
Cholesterol and Cholesterol-Lowering Medications in COVID-19—An Unresolved Matter



Abstract
:1. Introduction
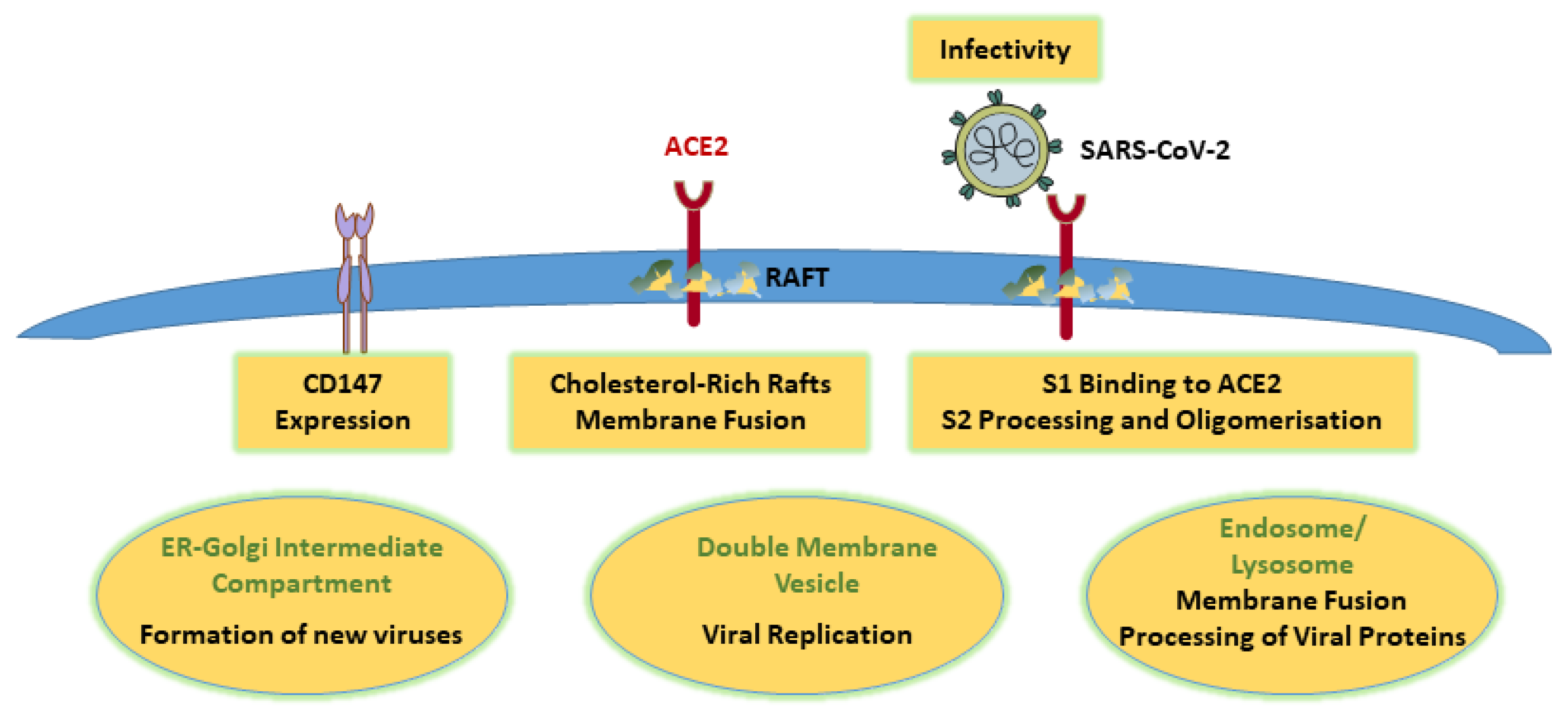
2. The Multiple Roles of Cholesterol for SARS-CoV-2 Infection and Propagation
3. Cholesterol-Containing Lipoproteins and Other Risk Factors Contributing to COVID-19 Severity
4. The Association of Low Plasma Cholesterol Levels with Severe COVID-19
5. Studies Challenging the Association of Low Plasma Cholesterol Levels and COVID-19 Severity
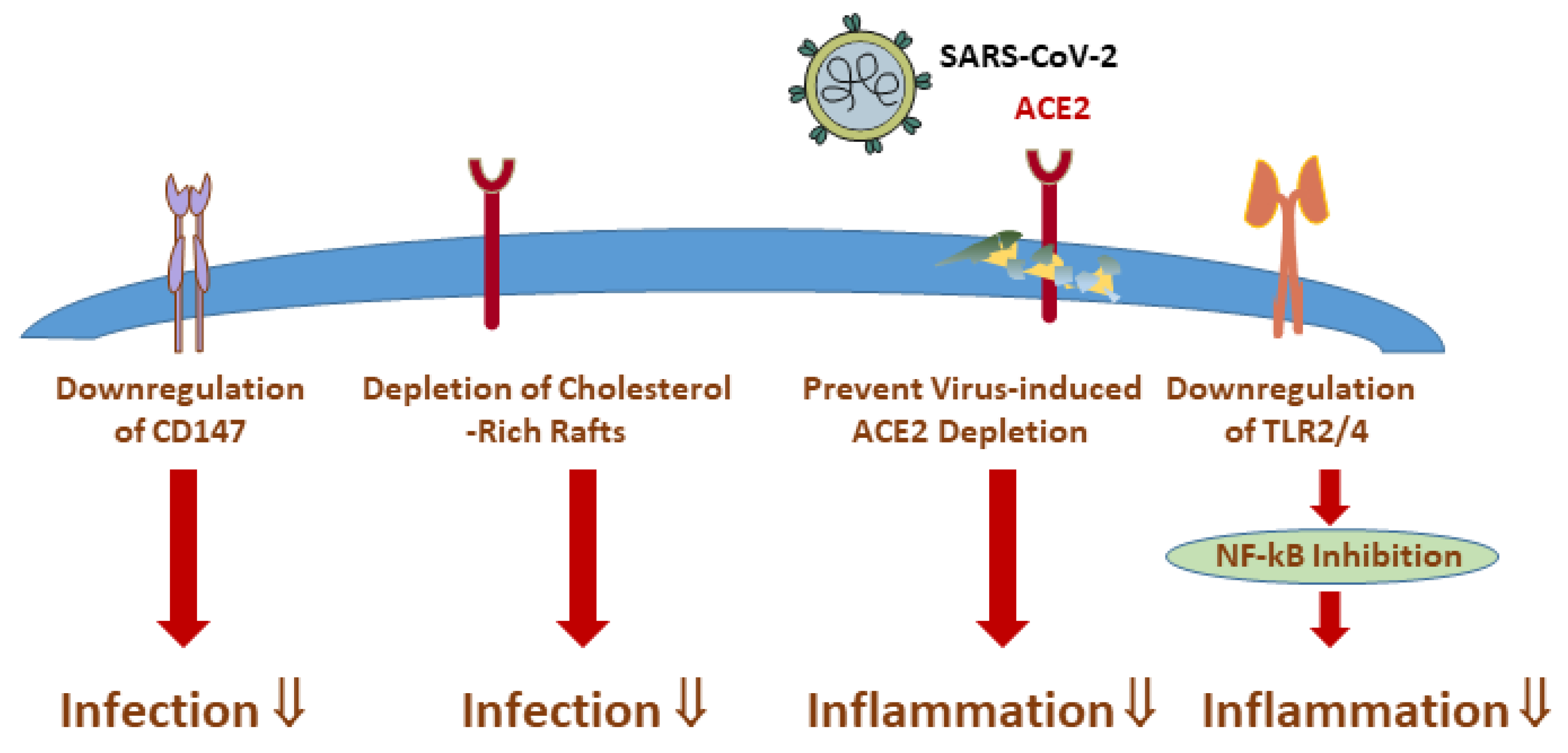
6. Statins Offer Multiple Modes of Action with the Potential to Lower COVID-19 Severity
7. Statins Influence COVID-19 Severity and Outcome
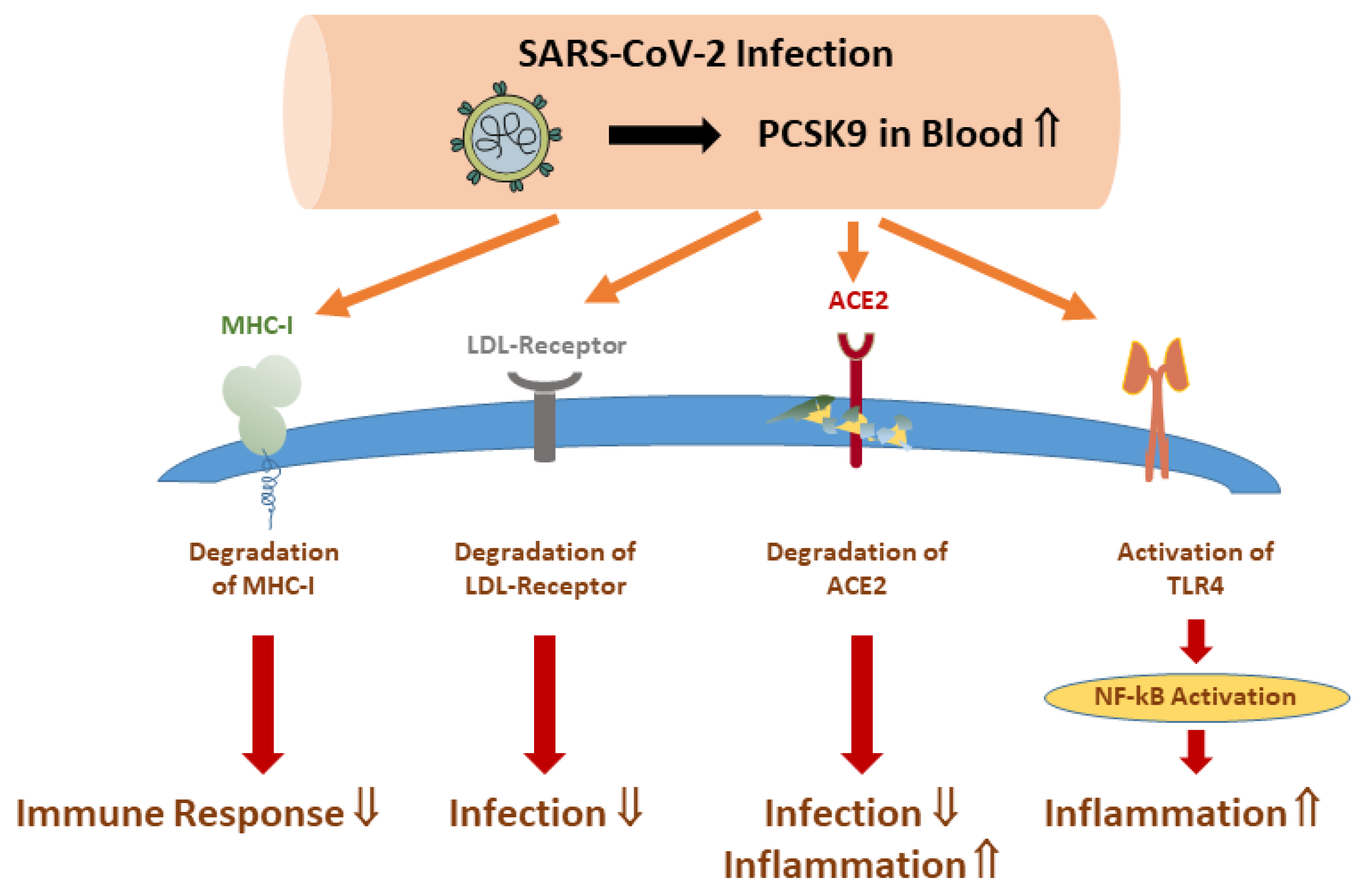
8. PCSK9 and COVID-19 Disease
9. Novel Drugs to Treat Dyslipidaemia and COVID-19 Disease
10. SARS-CoV-2 Infection Modulates Cholesterol-Sensitive Pulmonary Functions
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shappell, C.N.; Klompas, M.; Chan, C.; Chen, T.; Kanjilal, S.; McKenna, C.; Rhee, C.; CDC Prevention Epicenters Program. Use of Electronic Clinical Data to Track Incidence and Mortality for SARS-CoV-2-Associated Sepsis. JAMA Netw. Open 2023, 6, e2335728. [Google Scholar] [CrossRef] [PubMed]
- Cesar-Silva, D.; Pereira-Dutra, F.S.; Moraes Giannini, A.L.; Jacques, G.d.A.C. The Endolysosomal System: The Acid Test for SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 4576. [Google Scholar] [CrossRef]
- Grewal, T.; Nguyen, M.K.L.; Buechler, C. Cholesterol and COVID-19-therapeutic opportunities at the host/virus interface during cell entry. Life Sci. Alliance 2024, 7, e202302453. [Google Scholar] [CrossRef] [PubMed]
- Uppal, S.; Postnikova, O.; Villasmil, R.; Rogozin, I.B.; Bocharov, A.V.; Eggerman, T.L.; Poliakov, E.; Redmond, T.M. Low-Density Lipoprotein Receptor (LDLR) Is Involved in Internalization of Lentiviral Particles Pseudotyped with SARS-CoV-2 Spike Protein in Ocular Cells. Int. J. Mol. Sci. 2023, 24, 11860. [Google Scholar] [CrossRef]
- Sakurai, Y.; Okada, S.; Ozeki, T.; Yoshikawa, R.; Kinoshita, T.; Yasuda, J. SARS-CoV-2 Omicron subvariants progressively adapt to human cells with altered host cell entry. mSphere 2024, 9, e0033824. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, H.; Li, X.; Wang, H. Spike protein mediated membrane fusion during SARS-CoV-2 infection. J. Med. Virol. 2023, 95, e282122023. [Google Scholar] [CrossRef]
- Lim, S.; Zhang, M.; Chang, T.L. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses 2022, 14, 2535. [Google Scholar] [CrossRef]
- Hilser, J.R.; Han, Y.; Biswas, S.; Gukasyan, J.; Cai, Z.; Zhu, R.; Tang, W.H.W.; Deb, A.; Lusis, A.J.; Hartiala, J.A.; et al. Association of serum HDL-cholesterol and apolipoprotein A1 levels with risk of severe SARS-CoV-2 infection. J. Lipid Res. 2021, 62, 100061. [Google Scholar] [CrossRef]
- Scalsky, R.J.; Chen, Y.J.; Desai, K.; O’Connell, J.R.; Perry, J.A.; Hong, C.C. Baseline cardiometabolic profiles and SARS-CoV-2 infection in the UK Biobank. PLoS ONE 2021, 16, e0248602. [Google Scholar] [CrossRef]
- Al Youha, S.; Alowaish, O.; Ibrahim, I.K.; Alghounaim, M.; Abu-Sheasha, G.A.; Fakhra, Z.; Al Hendi, S.; AlQabandi, Y.; Almazeedi, S.; Al Asoomi, F.; et al. Factors associated with SARS-CoV-2 infection amongst healthcare workers in a COVID-19 designated hospital. J. Infect. Public Health 2021, 14, 1226–1232. [Google Scholar] [CrossRef]
- Hariyanto, T.I.; Kurniawan, A. Dyslipidemia is associated with severe coronavirus disease 2019 (COVID-19) infection. Diabetes Metab. Syndr. 2020, 14, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; He, J.; Dong, C.; Li, B.; Ma, Z.; Li, B.; Huang, T.; Fan, J.; He, G.; Zhao, X. Altered Lipid Profile Is a Risk Factor for the Poor Progression of COVID-19: From Two Retrospective Cohorts. Front. Cell Infect. Microbiol. 2021, 11, 712530. [Google Scholar] [CrossRef]
- Barman, H.A.; Pala, A.S.; Dogan, O.; Atici, A.; Yumuk, M.T.; Alici, G.; Sit, O.; Gungor, B.; Dogan, S.M. Prognostic significance of temporal changes of lipid profile in COVID-19 patients. Obes. Med. 2021, 28, 100373. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. The bidirectional interaction of COVID-19 infections and lipoproteins. Best Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101751. [Google Scholar] [CrossRef]
- Wang, G.; Deng, J.; Li, J.; Wu, C.; Dong, H.; Wu, S.; Zhong, Y. The Role of High-Density Lipoprotein in COVID-19. Front. Pharmacol. 2021, 12, 720283. [Google Scholar] [CrossRef]
- Chidambaram, V.; Shanmugavel Geetha, H.; Kumar, A.; Majella, M.G.; Sivakumar, R.K.; Voruganti, D.; Mehta, J.L.; Karakousis, P.C. Association of Lipid Levels With COVID-19 Infection, Disease Severity and Mortality: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 862999. [Google Scholar] [CrossRef]
- Kukla, M.; Menzyk, T.; Dembinski, M.; Winiarski, M.; Garlicki, A.; Bociaga-Jasik, M.; Skonieczna, M.; Hudy, D.; Maziarz, B.; Kusnierz-Cabala, B.; et al. Anti-inflammatory adipokines: Chemerin, vaspin, omentin concentrations and SARS-CoV-2 outcomes. Sci. Rep. 2021, 11, 21514. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Macchi, C.; Iodice, S.; Tersalvi, G.; Rota, I.; Ghidini, S.; Terranova, L.; Valenti, L.; Amati, F.; Aliberti, S.; et al. Prognostic parameters of in-hospital mortality in COVID-19 patients-An Italian experience. Eur. J. Clin. Invest. 2021, 51, e13629. [Google Scholar] [CrossRef]
- Sampedro-Nunez, M.; Aguirre-Moreno, N.; Garcia-Fraile Fraile, L.; Jimenez-Blanco, S.; Knott-Torcal, C.; Sanz-Martin, P.; Fernandez-Jimenez, G.; Marazuela, M. Finding answers in lipid profile in COVID-19 patients. Endocrine 2021, 74, 443–454. [Google Scholar] [CrossRef]
- Bianconi, V.; Mannarino, M.R.; Cosentini, E.; Figorilli, F.; Colangelo, C.; Cellini, G.; Braca, M.; Lombardini, R.; Paltriccia, R.; Sahebkar, A.; et al. The impact of statin therapy on in-hospital prognosis and endothelial function of patients at high-to-very high cardiovascular risk admitted for COVID-19. J. Med. Virol. 2023, 95, e28678. [Google Scholar] [CrossRef]
- Chow, R.; Im, J.; Chiu, N.; Chiu, L.; Aggarwal, R.; Lee, J.; Choi, Y.G.; Prsic, E.H.; Shin, H.J. The protective association between statins use and adverse outcomes among COVID-19 patients: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0253576. [Google Scholar] [CrossRef]
