
Generation and Characterization of hiPS Lines from Three Patients Affected by Different Forms of HPDL-Related Neurological Disorders

 , , ,
, , ,
Abstract
1. Introduction
2. Results
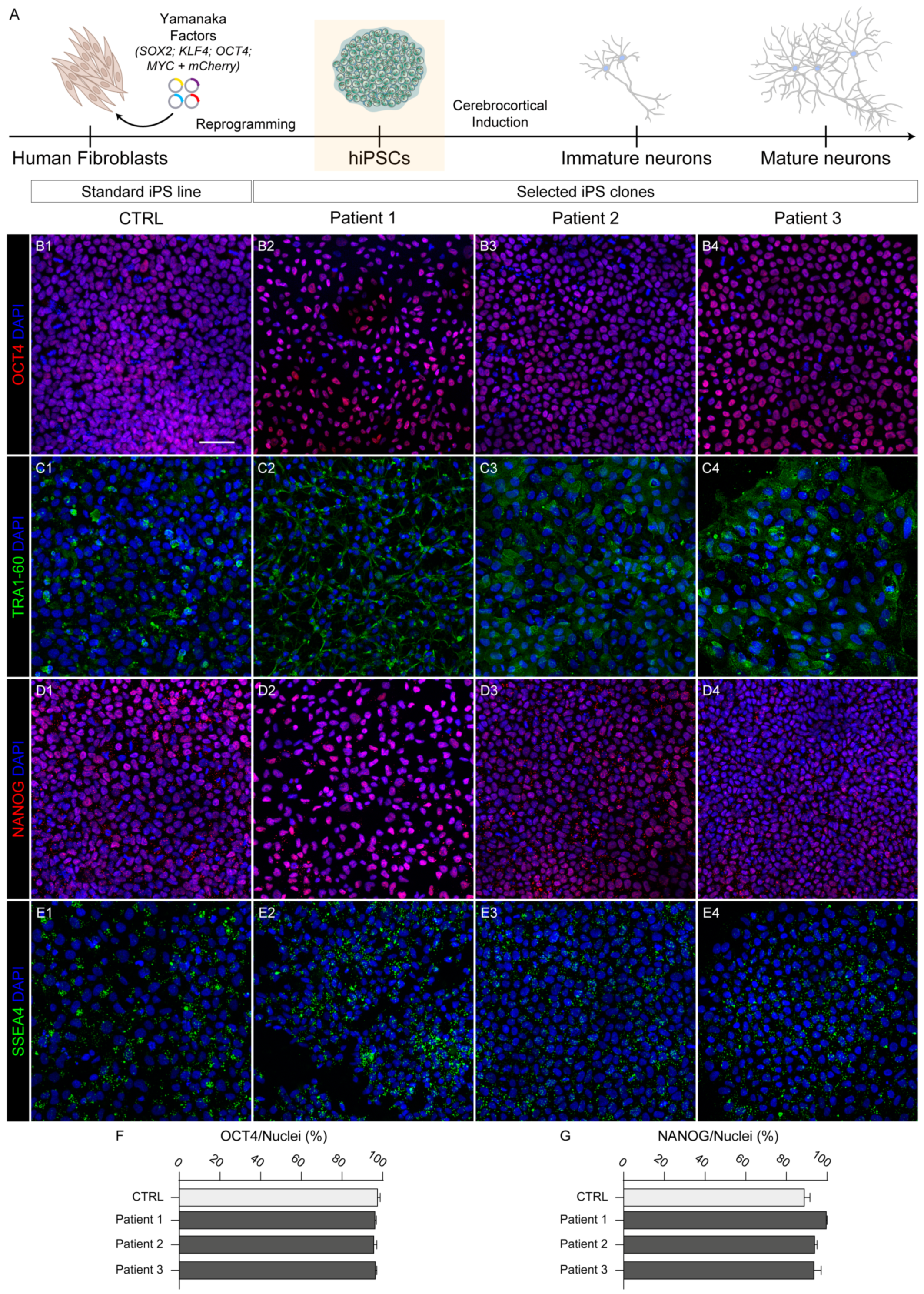
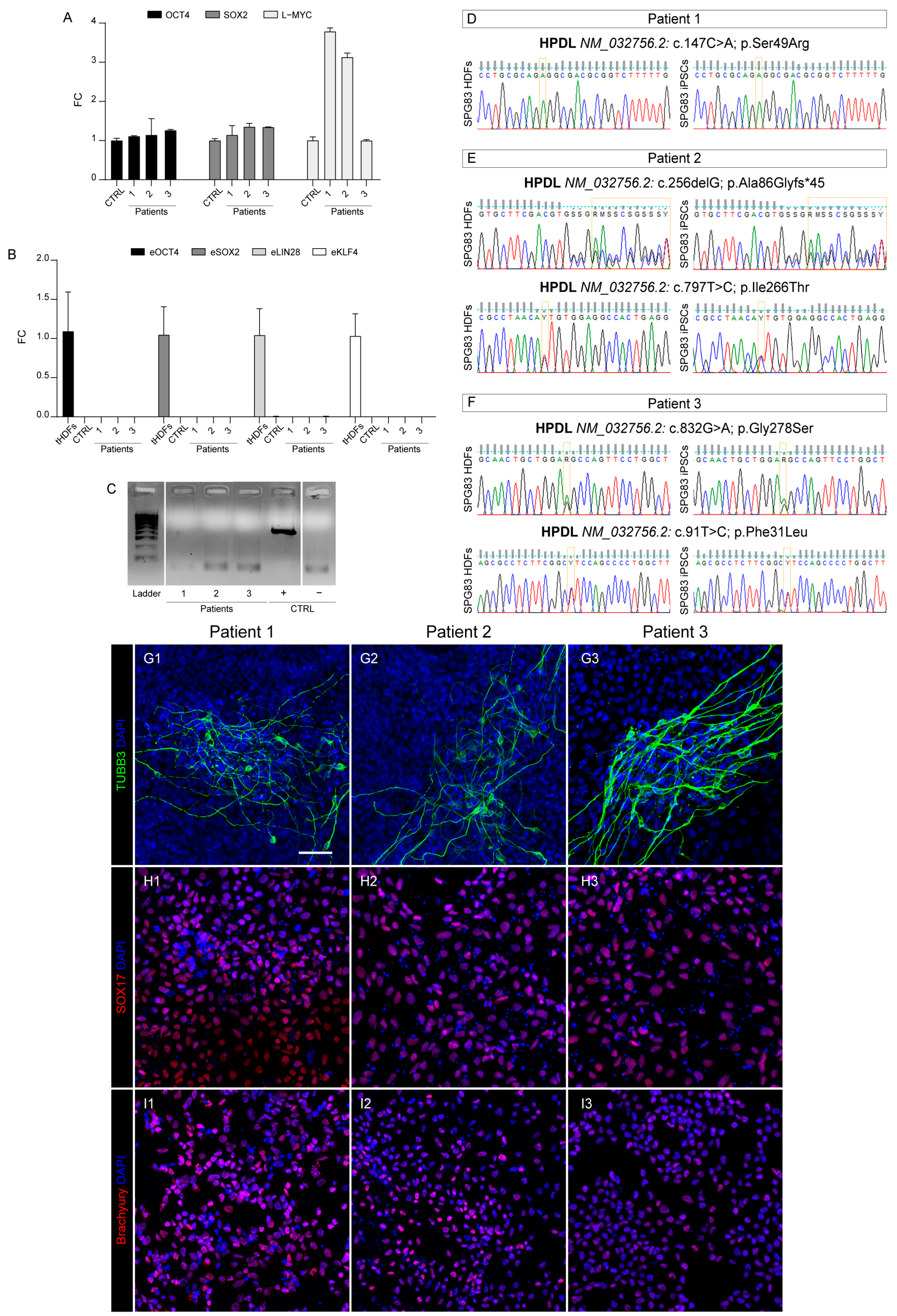
