al. GTP-cyclohydrolase deficiency induced peripheral and deep microcirculation dysfunction with age. Microvasc. Res. 2021, 133, 104078. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, J.Q.; Dai, X.Y.; Li, J.F.; Gao, F.Y.; Zhang, X.F.; Tian, Y.J.; Shi, W.J.; Zhu, J.B.; Chen, J.K. Endothelium-Specific GTP Cyclohydrolase I Overexpression Restores Endothelial Function in Aged Mice. J. Vasc. Res. 2021, 58, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Drera, A.; Rodella, L.; Brangi, E.; Riccardi, M.; Vizzardi, E. Endothelial Dysfunction in Heart Failure: What Is Its Role? J. Clin. Med. 2024, 13, 2534. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Carracedo, R.; Tesoro, L.; Hernandez, I.; Diez-Mata, J.; Filice, M.; Toro, R.; Rodriguez-Piñero, M.; Zamorano, J.L.; Saura, M.; Zaragoza, C. Non-Invasive Detection of Extracellular Matrix Metalloproteinase Inducer EMMPRIN, a New Therapeutic Target against Atherosclerosis, Inhibited by Endothelial Nitric Oxide. Int. J. Mol. Sci. 2018, 19, 3248. [Google Scholar] [CrossRef]
- Su, Z.; Lin, M.; Zhang, H.; Li, J.; Wu, M.; Lv, H.; Wang, J.; Xie, S. The Release of Cyclophilin A from Rapamycin-Stimulated Vascular Smooth Muscle Cells Mediated by Myosin II Activation: Involvement of Apoptosis but Not Autophagy. J. Vasc. Res. 2020, 57, 254–260. [Google Scholar] [CrossRef]
- Lee, G.H.; Hoang, T.H.; Jung, E.S.; Jung, S.J.; Han, S.K.; Chung, M.J.; Chae, S.W.; Chae, H.J. Anthocyanins attenuate endothelial dysfunction through regulation of uncoupling of nitric oxide synthase in aged rats. Aging Cell. 2020, 19, e13279. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef]
- Diaz-Del Cerro, E.; Martinez de Toda, I.; Félix, J.; Baca, A.; De la Fuente, M. Components of the Glutathione Cycle as Markers of Biological Age: An Approach to Clinical Application in Aging. Antioxidants 2023, 12, 1529. [Google Scholar] [CrossRef]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, H.; Rizzo, A.N.; Zhang, W.; Mai, Y.; Ye, M. Endothelial nitric oxide synthase dimerization is regulated by heat shock protein 90 rather than by phosphorylation. PLoS ONE 2014, 9, e105479. [Google Scholar] [CrossRef] [PubMed]
- Oberhuber, R.; Riede, G.; Cardini, B.; Bernhard, D.; Messner, B.; Watschinger, K.; Steger, C.; Brandacher, G.; Pratschke, J.; Golderer, G.; et al. Impaired Endothelial Nitric Oxide Synthase Homodimer Formation Triggers Development of Transplant Vasculopathy—Insights from a Murine Aortic Transplantation Model. Sci. Rep. 2016, 6, 37917. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.; Unbehaun, A.; Schellenberg, B.; Mayer, B.; Werner-Felmayer, G.; Werner, E.R. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J. Biol. Chem. 2001, 276, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: Implications for uncoupling endothelial nitric oxide synthase. Biochem. Pharmacol. 2005, 70, 343–354. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Sarad, K.; Jankowska, U.; Skupien-Rabian, B.; Babler, A.; Kramann, R.; Dulak, J.; Jaźwa-Kusior, A. Senescence of endothelial cells promotes phenotypic changes in adventitial fibroblasts: Possible implications for vascular aging. Mol. Cell. Biochem. 2024. [Google Scholar] [CrossRef]
- Manzo, O.L.; Nour, J.; Sasset, L.; Marino, A.; Rubinelli, L.; Palikhe, S.; Smimmo, M.; Hu, Y.; Bucci, M.R.; Borczuk, A.; et al. Rewiring Endothelial Sphingolipid Metabolism to Favor S1P Over Ceramide Protects From Coronary Atherosclerosis. Circ. Res. 2024, 134, 990–1005. [Google Scholar] [CrossRef]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef]
- Xia, W.; Zhang, M.; Liu, C.; Wang, S.; Xu, A.; Xia, Z.; Pang, L.; Cai, Y. Exploring the therapeutic potential of tetrahydrobiopterin for heart failure with preserved ejection fraction: A path forward. Life Sci. 2024, 345, 122594. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Settergren, M.; Böhm, F.; Malmström, R.E.; Channon, K.M.; Pernow, J. L-arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis 2009, 204, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Matsuoka, H.; Miyazaki, H.; Usui, M.; Okuda, S.; Imaizumi, T. Tetrahydrobiopterin restores endothelial function in long-term smokers. J. Am. Coll. Cardiol. 2000, 35, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; d’Uscio, L.V.; Nath, K.A. Vascular protection by tetrahydrobiopterin: Progress and therapeutic prospects. Trends Pharmacol. Sci. 2009, 30, 48–54. [Google Scholar] [CrossRef]