- Lao, U.S.; Law, C.F.; Baptista-Hon, D.T.; Tomlinson, B. Systematic Review and Meta-Analysis of Statin Use and Mortality, Intensive Care Unit Admission and Requirement for Mechanical Ventilation in COVID-19 Patients. J. Clin. Med. 2022, 11, 5454. [Google Scholar] [CrossRef] [PubMed]
- Hariyanto, T.I.; Kurniawan, A. Statin therapy did not improve the in-hospital outcome of coronavirus disease 2019 (COVID-19) infection. Diabetes Metab. Syndr. 2020, 14, 1613–1615. [Google Scholar] [CrossRef]
- Barrantes, F.J. The constellation of cholesterol-dependent processes associated with SARS-CoV-2 infection. Prog. Lipid Res. 2022, 87, 101166. [Google Scholar] [CrossRef]
- Mao, S.; Ren, J.; Xu, Y.; Lin, J.; Pan, C.; Meng, Y.; Xu, N. Studies in the antiviral molecular mechanisms of 25-hydroxycholesterol: Disturbing cholesterol homeostasis and post-translational modification of proteins. Eur. J. Pharmacol. 2022, 926, 175033. [Google Scholar] [CrossRef]
- Palacios-Rapalo, S.N.; De Jesús-González, L.A.; Cordero-Rivera, C.D.; Farfan-Morales, C.N.; Osuna-Ramos, J.F.; Martinez-Mier, G.; Quistián-Galván, J.; Muñoz-Pérez, A.; Bernal-Dolores, V.; del Angel, R.M.; et al. Cholesterol-Rich Lipid Rafts as Platforms for SARS-CoV-2 Entry. Front. Immunol. 2021, 12, 796855. [Google Scholar] [CrossRef]
- Tang, Y.; Hu, L.; Liu, Y.; Zhou, B.; Qin, X.; Ye, J.; Shen, M.; Wu, Z.; Zhang, P. Possible mechanisms of cholesterol elevation aggravating COVID-19. Int. J. Med. Sci. 2021, 18, 3533–3543. [Google Scholar] [CrossRef] [PubMed]
- Makhoul, E.; Aklinski, J.L.; Miller, J.; Leonard, C.; Backer, S.; Kahar, P.; Parmar, M.S.; Khanna, D. A Review of COVID-19 in Relation to Metabolic Syndrome: Obesity, Hypertension, Diabetes, and Dyslipidemia. Cureus 2022, 14, e27438. [Google Scholar] [CrossRef]
- Abou-Hamdan, M.; Saleh, R.; Mani, S.; Dournaud, P.; Metifiot, M.; Blondot, M.L.; Andreola, M.L.; Abdel-Sater, F.; De Reggi, M.; Gressens, P.; et al. Potential antiviral effects of pantethine against SARS-CoV-2. Sci. Rep. 2023, 13, 2237. [Google Scholar] [CrossRef]
- Enrich, C.; Rentero, C.; Hierro, A.; Grewal, T. Role of cholesterol in SNARE-mediated trafficking on intracellular membranes. J. Cell Sci. 2015, 128, 1071–1081. [Google Scholar] [CrossRef]
- Mesmin, B.; Maxfield, F.R. Intracellular sterol dynamics. Biochim. Biophys. Acta 2009, 1791, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Jablonski, S.M.; Jablonski, J.; Hobson, R.; Valente, S.; Reddy, C.B.; Hansen, S.B. The role of high cholesterol in SARS-CoV-2 infectivity. J. Biol. Chem. 2023, 299, 104763. [Google Scholar] [CrossRef]
- Wing, P.A.C.; Schmidt, N.M.; Peters, R.; Erdmann, M.; Brown, R.; Wang, H.; Swadling, L.; Investigators, C.O.; Newman, J.; Thakur, N.; et al. An ACAT inhibitor suppresses SARS-CoV-2 replication and boosts antiviral T cell activity. PLoS Pathog. 2023, 19, e1011323. [Google Scholar] [CrossRef]
- Yuan, Z.; Pavel, M.A.; Wang, H.; Kwachukwu, J.C.; Mediouni, S.; Jablonski, J.A.; Nettles, K.W.; Reddy, C.B.; Valente, S.T.; Hansen, S.B. Hydroxychloroquine blocks SARS-CoV-2 entry into the endocytic pathway in mammalian cell culture. Commun. Biol. 2022, 5, 958. [Google Scholar] [CrossRef]
- Kovacs, T.; Kurtan, K.; Varga, Z.; Nagy, P.; Panyi, G.; Zakany, F. Veklury(R) (remdesivir) formulations inhibit initial membrane-coupled events of SARS-CoV-2 infection due to their sulfobutylether-beta-cyclodextrin content. Br. J. Pharmacol. 2023, 180, 2064–2084. [Google Scholar] [CrossRef]
- Li, G.M.; Li, Y.G.; Yamate, M.; Li, S.M.; Ikuta, K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007, 9, 96–102. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Hui, H.; Tiwari, S.K.; Zhang, Q.; Croker, B.A.; Rawlings, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020, 39, e106057. [Google Scholar] [CrossRef] [PubMed]
- Murae, M.; Sakai, S.; Miyata, N.; Shimizu, Y.; Okemoto-Nakamura, Y.; Kishimoto, T.; Ogawa, M.; Tani, H.; Tanaka, K.; Noguchi, K.; et al. Inhibition Mechanism of SARS-CoV-2 Infection by a Cholesterol Derivative, Nat-20(S)-yne. Biol. Pharm. Bull. 2024, 47, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Kulma, M.; Sakanovic, A.; Bedina-Zavec, A.; Caserman, S.; Omersa, N.; Solinc, G.; Orehek, S.; Hafner-Bratkovic, I.; Kuhar, U.; Slavec, B.; et al. Sequestration of membrane cholesterol by cholesterol-binding proteins inhibits SARS-CoV-2 entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2024, 716, 149954. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, L.; Qin, C.; Huo, F.; Liang, X.; Yang, X.; Zhang, K.; Lin, P.; Liu, J.; Feng, Z.; et al. CD147 contributes to SARS-CoV-2-induced pulmonary fibrosis. Signal Transduct. Target. Ther. 2022, 7, 382. [Google Scholar] [CrossRef]
- Kanyenda, L.J.; Verdile, G.; Martins, R.; Meloni, B.P.; Chieng, J.; Mastaglia, F.; Laws, S.M.; Anderton, R.S.; Boulos, S. Is cholesterol and amyloid-beta stress induced CD147 expression a protective response? Evidence that extracellular cyclophilin a mediated neuroprotection is reliant on CD147. J. Alzheimers Dis. 2014, 39, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Ripa, I.; Andreu, S.; Lopez-Guerrero, J.A.; Bello-Morales, R. Membrane Rafts: Portals for Viral Entry. Front. Microbiol. 2021, 12, 631274. [Google Scholar] [CrossRef]
- Sanders, D.W.; Jumper, C.C.; Ackerman, P.J.; Bracha, D.; Donlic, A.; Kim, H.; Kenney, D.; Castello-Serrano, I.; Suzuki, S.; Tamura, T.; et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. eLife 2021, 10, e65962. [Google Scholar] [CrossRef] [PubMed]
- Meher, G.; Bhattacharjya, S.; Chakraborty, H. Membrane cholesterol regulates the oligomerization and fusogenicity of SARS-CoV fusion peptide: Implications in viral entry. Phys. Chem. Chem. Phys. 2023, 25, 7815–7824. [Google Scholar] [CrossRef] [PubMed]
- Essalmani, R.; Andreo, U.; Evagelidis, A.; Le Devehat, M.; Pereira Ramos, O.H.; Fruchart Gaillard, C.; Susan-Resiga, D.; Cohen, E.A.; Seidah, N.G. SKI-1/S1P Facilitates SARS-CoV-2 Spike Induced Cell-to-Cell Fusion via Activation of SREBP-2 and Metalloproteases, Whereas PCSK9 Enhances the Degradation of ACE2. Viruses 2023, 15, 360. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Schloer, S.; Goretzko, J.; Pleschka, S.; Ludwig, S.; Rescher, U. Combinatory Treatment with Oseltamivir and Itraconazole Targeting Both Virus and Host Factors in Influenza A Virus Infection. Viruses 2020, 12, 703. [Google Scholar] [CrossRef]
- Kuhnl, A.; Musiol, A.; Heitzig, N.; Johnson, D.E.; Ehrhardt, C.; Grewal, T.; Gerke, V.; Ludwig, S.; Rescher, U. Late Endosomal/Lysosomal Cholesterol Accumulation Is a Host Cell-Protective Mechanism Inhibiting Endosomal Escape of Influenza A Virus. mBio 2018, 9, 01345-18. [Google Scholar] [CrossRef]
- Musiol, A.; Gran, S.; Ehrhardt, C.; Ludwig, S.; Grewal, T.; Gerke, V.; Rescher, U. Annexin A6-balanced late endosomal cholesterol controls influenza A replication and propagation. mBio 2013, 4, e00608-13. [Google Scholar] [CrossRef] [PubMed]
- Schloer, S.; Goretzko, J.; Kuhnl, A.; Brunotte, L.; Ludwig, S.; Rescher, U. The clinically licensed antifungal drug itraconazole inhibits influenza virus in vitro and in vivo. Emerg. Microbes Infect. 2019, 8, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bosch, B.J.; Li, F.; Li, W.; Lee, K.H.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006, 281, 3198–3203. [Google Scholar] [CrossRef]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef]
- Datta, G.; Rezagholizadeh, N.; Hasler, W.A.; Khan, N.; Chen, X. SLC38A9 regulates SARS-CoV-2 viral entry. iScience 2024, 27, 110387. [Google Scholar] [CrossRef]
- Ray, L.B. Lysosomal cholesterol activates mTORC1. Science 2017, 355, 1277–1279. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef]
- Elste, J.; Cast, N.; Udawatte, S.; Adhikari, K.; Payen, S.H.; Verma, S.C.; Shukla, D.; Swanson-Mungerson, M.; Tiwari, V. Co-Expression of Niemann-Pick Type C1-Like1 (NPC1L1) with ACE2 Receptor Synergistically Enhances SARS-CoV-2 Entry and Fusion. Biomedicines 2024, 12, 821. [Google Scholar] [CrossRef]
- Sudhop, T.; Reber, M.; Tribble, D.; Sapre, A.; Taggart, W.; Gibbons, P.; Musliner, T.; von Bergmann, K.; Lutjohann, D. Changes in cholesterol absorption and cholesterol synthesis caused by ezetimibe and/or simvastatin in men. J. Lipid Res. 2009, 50, 2117–2123. [Google Scholar] [CrossRef]
- Israel, A.; Schaffer, A.A.; Cicurel, A.; Cheng, K.; Sinha, S.; Schiff, E.; Feldhamer, I.; Tal, A.; Lavie, G.; Ruppin, E. Identification of drugs associated with reduced severity of COVID-19—A case-control study in a large population. Elife 2021, 10, e68165. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Li, P.; Niemeyer, D.; Richter, A.; Pusl, K.; von Brunn, B.; Ru, Y.; Xiang, C.; Schwinghammer, S.; Liu, J.; et al. Oxysterole-binding protein targeted by SARS-CoV-2 viral proteins regulates coronavirus replication. Front. Cell Infect. Microbiol. 2024, 14, 1383917. [Google Scholar] [CrossRef]
- Nagy, P.D.; Strating, J.R.; van Kuppeveld, F.J. Building Viral Replication Organelles: Close Encounters of the Membrane Types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef]
- Muller, C.; Hardt, M.; Schwudke, D.; Neuman, B.W.; Pleschka, S.; Ziebuhr, J. Inhibition of Cytosolic Phospholipase A (2) alpha Impairs an Early Step of Coronavirus Replication in Cell Culture. J. Virol. 2018, 92, e01463-17. [Google Scholar] [CrossRef]
- Cubells, L.; Vila de Muga, S.; Tebar, F.; Bonventre, J.V.; Balsinde, J.; Pol, A.; Grewal, T.; Enrich, C. Annexin A6-induced inhibition of cytoplasmic phospholipase A2 is linked to caveolin-1 export from the Golgi. J. Biol. Chem. 2008, 283, 10174–10183. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Sergeeva, O.; Turelli, P.; Qing, E.; Kunz, B.; Raclot, C.; Paz Montoya, J.; Abriata, L.A.; Gallagher, T.; et al. S-acylation controls SARS-CoV-2 membrane lipid organization and enhances infectivity. Dev. Cell 2021, 56, 2790–2807.e8. [Google Scholar] [CrossRef]
- Lenard, J.; Compans, R.W. The membrane structure of lipid-containing viruses. Biochim. Biophys. Acta 1974, 344, 51–94. [Google Scholar] [CrossRef]
- Barman, S.; Nayak, D.P. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J. Virol. 2007, 81, 12169–12178. [Google Scholar] [CrossRef]
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255. [Google Scholar] [CrossRef]
- Nestel, P.J. Cellular Control of Cholesterol-Metabolism. Pathology 1978, 10, 184. [Google Scholar] [CrossRef]
- Bays, H.E.; Kirkpatrick, C.F.; Maki, K.C.; Toth, P.P.; Morgan, R.T.; Tondt, J.; Christensen, S.M.; Dixon, D.L.; Jacobson, T.A. Obesity, dyslipidemia, and cardiovascular disease: A joint expert review from the Obesity Medicine Association and the National Lipid Association 2024. J. Clin. Lipidol. 2024, 18, e320–e350. [Google Scholar] [CrossRef]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef]
- Hofmaenner, D.A.; Arina, P.; Kleyman, A.; Page Black, L.; Salomao, R.; Tanaka, S.; Guirgis, F.W.; Arulkumaran, N.; Singer, M. Association Between Hypocholesterolemia and Mortality in Critically Ill Patients with Sepsis: A Systematic Review and Meta-Analysis. Crit. Care Explor. 2023, 5, e0860. [Google Scholar] [CrossRef]
- Holven, K.B.; Roeters van Lennep, J. Sex differences in lipids: A life course approach. Atherosclerosis 2023, 384, 117270. [Google Scholar] [CrossRef]
- Massy, Z.A.; de Zeeuw, D. LDL cholesterol in CKD--to treat or not to treat? Kidney Int. 2013, 84, 451–456. [Google Scholar] [CrossRef]
- Luxenburger, H.; Thimme, R. SARS-CoV-2 and the liver: Clinical and immunological features in chronic liver disease. Gut 2023, 72, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E. Infection of liver hepatocytes with SARS-CoV-2. Nat. Metab. 2022, 4, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Bauer, S. ATP binding cassette transporter A1 (ABCA1) associated proteins: Potential drug targets in the metabolic syndrome and atherosclerotic disease? Curr. Pharm. Biotechnol. 2012, 13, 319–330. [Google Scholar] [CrossRef]
- Helou, M.; Nasr, J.; El Osta, N.; Jabbour, E.; Husni, R. Liver manifestations in COVID-19 patients: A review article. World J. Clin. Cases 2023, 11, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver–guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, M.; Yang, H. Cirrhosis is an independent predictor for COVID-19 mortality: A meta-analysis of confounding cofactors-controlled data. J. Hepatol. 2023, 78, e28–e31. [Google Scholar] [CrossRef]
- Oh, J.S.; Choi, J.S.; Lee, Y.H.; Ko, K.O.; Lim, J.W.; Cheon, E.J.; Lee, G.M.; Yoon, J.M. The Relationships between Respiratory Virus Infection and Aminotransferase in Children. Pediatr. Gastroenterol. Hepatol. Nutr. 2016, 19, 243–250. [Google Scholar] [CrossRef]
- Takemura, K.; Board, P.G.; Koga, F. A Systematic Review of Serum gamma-Glutamyltransferase as a Prognostic Biomarker in Patients with Genitourinary Cancer. Antioxidants 2021, 10, 549. [Google Scholar] [CrossRef]