- Maglione, M.; Oberhuber, R.; Cardini, B.; Watschinger, K.; Hermann, M.; Obrist, P.; Hengster, P.; Mark, W.; Schneeberger, S.; Werner-Felmayer, G.; et al. Donor pretreatment with tetrahydrobiopterin saves pancreatic isografts from ischemia reperfusion injury in a mouse model. Am. J. Transplant. 2010, 10, 2231–2240. [Google Scholar] [CrossRef]
- Xie, L.; Talukder, M.A.; Sun, J.; Varadharaj, S.; Zweier, J.L. Liposomal tetrahydrobiopterin preserves eNOS coupling in the post-ischemic heart conferring in vivo cardioprotection. J. Mol. Cell. Cardiol. 2015, 86, 14–22. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108–1119. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Ramprasath, T.; Zou, M.H. Oxidative Stress, GTPCH1, and Endothelial Nitric Oxide Synthase Uncoupling in Hypertension. Antioxid. Redox Signal. 2021, 34, 750–764. [Google Scholar] [CrossRef]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.H. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, X.; Yao, Q.; Liu, J.; Tian, Y.; Yang, C.; Xiao, L.; Wang, N. PPARδ agonist prevents endothelial dysfunction via induction of dihydrofolate reductase gene and activation of tetrahydrobiopterin salvage pathway. Br. J. Pharmacol. 2019, 176, 2945–2961. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Tsutsui, M. Nitric oxide synthases in the pathogenesis of cardiovascular disease: Lessons from genetically modified mice. Pflugers Arch. 2010, 459, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Zaragoza, C.; Bao, C.; Herranz, B.; Rodriguez-Puyol, M.; Lowenstein, C.J. Stat3 mediates interleukin-6 [correction of interelukin-6] inhibition of human endothelial nitric-oxide synthase expression. J. Biol. Chem. 2006, 281, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Förstermann, U.; Li, H. Resveratrol and endothelial nitric oxide. Molecules 2014, 19, 16102–16121. [Google Scholar] [CrossRef]
- Ladurner, A.; Schmitt, C.A.; Schachner, D.; Atanasov, A.G.; Werner, E.R.; Dirsch, V.M.; Heiss, E.H. Ascorbate stimulates endothelial nitric oxide synthase enzyme activity by rapid modulation of its phosphorylation status. Free Radic. Biol. Med. 2012, 52, 2082–2090. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Cho, L.; Brennan, D.M.; Hazen, S.L. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J. Am. Coll. Cardiol. 2009, 53, 2061–2067. [Google Scholar] [CrossRef]
- Xiong, Y.; Fru, M.F.; Yu, Y.; Montani, J.P.; Ming, X.F.; Yang, Z. Long term exposure to L-arginine accelerates endothelial cell senescence through arginase-II and S6K1 signaling. Aging 2014, 6, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Tsujioka, H.; Ikejima, H.; Kuroi, A.; Takarada, S.; Kitabata, H.; Tanimoto, T.; Muragaki, Y.; Mochizuki, S.; Goto, M.; et al. Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension 2008, 52, 563–572. [Google Scholar] [CrossRef]
- Nussberger, J.; Aubert, J.F.; Bouzourene, K.; Pellegrin, M.; Hayoz, D.; Mazzolai, L. Renin inhibition by aliskiren prevents atherosclerosis progression: Comparison with irbesartan, atenolol, and amlodipine. Hypertension 2008, 51, 1306–1311. [Google Scholar] [CrossRef]
- Imanishi, T.; Ikejima, H.; Tsujioka, H.; Kuroi, A.; Kobayashi, K.; Muragaki, Y.; Mochizuki, S.; Goto, M.; Yoshida, K.; Akasaka, T. Addition of eplerenone to an angiotensin-converting enzyme inhibitor effectively improves nitric oxide bioavailability. Hypertension 2008, 51, 734–741. [Google Scholar] [CrossRef]
- Hattori, Y.; Nakanishi, N.; Akimoto, K.; Yoshida, M.; Kasai, K. HMG-CoA reductase inhibitor increases GTP cyclohydrolase I mRNA and tetrahydrobiopterin in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.H.; Köhler, T.; Rückschloss, U.; Just, I.; Hecker, M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 61–69. [Google Scholar] [CrossRef]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Karimi Galougahi, K.; Zhang, Y.; Kienzle, V.; Liu, C.C.; Quek, L.E.; Patel, S.; Lau, E.; Cordina, R.L.; Figtree, G.A.; Celermajer, D.S. β3 adrenergic agonism: A novel pathway which improves right ventricular-pulmonary arterial hemodynamics in pulmonary arterial hypertension. Physiol. Rep. 2023, 11, e15549. [Google Scholar] [CrossRef]