- Moon, A.M.; Barritt, A.S., 4th. Elevated Liver Enzymes in Patients with COVID-19: Look, but Not Too Hard. Dig. Dis. Sci. 2021, 66, 1767–1769. [Google Scholar] [CrossRef]
- Agarwal, A.; Chen, A.; Ravindran, N.; To, C.; Thuluvath, P.J. Gastrointestinal and Liver Manifestations of COVID-19. J. Clin. Exp. Hepatol. 2020, 10, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.G.; Geier, A.; Merle, U. Non-alcoholic fatty liver disease and COVID-19: Harmless companions or disease intensifier? World J. Gastroenterol. 2023, 29, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Ekpanyapong, S.; Reddy, K.R. Liver and Biliary Tract Disease in Patients with Coronavirus disease-2019 Infection. Gastroenterol. Clin. North. Am. 2023, 52, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Anderson, J.L.C.; Gruppen, E.G.; Lei, Y.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. High-Density Lipoprotein Anti-Inflammatory Capacity and Incident Cardiovascular Events. Circulation 2021, 143, 1935–1945. [Google Scholar] [CrossRef]
- Trinder, M.; Walley, K.R.; Boyd, J.H.; Brunham, L.R. Causal Inference for Genetically Determined Levels of High-Density Lipoprotein Cholesterol and Risk of Infectious Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 267–278. [Google Scholar] [CrossRef]
- Grion, C.M.; Cardoso, L.T.; Perazolo, T.F.; Garcia, A.S.; Barbosa, D.S.; Morimoto, H.K.; Matsuo, T.; Carrilho, A.J. Lipoproteins and CETP levels as risk factors for severe sepsis in hospitalized patients. Eur. J. Clin. Invest. 2010, 40, 330–338. [Google Scholar] [CrossRef]
- Stiekema, L.C.A.; Willemsen, L.; Kaiser, Y.; Prange, K.H.M.; Wareham, N.J.; Boekholdt, S.M.; Kuijk, C.; de Winther, M.P.J.; Voermans, C.; Nahrendorf, M.; et al. Impact of cholesterol on proinflammatory monocyte production by the bone marrow. Eur. Heart J. 2021, 42, 4309–4320. [Google Scholar] [CrossRef]
- Al-Banna, N.; Lehmann, C. Oxidized LDL and LOX-1 in experimental sepsis. Mediat. Inflamm. 2013, 2013, 761789. [Google Scholar] [CrossRef]
- Netea, M.G.; Demacker, P.N.; Kullberg, B.J.; Boerman, O.C.; Verschueren, I.; Stalenhoef, A.F.; van der Meer, J.W. Low-density lipoprotein receptor-deficient mice are protected against lethal endotoxemia and severe gram-negative infections. J. Clin. Investig. 1996, 97, 1366–1372. [Google Scholar] [CrossRef]
- Booth, A.; Reed, A.B.; Ponzo, S.; Yassaee, A.; Aral, M.; Plans, D.; Labrique, A.; Mohan, D. Population risk factors for severe disease and mortality in COVID-19: A global systematic review and meta-analysis. PLoS ONE 2021, 16, e0247461. [Google Scholar] [CrossRef] [PubMed]
- Sieurin, J.; Branden, G.; Magnusson, C.; Hergens, M.P.; Kosidou, K. A population-based cohort study of sex and risk of severe outcomes in COVID-19. Eur. J. Epidemiol. 2022, 37, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Scheidt-Nave, C.; Du, Y.; Knopf, H.; Schienkiewitz, A.; Ziese, T.; Nowossadeck, E.; Gösswald, A.; Busch, M.A. Prevalence of dyslipidemia among adults in Germany. Results of the German Health Interview and Examination Survey for Adults (DEGS1). Bundesgesundheitsblatt-Gesundheitsforschung-Gesundheitsschutz 2013, 56, 661–667. [Google Scholar] [CrossRef]
- Guadamuz, J.S.; Shooshtari, A.; Qato, D.M. Global, regional and national trends in statin utilisation in high-income and low/middle-income countries, 2015–2020. BMJ Open 2022, 12, e061350. [Google Scholar] [CrossRef]
- Li, Y.; Ashcroft, T.; Chung, A.; Dighero, I.; Dozier, M.; Horne, M.; McSwiggan, E.; Shamsuddin, A.; Nair, H. Risk factors for poor outcomes in hospitalised COVID-19 patients: A systematic review and meta-analysis. J. Glob. Health 2021, 11, 10001. [Google Scholar] [CrossRef]
- Grewal, T.; Buechler, C. Adipokines as Diagnostic and Prognostic Markers for the Severity of COVID-19. Biomedicines 2023, 11, 1302. [Google Scholar] [CrossRef]
- Atmosudigdo, I.S.; Lim, M.A.; Radi, B.; Henrina, J.; Yonas, E.; Vania, R.; Pranata, R. Dyslipidemia Increases the Risk of Severe COVID-19: A Systematic Review, Meta-analysis, and Meta-regression. Clin. Med. Insights Endocrinol. Diabetes 2021, 14, 1179551421990675. [Google Scholar] [CrossRef]
- Mostaza, J.M.; Salinero-Fort, M.A.; Cardenas-Valladolid, J.; Rodriguez-Artalejo, F.; Diaz-Almiron, M.; Vich-Perez, P.; San Andres-Rebollo, F.J.; Vicente, I.; Lahoz, C. Pre-infection HDL-cholesterol levels and mortality among elderly patients infected with SARS-CoV-2. Atherosclerosis 2022, 341, 13–19. [Google Scholar] [CrossRef]
- Hofmaenner, D.A.; Kleyman, A.; Press, A.; Bauer, M.; Singer, M. The Many Roles of Cholesterol in Sepsis: A Review. Am. J. Respir. Crit. Care Med. 2022, 205, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Van Wyngene, L.; Vandewalle, J.; Libert, C. Reprogramming of basic metabolic pathways in microbial sepsis: Therapeutic targets at last? EMBO Mol. Med. 2018, 10, e8712. [Google Scholar] [CrossRef]
- Zinellu, A.; Paliogiannis, P.; Fois, A.G.; Solidoro, P.; Carru, C.; Mangoni, A.A. Cholesterol and Triglyceride Concentrations, COVID-19 Severity, and Mortality: A Systematic Review and Meta-Analysis with Meta-Regression. Front. Public. Health 2021, 9, 705916. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zeng, W.; Su, J.; Wan, H.; Yu, X.; Cao, X.; Tan, W.; Wang, H. Hypolipidemia is associated with the severity of COVID-19. J. Clin. Lipidol. 2020, 14, 297–304. [Google Scholar] [CrossRef]
- Zhao, M.; Luo, Z.; He, H.; Shen, B.; Liang, J.; Zhang, J.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; et al. Decreased Low-Density Lipoprotein Cholesterol Level Indicates Poor Prognosis of Severe and Critical COVID-19 Patients: A Retrospective, Single-Center Study. Front. Med. 2021, 8, 585851. [Google Scholar] [CrossRef]
- Henriquez-Camacho, C.; Losa, J. Biomarkers for sepsis. Biomed. Res. Int. 2014, 2014, 547818. [Google Scholar] [CrossRef]
- Stasi, A.; Franzin, R.; Fiorentino, M.; Squiccimarro, E.; Castellano, G.; Gesualdo, L. Multifaced Roles of HDL in Sepsis and SARS-CoV-2 Infection: Renal Implications. Int. J. Mol. Sci. 2021, 22, 5980. [Google Scholar] [CrossRef]
- Alkhouli, M.; Nanjundappa, A.; Annie, F.; Bates, M.C.; Bhatt, D.L. Sex Differences in Case Fatality Rate of COVID-19: Insights from a Multinational Registry. Mayo Clin. Proc. 2020, 95, 1613–1620. [Google Scholar] [CrossRef]
- Vahidy, F.S.; Pan, A.P.; Ahnstedt, H.; Munshi, Y.; Choi, H.A.; Tiruneh, Y.; Nasir, K.; Kash, B.A.; Andrieni, J.D.; McCullough, L.D. Sex differences in susceptibility, severity, and outcomes of coronavirus disease 2019: Cross-sectional analysis from a diverse US metropolitan area. PLoS ONE 2021, 16, e0245556. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex differences in lipid and lipoprotein metabolism. Mol. Metab. 2018, 15, 45–55. [Google Scholar] [CrossRef]
- Xu, W.; Guan, H.; Gao, D.; Pan, J.; Wang, Z.; Alam, M.; Lian, J.; Zhou, J. Sex-specific association of monocyte count to high-density lipoprotein ratio with SYNTAX score in patients with suspected stable coronary artery disease. Medicine 2019, 98, e17536. [Google Scholar] [CrossRef]
- Rauschert, S.; Uhl, O.; Koletzko, B.; Mori, T.A.; Beilin, L.J.; Oddy, W.H.; Hellmuth, C. Sex differences in the association of phospholipids with components of the metabolic syndrome in young adults. Biol. Sex. Differ. 2017, 8, 10. [Google Scholar] [CrossRef]
- Peschel, G.; Grimm, J.; Muller, M.; Horing, M.; Krautbauer, S.; Weigand, K.; Liebisch, G.; Buechler, C. Sex-specific changes in triglyceride profiles in liver cirrhosis and hepatitis C virus infection. Lipids Health Dis. 2022, 21, 106. [Google Scholar] [CrossRef]
- Escarcega, R.D.; Honarpisheh, P.; Colpo, G.D.; Ahnstedt, H.W.; Couture, L.; Juneja, S.; Torres, G.; Ortiz, G.J.; Sollome, J.; Tabor, N.; et al. Sex differences in global metabolomic profiles of COVID-19 patients. Cell Death Dis. 2022, 13, 461. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Kano, K.; Hara, M.; Tsukamoto, K.; Aoki, J.; Yatomi, Y. Regulation of plasma glycero-lysophospholipid levels by lipoprotein metabolism. Biochem. J. 2019, 476, 3565–3581. [Google Scholar] [CrossRef]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging roles of lysophospholipids in health and disease. Prog. Lipid Res. 2020, 80, 101068. [Google Scholar] [CrossRef]
- Duong, C.Q.; Bared, S.M.; Abu-Khader, A.; Buechler, C.; Schmitz, A.; Schmitz, G. Expression of the lysophospholipid receptor family and investigation of lysophospholipid-mediated responses in human macrophages. Biochim. Biophys. Acta 2004, 1682, 112–119. [Google Scholar] [CrossRef]
- Graler, M.H.; Goetzl, E.J. Lysophospholipids and their G protein-coupled receptors in inflammation and immunity. Biochim. Biophys. Acta 2002, 1582, 168–174. [Google Scholar] [CrossRef]
- Bozelli, J.C., Jr.; Azher, S.; Epand, R.M. Plasmalogens and Chronic Inflammatory Diseases. Front. Physiol. 2021, 12, 730829. [Google Scholar] [CrossRef]
- Godlewska, U.; Bulanda, E.; Wypych, T.P. Bile acids in immunity: Bidirectional mediators between the host and the microbiota. Front. Immunol. 2022, 13, 949033. [Google Scholar] [CrossRef]
- Valdes, A.; Moreno, L.O.; Rello, S.R.; Orduna, A.; Bernardo, D.; Cifuentes, A. Metabolomics study of COVID-19 patients in four different clinical stages. Sci. Rep. 2022, 12, 1650. [Google Scholar] [CrossRef]
- Huang, X.; Liu, X.; Li, Z. Bile acids and coronavirus disease 2019. Acta. Pharm. Sin. B 2024, 14, 1939–1950. [Google Scholar] [CrossRef]
- Patel, P.N.; Shah, R.Y.; Ferguson, J.F.; Reilly, M.P. Human experimental endotoxemia in modeling the pathophysiology, genomics, and therapeutics of innate immunity in complex cardiometabolic diseases. Arter. Thromb. Vasc. Biol. 2015, 35, 525–534. [Google Scholar] [CrossRef]
- Marcello, A.; Civra, A.; Milan Bonotto, R.; Nascimento Alves, L.; Rajasekharan, S.; Giacobone, C.; Caccia, C.; Cavalli, R.; Adami, M.; Brambilla, P.; et al. The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-CoV-2 and is markedly decreased in COVID-19 patients. Redox Biol. 2020, 36, 101682. [Google Scholar] [CrossRef] [PubMed]
- Saballs, S.M.; Parra, S.; Martinez, N.; Amigo, N.; Cabau, L.; Iftimie, S.; Pavon, R.; Gavaldo, X.; Correig, X.; Paredes, S.; et al. Lipidomic and Metabolomic Changes in Community-acquired and COVID-19 Pneumonia. J. Lipid Res. 2024, 65, 100622. [Google Scholar] [CrossRef]
- Li, G.; Du, L.; Cao, X.; Wei, X.; Jiang, Y.; Lin, Y.; Nguyen, V.; Tan, W.; Wang, H. Follow-up study on serum cholesterol profiles and potential sequelae in recovered COVID-19 patients. BMC Infect. Dis. 2021, 21, 299. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Risks and burdens of incident dyslipidaemia in long COVID: A cohort study. Lancet Diabetes Endocrinol. 2023, 11, 120–128. [Google Scholar] [CrossRef]
- Mietus-Snyder, M.; Suslovic, W.; Delaney, M.; Playford, M.P.; Ballout, R.A.; Barber, J.R.; Otvos, J.D.; DeBiasi, R.L.; Mehta, N.N.; Remaley, A.T. Changes in HDL cholesterol, particles, and function associate with pediatric COVID-19 severity. Front. Cardiovasc. Med. 2022, 9, 1033660. [Google Scholar] [CrossRef]
- Uysal, P.; Yuksel, A.; Durmus, S.; Cuhadaroglu, C.; Gelisgen, R.; Uzun, H. Can circulating oxidative stress-related biomarkers be used as an early prognostic marker for COVID-19? Front. Med. 2023, 10, 1041115. [Google Scholar] [CrossRef]
- Lavis, P.; Morra, S.; Orte Cano, C.; Albayrak, N.; Corbiere, V.; Olislagers, V.; Dauby, N.; Del Marmol, V.; Marchant, A.; Decaestecker, C.; et al. Chemerin plasma levels are increased in COVID-19 patients and are an independent risk factor of mortality. Front. Immunol. 2022, 13, 941663. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, L.; Liang, B. COVID-19 susceptibility and severity for dyslipidemia: A mendelian randomization investigation. Heliyon 2023, 9, e20247. [Google Scholar] [CrossRef]
- Kazenwadel, J.; Berezhnoy, G.; Cannet, C.; Schafer, H.; Geisler, T.; Rohlfing, A.K.; Gawaz, M.; Merle, U.; Trautwein, C. Stratification of hypertension and SARS-CoV-2 infection by quantitative NMR spectroscopy of human blood serum. Commun. Med. 2023, 3, 145. [Google Scholar] [CrossRef]
- Caterino, M.; Gelzo, M.; Sol, S.; Fedele, R.; Annunziata, A.; Calabrese, C.; Fiorentino, G.; D’Abbraccio, M.; Dell’Isola, C.; Fusco, F.M.; et al. Dysregulation of lipid metabolism and pathological inflammation in patients with COVID-19. Sci. Rep. 2021, 11, 2941. [Google Scholar] [CrossRef] [PubMed]
- Mester, P.; Amend, P.; Schmid, S.; Muller, M.; Buechler, C.; Pavel, V. Plasma Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) as a Possible Biomarker for Severe COVID-19. Viruses 2023, 15, 1511. [Google Scholar] [CrossRef]
- Grewal, T.; Buechler, C. Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond. Int. J. Mol. Sci. 2022, 23, 1070. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Aleya, L.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Total Environ. 2022, 808, 152072. [Google Scholar] [CrossRef]