- Bivalacqua, T.J.; Musicki, B.; Hsu, L.L.; Berkowitz, D.E.; Champion, H.C.; Burnett, A.L. Sildenafil citrate-restored eNOS and PDE5 regulation in sickle cell mouse penis prevents priapism via control of oxidative/nitrosative stress. PLoS ONE 2013, 8, e68028. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Wei, R.; Ke, J.; Yang, J.; Liu, Y.; Wang, X.; Wang, G.; Hong, T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J. Diabetes Complicat. 2016, 30, 1017–1024. [Google Scholar] [CrossRef]
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor Mediates Cardiovascular Protection by Liraglutide in Mice with Experimental Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Hale, A.B.; Patel, J.; Chuaiphichai, S.; Al Haj Zen, A.; Rashbrook, V.S.; Trelfa, L.; Crabtree, M.J.; McNeill, E.; Channon, K.M. Roles for endothelial cell and macrophage Gch1 and tetrahydrobiopterin in atherosclerosis progression. Cardiovasc. Res. 2018, 114, 1385–1399. [Google Scholar] [CrossRef]
- Sen, A.; Singh, A.; Roy, A.; Mohanty, S.; Naik, N.; Kalaivani, M.; Ramakrishnan, L. Role of endothelial colony forming cells (ECFCs) Tetrahydrobiopterin (BH4) in determining ECFCs functionality in coronary artery disease (CAD) patients. Sci. Rep. 2022, 12, 3076. [Google Scholar] [CrossRef]
- Novoa, U.; Soto, K.; Valdés, C.; Villaseñor, J.; Treuer, A.V.; González, D.R. Tetrahydrobiopterin (BH4) Supplementation Prevents the Cardiorenal Effects of Diabetes in Mice by Reducing Oxidative Stress, Inflammation and Fibrosis. Biomedicines 2022, 10, 2479. [Google Scholar] [CrossRef]
- He, T.; Smith, L.A.; Lu, T.; Joyner, M.J.; Katusic, Z.S. Activation of peroxisome proliferator-activated receptor-δ enhances regenerative capacity of human endothelial progenitor cells by stimulating biosynthesis of tetrahydrobiopterin. Hypertension 2011, 58, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.A.; Heaps, C.L.; Wu, G.; Labhasetwar, V.; Meininger, C.J. Nanoparticle-mediated delivery of tetrahydrobiopterin restores endothelial function in diabetic rats. Nitric Oxide 2024, 148, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wu, Y.; Zhang, Y.; Youn, J.Y.; Cai, H. Combination of folic acid with nifedipine is completely effective in attenuating aortic aneurysm formation as a novel oral medication. Redox Biol. 2022, 58, 102521. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, F.; Contartese, D.; Di Sarno, L.; Borsari, V.; Fini, M.; Giavaresi, G. In Vitro Models of Cell Senescence: A Systematic Review on Musculoskeletal Tissues and Cells. Int. J. Mol. Sci. 2023, 24, 15617. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Navarro, I.; Botana, L.; Diez-Mata, J.; Tesoro, L.; Jimenez-Guirado, B.; Gonzalez-Cucharero, C.; Alcharani, N.; Zamorano, J.L.; Saura, M.; Zaragoza, C. Replicative Endothelial Cell Senescence May Lead to Endothelial Dysfunction by Increasing the BH2/BH4 Ratio Induced by Oxidative Stress, Reducing BH4 Availability, and Decreasing the Expression of eNOS. Int. J. Mol. Sci. 2024, 25, 9890. https://doi.org/10.3390/ijms25189890
Hernandez-Navarro I, Botana L, Diez-Mata J, Tesoro L, Jimenez-Guirado B, Gonzalez-Cucharero C, Alcharani N, Zamorano JL, Saura M, Zaragoza C. Replicative Endothelial Cell Senescence May Lead to Endothelial Dysfunction by Increasing the BH2/BH4 Ratio Induced by Oxidative Stress, Reducing BH4 Availability, and Decreasing the Expression of eNOS. International Journal of Molecular Sciences. 2024; 25(18):9890. https://doi.org/10.3390/ijms25189890
Chicago/Turabian StyleHernandez-Navarro, Ignacio, Laura Botana, Javier Diez-Mata, Laura Tesoro, Beatriz Jimenez-Guirado, Claudia Gonzalez-Cucharero, Nunzio Alcharani, Jose Luis Zamorano, Marta Saura, and Carlos Zaragoza. 2024. "Replicative Endothelial Cell Senescence May Lead to Endothelial Dysfunction by Increasing the BH2/BH4 Ratio Induced by Oxidative Stress, Reducing BH4 Availability, and Decreasing the Expression of eNOS" International Journal of Molecular Sciences 25, no. 18: 9890. https://doi.org/10.3390/ijms25189890
APA StyleHernandez-Navarro, I., Botana, L., Diez-Mata, J., Tesoro, L., Jimenez-Guirado, B., Gonzalez-Cucharero, C., Alcharani, N., Zamorano, J. L., Saura, M., & Zaragoza, C. (2024). Replicative Endothelial Cell Senescence May Lead to Endothelial Dysfunction by Increasing the BH2/BH4 Ratio Induced by Oxidative Stress, Reducing BH4 Availability, and Decreasing the Expression of eNOS. International Journal of Molecular Sciences, 25(18), 9890. https://doi.org/10.3390/ijms25189890