- Bakillah, A.; Hejji, F.A.; Almasaud, A.; Jami, H.A.; Hawwari, A.; Qarni, A.A.; Iqbal, J.; Alharbi, N.K. Lipid Raft Integrity and Cellular Cholesterol Homeostasis Are Critical for SARS-CoV-2 Entry into Cells. Nutrients 2022, 14, 3417. [Google Scholar] [CrossRef] [PubMed]
- Fiore, D.; Proto, M.C.; Franceschelli, S.; Pascale, M.; Bifulco, M.; Gazzerro, P. In Vitro Evidence of Statins’ Protective Role against COVID-19 Hallmarks. Biomedicines 2022, 10, 2123. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.; Temerozo, J.R.; Pereira-Dutra, F.S.; Ferreira, A.C.; Mattos, M.; Goncalves, B.S.; Sacramento, C.Q.; Palhinha, L.; Cunha-Fernandes, T.; Dias, S.S.G.; et al. Simvastatin Downregulates the SARS-CoV-2-Induced Inflammatory Response and Impairs Viral Infection Through Disruption of Lipid Rafts. Front. Immunol. 2022, 13, 820131. [Google Scholar] [CrossRef]
- De Spiegeleer, A.; Bronselaer, A.; Teo, J.T.; Byttebier, G.; De Tre, G.; Belmans, L.; Dobson, R.; Wynendaele, E.; Van De Wiele, C.; Vandaele, F.; et al. The Effects of ARBs, ACEis, and Statins on Clinical Outcomes of COVID-19 Infection Among Nursing Home Residents. J. Am. Med. Dir. Assoc. 2020, 21, 909–914.e2. [Google Scholar] [CrossRef]
- Silhol, F.; Sarlon, G.; Deharo, J.C.; Vaisse, B. Downregulation of ACE2 induces overstimulation of the renin-angiotensin system in COVID-19: Should we block the renin-angiotensin system? Hypertens. Res. 2020, 43, 854–856. [Google Scholar] [CrossRef]
- Sasidhar, M.V.; Chevooru, S.K.; Eickelberg, O.; Hartung, H.P.; Neuhaus, O. Downregulation of monocytic differentiation via modulation of CD147 by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. PLoS ONE 2017, 12, e0189701. [Google Scholar] [CrossRef]
- Zivkovic, S.; Maric, G.; Cvetinovic, N.; Lepojevic-Stefanovic, D.; Bozic Cvijan, B. Anti-Inflammatory Effects of Lipid-Lowering Drugs and Supplements-A Narrative Review. Nutrients 2023, 15, 1517. [Google Scholar] [CrossRef]
- Smaldone, C.; Brugaletta, S.; Pazzano, V.; Liuzzo, G. Immunomodulator activity of 3-hydroxy-3-methilglutaryl-CoA inhibitors. Cardiovasc. Hematol. Agents Med. Chem. 2009, 7, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorff, A.; Muth, H.; Parviz, B.; Staubitz, A.; Haberbosch, W.; Tillmanns, H.; Holschermann, H. Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int. J. Clin. Pharmacol. Ther. 2003, 41, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.B.; Libby, P.; Liao, J.K. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J. Biol. Chem. 1995, 270, 14214–14219. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Hoshi, K.; Ichihara, K. Fluvastatin, an inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase, scavenges free radicals and inhibits lipid peroxidation in rat liver microsomes. Eur. J. Pharmacol. 1998, 361, 143–149. [Google Scholar] [CrossRef]
- Ferrari, F.; Martins, V.M.; Teixeira, M.; Santos, R.D.; Stein, R. COVID-19 and Thromboinflammation: Is There a Role for Statins? Clinics 2021, 76, e2518. [Google Scholar] [CrossRef]
- Reiner, Z.; Hatamipour, M.; Banach, M.; Pirro, M.; Al-Rasadi, K.; Jamialahmadi, T.; Radenkovic, D.; Montecucco, F.; Sahebkar, A. Statins and the COVID-19 main protease: In silico evidence on direct interaction. Arch. Med. Sci. 2020, 16, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Ghosh Dastidar, D.; Roy, K.; Ghosh, A.; Mukhopadhyay, D.; Sikdar, N.; Biswas, N.K.; Chakrabarti, G.; Das, A. Computational prediction of the molecular mechanism of statin group of drugs against SARS-CoV-2 pathogenesis. Sci. Rep. 2022, 12, 6241. [Google Scholar] [CrossRef]
- Chen, Y.; Ku, H.; Zhao, L.; Wheeler, D.C.; Li, L.C.; Li, Q.; Varghese, Z.; Moorhead, J.F.; Powis, S.H.; Huang, A.; et al. Inflammatory stress induces statin resistance by disrupting 3-hydroxy-3-methylglutaryl-CoA reductase feedback regulation. Arter. Thromb. Vasc. Biol. 2014, 34, 365–376. [Google Scholar] [CrossRef]
- Chen, Y.; Ruan, X.Z.; Li, Q.; Huang, A.; Moorhead, J.F.; Powis, S.H.; Varghese, Z. Inflammatory cytokines disrupt LDL-receptor feedback regulation and cause statin resistance: A comparative study in human hepatic cells and mesangial cells. Am. J. Physiol. Ren. Physiol. 2007, 293, F680–F687. [Google Scholar] [CrossRef]
- Ahmadi, Y.; Fard, J.K.; Ghafoor, D.; Eid, A.H.; Sahebkar, A. Paradoxical effects of statins on endothelial and cancer cells: The impact of concentrations. Cancer Cell Int. 2023, 23, 43. [Google Scholar] [CrossRef]
- Hussien, N.R.; Al-Niemi, M.S.; Al-Kuraishy, H.M.; Al-Gareeb, A.I. Statins and COVID-19: The Neglected Front of bidirectional effects. J. Pak. Med. Assoc. 2021, 7 (Suppl. S8), 133–136. [Google Scholar]
- Tani, S.; Takahashi, A.; Nagao, K.; Hirayama, A. Contribution of apolipoprotein A-I to the reduction in high-sensitivity C-reactive protein levels by different statins: Comparative study of pitavastatin and atorvastatin. Heart Vessel. 2015, 30, 762–770. [Google Scholar] [CrossRef]
- Yan, Y.J.; Li, Y.; Lou, B.; Wu, M.P. Beneficial effects of ApoA-I on LPS-induced acute lung injury and endotoxemia in mice. Life Sci. 2006, 79, 210–215. [Google Scholar] [CrossRef]
- Buechler, C.; Pohl, R.; Aslanidis, C. Pro-Resolving Molecules-New Approaches to Treat Sepsis? Int. J. Mol. Sci. 2017, 18, 476. [Google Scholar] [CrossRef]
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol. 2019, 7, 108. [Google Scholar] [CrossRef]
- Shen, L.; Qiu, L.; Wang, L.; Huang, H.; Liu, D.; Xiao, Y.; Liu, Y.; Jin, J.; Liu, X.; Wang, D.W.; et al. Statin Use and In-hospital Mortality in Patients with COVID-19 and Coronary Heart Disease. Sci. Rep. 2021, 11, 23874. [Google Scholar] [CrossRef] [PubMed]
- Sadeghdoust, M.; Aligolighasemabadi, F.; Dehesh, T.; Taefehshokr, N.; Sadeghdoust, A.; Kotfis, K.; Hashemiattar, A.; Ravandi, A.; Aligolighasemabadi, N.; Vakili, O.; et al. The Effects of Statins on Respiratory Symptoms and Pulmonary Fibrosis in COVID-19 Patients with Diabetes Mellitus: A Longitudinal Multicenter Study. Arch. Immunol. Ther. Exp. 2023, 71, 8. [Google Scholar] [CrossRef]
- McAlister, F.A.; Wang, T.; Wang, X.; Chu, A.; Goodman, S.G.; van Diepen, S.; Jackevicius, C.A.; Kaul, P.; Udell, J.; Ko, D.T.; et al. Statins and SARS-CoV-2 Infection: Results of a Population-Based Prospective Cohort Study of 469 749 Adults from 2 Canadian Provinces. J. Am. Heart Assoc. 2021, 10, e022330. [Google Scholar] [CrossRef]
- Bouillon, K.; Baricault, B.; Semenzato, L.; Botton, J.; Bertrand, M.; Drouin, J.; Dray-Spira, R.; Weill, A.; Zureik, M. Association of Statins for Primary Prevention of Cardiovascular Diseases with Hospitalization for COVID-19: A Nationwide Matched Population-Based Cohort Study. J. Am. Heart Assoc. 2022, 11, e023357. [Google Scholar] [CrossRef]
- Golomb, B.A.; Han, J.H.; Langsjoen, P.H.; Dinkeloo, E.; Zemljic-Harpf, A.E. Statin Use in Relation to COVID-19 and Other Respiratory Infections: Muscle and Other Considerations. J. Clin. Med. 2023, 12, 4659. [Google Scholar] [CrossRef] [PubMed]
- Pienkos, S.M.; Moore, A.R.; Guan, J.; Levitt, J.E.; Matthay, M.A.; Baron, R.M.; Conlon, J.; McAuley, D.F.; O’Kane, C.M.; Rogers, A.J. Effect of total cholesterol and statin therapy on mortality in ARDS patients: A secondary analysis of the SAILS and HARP-2 trials. Crit. Care 2023, 27, 126. [Google Scholar] [CrossRef]
- Kim, S.W.; Kang, H.J.; Jhon, M.; Kim, J.W.; Lee, J.Y.; Walker, A.J.; Agustini, B.; Kim, J.M.; Berk, M. Statins and Inflammation: New Therapeutic Opportunities in Psychiatry. Front. Psychiatry 2019, 10, 103. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; Doring, Y.; van der Vorst, E.P.C. PCSK9: A Multi-Faceted Protein That Is Involved in Cardiovascular Biology. Biomedicines 2021, 9, 793. [Google Scholar] [CrossRef]
- Dubuc, G.; Chamberland, A.; Wassef, H.; Davignon, J.; Seidah, N.G.; Bernier, L.; Prat, A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2004, 24, 1454–1459. [Google Scholar] [CrossRef]
- Nozue, T. Lipid Lowering Therapy and Circulating PCSK9 Concentration. J. Atheroscler. Thromb. 2017, 24, 895–907. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Grin, P.M.; Khan, M.; Prat, A.; Zhou, J.; Fox-Robichaud, A.E.; Seidah, N.G.; Liaw, P.C. Differential Expression of PCSK9 Modulates Infection, Inflammation, and Coagulation in a Murine Model of Sepsis. Shock 2016, 46, 672–680. [Google Scholar] [CrossRef]
- Lei, L.; Li, X.; Yuan, Y.J.; Chen, Z.L.; He, J.H.; Wu, J.H.; Cai, X.S. Inhibition of proprotein convertase subtilisin/kexin type 9 attenuates 2,4,6-trinitrobenzenesulfonic acid-induced colitis via repressing toll-like receptor 4/nuclear factor-kappa B. Kaohsiung J. Med. Sci. 2020, 36, 705–711. [Google Scholar] [CrossRef]
- Boyd, J.H.; Fjell, C.D.; Russell, J.A.; Sirounis, D.; Cirstea, M.S.; Walley, K.R. Increased Plasma PCSK9 Levels Are Associated with Reduced Endotoxin Clearance and the Development of Acute Organ Failures during Sepsis. J. Innate Immun. 2016, 8, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.P.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.A.; Fjell, C.D.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar] [CrossRef]
- Mitchell, K.A.; Moore, J.X.; Rosenson, R.S.; Irvin, R.; Guirgis, F.W.; Shapiro, N.; Safford, M.; Wang, H.E. PCSK9 loss-of-function variants and risk of infection and sepsis in the Reasons for Geographic and Racial Differences in Stroke (REGARDS) cohort. PLoS ONE 2019, 14, e0210808. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, W.; Burgner, D.; Tonkin, A.; Zhu, C.; Sun, C.; Magnussen, C.G.; Ernst, M.E.; Breslin, M.; Nicholls, S.J.; et al. The association between PCSK9 inhibitor use and sepsis—A systematic review and meta-analysis of 20 double-blind, randomized, placebo-controlled trials. Am. J. Med. 2023, 136, 558–567. [Google Scholar] [CrossRef]
- Scalise, V.; Sanguinetti, C.; Neri, T.; Cianchetti, S.; Lai, M.; Carnicelli, V.; Celi, A.; Pedrinelli, R. PCSK9 Induces Tissue Factor Expression by Activation of TLR4/NFkB Signaling. Int. J. Mol. Sci. 2021, 22, 12640. [Google Scholar] [CrossRef]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef]
- Moriyama, M.; Lucas, C.; Monteiro, V.S.; Yale SARS-CoV-2 Genomic Surveillance Initiative; Iwasaki, A. Enhanced inhibition of MHC-I expression by SARS-CoV-2 Omicron subvariants. Proc. Natl. Acad. Sci. USA 2023, 120, e2221652120. [Google Scholar] [CrossRef]
- Navarese, E.P.; Podhajski, P.; Gurbel, P.A.; Grzelakowska, K.; Ruscio, E.; Tantry, U.; Magielski, P.; Kubica, A.; Niezgoda, P.; Adamski, P.; et al. PCSK9 Inhibition During the Inflammatory Stage of SARS-CoV-2 Infection. J. Am. Coll. Cardiol. 2023, 81, 224–234. [Google Scholar] [CrossRef]
- Elahi, R.; Hozhabri, S.; Moradi, A.; Siahmansouri, A.; Jahani Maleki, A.; Esmaeilzadeh, A. Targeting the cGAS-STING pathway as an inflammatory crossroad in coronavirus disease 2019 (COVID-19). Immunopharmacol. Immunotoxicol. 2023, 45, 639–649. [Google Scholar] [CrossRef]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct. Target. Ther. 2020, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Mester, P.; Amend, P.; Schmid, S.; Wenzel, J.J.; Horing, M.; Liebisch, G.; Krautbauer, S.; Muller, M.; Buechler, C.; Pavel, V. Proprotein Convertase Subtilisin/Kexin Type 9 Induction in COVID-19 Is Poorly Associated with Disease Severity and Cholesterol Levels. Infect. Dis. Rep. 2024, 16, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Xiao, J.; Ji, J.; Chen, L. Association of lipid-lowering drugs with COVID-19 outcomes from a Mendelian randomization study. Elife 2021, 10, e73873. [Google Scholar] [CrossRef]
- Mercep, I.; Strikic, D.; Sliskovic, A.M.; Reiner, Z. New Therapeutic Approaches in Treatment of Dyslipidaemia–A Narrative Review. Pharmaceuticals 2022, 15, 839. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Lincoff, A.M.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. New Engl. J. Med. 2023, 388, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Bloedon, L.T.; Lin, G.; Powell, H.A.; Louie, M.J.; Nicholls, S.J.; Lincoff, A.M.; Nissen, S.E. Safety of bempedoic acid in patients at high cardiovascular risk and with statin intolerance. J. Clin. Lipidol. 2024, 18, e59–e69. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; Mainardi, A.; d’Alessandro, M.; Cameli, P.; Bennett, D.; Bargagli, E.; Sestini, P. Common Molecular Pathways Between Post-COVID19 Syndrome and Lung Fibrosis: A Scoping Review. Front. Pharmacol. 2022, 13, 748931. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.D.; Sumeh, A.S.; Sheraz, M.; Kavitha, M.S.; Venmathi Maran, B.A.; Rodrigues, K.F. A mini-review on the impact of COVID 19 on vital organs. Biomed. Pharmacother. 2021, 143, 112158. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, F.; Chai, F.; Chen, X. Effect of statins on pulmonary function in patients with chronic obstructive pulmonary disease: A systematic review and meta-analysis of randomized controlled trials. J. Thorac. Dis. 2023, 15, 3944–3952. [Google Scholar] [CrossRef]
- Podolanczuk, A.J.; Raghu, G.; Tsai, M.Y.; Kawut, S.M.; Peterson, E.; Sonti, R.; Rabinowitz, D.; Johnson, C.; Barr, R.G.; Hinckley Stukovsky, K.; et al. Cholesterol, lipoproteins and subclinical interstitial lung disease: The MESA study. Thorax 2017, 72, 472–474. [Google Scholar] [CrossRef]
- Bates, S.R.; Tao, J.Q.; Collins, H.L.; Francone, O.L.; Rothblat, G.H. Pulmonary abnormalities due to ABCA1 deficiency in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L980–L989. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.; Shah, D.; Duong, M.; Penn, R.B.; Fessler, M.B.; Madenspacher, J.; Stafstrom, W.; Kavuru, M.; Lu, B.; Kallen, C.B.; et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 74–86. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, X.; Wu, G.; Shen, L.; Chen, B. Effect of lipid-bound apoA-I cysteine mutants on lipopolysaccharide-induced endotoxemia in mice. J. Lipid Res. 2008, 49, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Fessler, M.B. A New Frontier in Immunometabolism. Cholesterol in Lung Health and Disease. Ann. Am. Thorac. Soc. 2017, 14, S399–S405. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Preuss, I.; Ludwig, M.G.; Baumgarten, B.; Bassilana, F.; Gessier, F.; Seuwen, K.; Sailer, A.W. Transcriptional regulation and functional characterization of the oxysterol/EBI2 system in primary human macrophages. Biochem. Biophys. Res. Commun. 2014, 446, 663–668. [Google Scholar] [CrossRef]
- Foo, C.X.; Bartlett, S.; Chew, K.Y.; Ngo, M.D.; Bielefeldt-Ohmann, H.; Arachchige, B.J.; Matthews, B.; Reed, S.; Wang, R.; Smith, C.; et al. GPR183 antagonism reduces macrophage infiltration in influenza and SARS-CoV-2 infection. Eur. Respir. J. 2023, 61, 2201306. [Google Scholar] [CrossRef] [PubMed]
- Madenspacher, J.H.; Morrell, E.D.; Gowdy, K.M.; McDonald, J.G.; Thompson, B.M.; Muse, G.; Martinez, J.; Thomas, S.; Mikacenic, C.; Nick, J.A.; et al. Cholesterol 25-hydroxylase promotes efferocytosis and resolution of lung inflammation. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Edwards, P.A.; Kennedy, M.A.; Mak, P.A. LXRs; oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vasc. Pharmacol. 2002, 38, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, I.; Poti, F.; Pedrelli, M.; Favari, E.; Moleri, E.; Franceschini, G.; Calabresi, L.; Bernini, F. The LXR agonist T0901317 promotes the reverse cholesterol transport from macrophages by increasing plasma efflux potential. J. Lipid Res. 2008, 49, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef]
- Ohashi, H.; Wang, F.; Stappenbeck, F.; Tsuchimoto, K.; Kobayashi, C.; Saso, W.; Kataoka, M.; Yamasaki, M.; Kuramochi, K.; Muramatsu, M.; et al. Identification of Anti-Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Oxysterol Derivatives In Vitro. Int. J. Mol. Sci. 2021, 22, 3163. [Google Scholar] [CrossRef]
- Agudelo, C.W.; Samaha, G.; Garcia-Arcos, I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. 2020, 19, 122. [Google Scholar] [CrossRef]
- Calfee, C.S.; Delucchi, K.L.; Sinha, P.; Matthay, M.A.; Hackett, J.; Shankar-Hari, M.; McDowell, C.; Laffey, J.G.; O’Kane, C.M.; McAuley, D.F., on behalf of the Irish Critical Care Trials Group. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: Secondary analysis of a randomised controlled trial. Lancet Respir. Med. 2018, 6, 691–698. [Google Scholar] [CrossRef]
- Zapatero-Belinchon, F.J.; Moeller, R.; Lasswitz, L.; van Ham, M.; Becker, M.; Brogden, G.; Rosendal, E.; Bi, W.; Carriqui-Madronal, B.; Islam, K.; et al. Fluvastatin mitigates SARS-CoV-2 infection in human lung cells. iScience 2021, 24, 103469. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, R.; Takahashi, J.; Shirakura, K.; Funatsu, R.; Kosugi, K.; Deguchi, S.; Yamamoto, M.; Tsunoda, Y.; Morita, M.; Muraoka, K.; et al. SARS-CoV-2 disrupts respiratory vascular barriers by suppressing Claudin-5 expression. Sci. Adv. 2022, 8, eabo6783. [Google Scholar] [CrossRef] [PubMed]
- Liebhaber, M.I.; Wright, R.S.; Gelberg, H.J.; Dyer, Z.; Kupperman, J.L. Polymyalgia, hypersensitivity pneumonitis and other reactions in patients receiving HMG-CoA reductase inhibitors: A report of ten cases. Chest 1999, 115, 886–889. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies Showing Cholesterol-Related Associations with COVID-19 Severity | Studies Lacking Cholesterol-Related Associations with COVID-19 Severity |
|---|---|
| (i) Dyslipidaemia before infection increased the risk for SARS-CoV-2 infection [9,10]. | (i) Dyslipidaemia before infection was not associated with the risk for SARS-CoV-2 infection [11]. |
| (i) Dyslipidaemia before infection increased the risk for a severe disease course and mortality [9,10,12,13]. | (i) Dyslipidaemia before infection was not associated with the risk for a severe disease course and mortality [14]. |
| (ii) Low blood cholesterol in patients with COVID-19 was related to a severe disease course and mortality [15,16,17]. | (ii) Blood cholesterol in patients with COVID-19 was not related to a severe disease course and mortality [18,19,20]. |
| (iii) Preadmission statin treatment was associated with better outcomes among COVID-19 patients [21,22,23]. | (iii) Statin use before COVID-19 hospitalisation did not protect from fatal outcomes [24]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grewal, T.; Nguyen, M.K.L.; Buechler, C. Cholesterol and Cholesterol-Lowering Medications in COVID-19—An Unresolved Matter. Int. J. Mol. Sci. 2024, 25, 10489. https://doi.org/10.3390/ijms251910489
Grewal T, Nguyen MKL, Buechler C. Cholesterol and Cholesterol-Lowering Medications in COVID-19—An Unresolved Matter. International Journal of Molecular Sciences. 2024; 25(19):10489. https://doi.org/10.3390/ijms251910489
Chicago/Turabian StyleGrewal, Thomas, Mai Khanh Linh Nguyen, and Christa Buechler. 2024. "Cholesterol and Cholesterol-Lowering Medications in COVID-19—An Unresolved Matter" International Journal of Molecular Sciences 25, no. 19: 10489. https://doi.org/10.3390/ijms251910489
APA StyleGrewal, T., Nguyen, M. K. L., & Buechler, C. (2024). Cholesterol and Cholesterol-Lowering Medications in COVID-19—An Unresolved Matter. International Journal of Molecular Sciences, 25(19), 10489. https://doi.org/10.3390/ijms251910489