2.1. Reprogramming and Characterization
2.2. Mycoplasma
2.3. Genome Identity
2.4. Pluripotency Capability
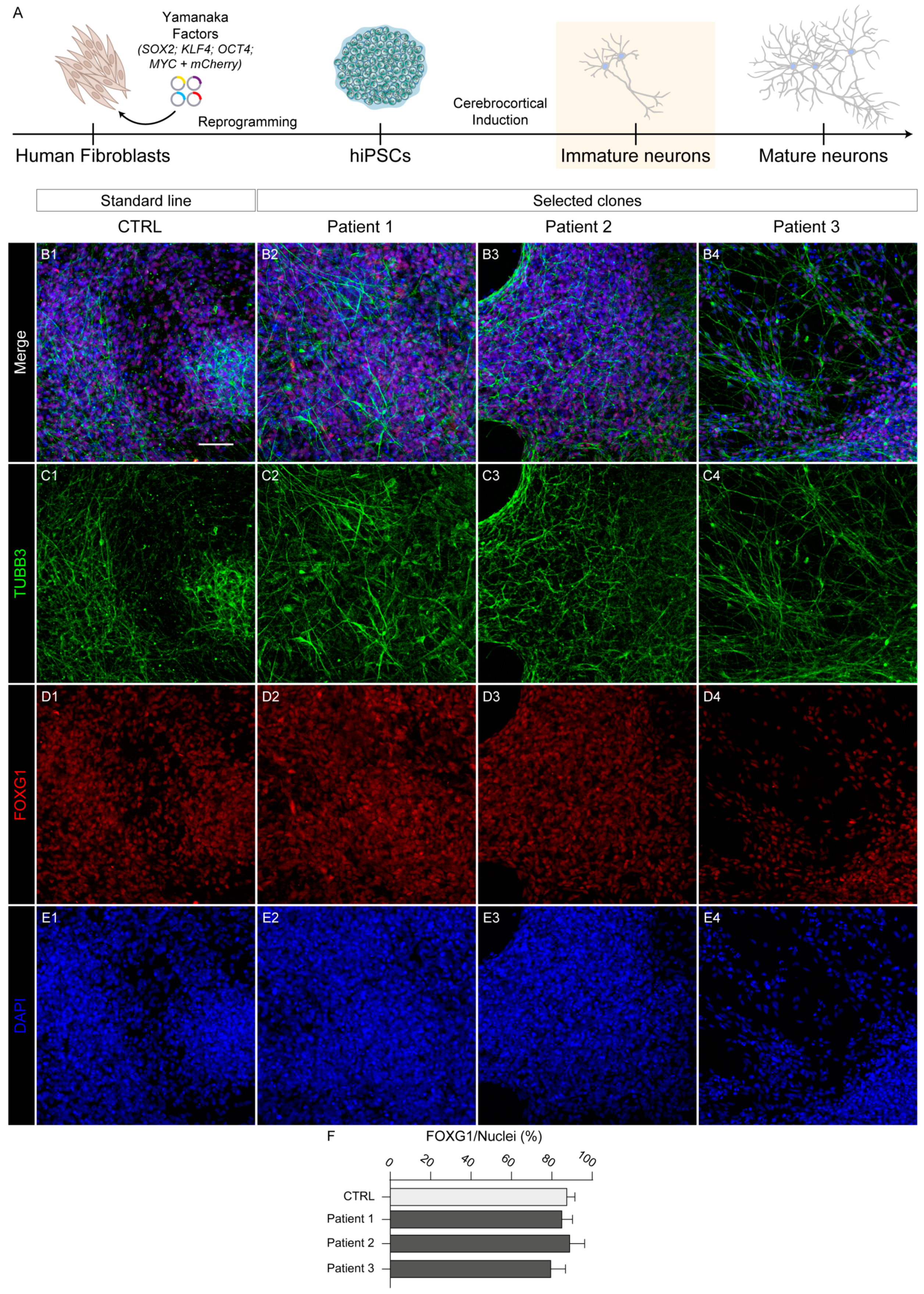
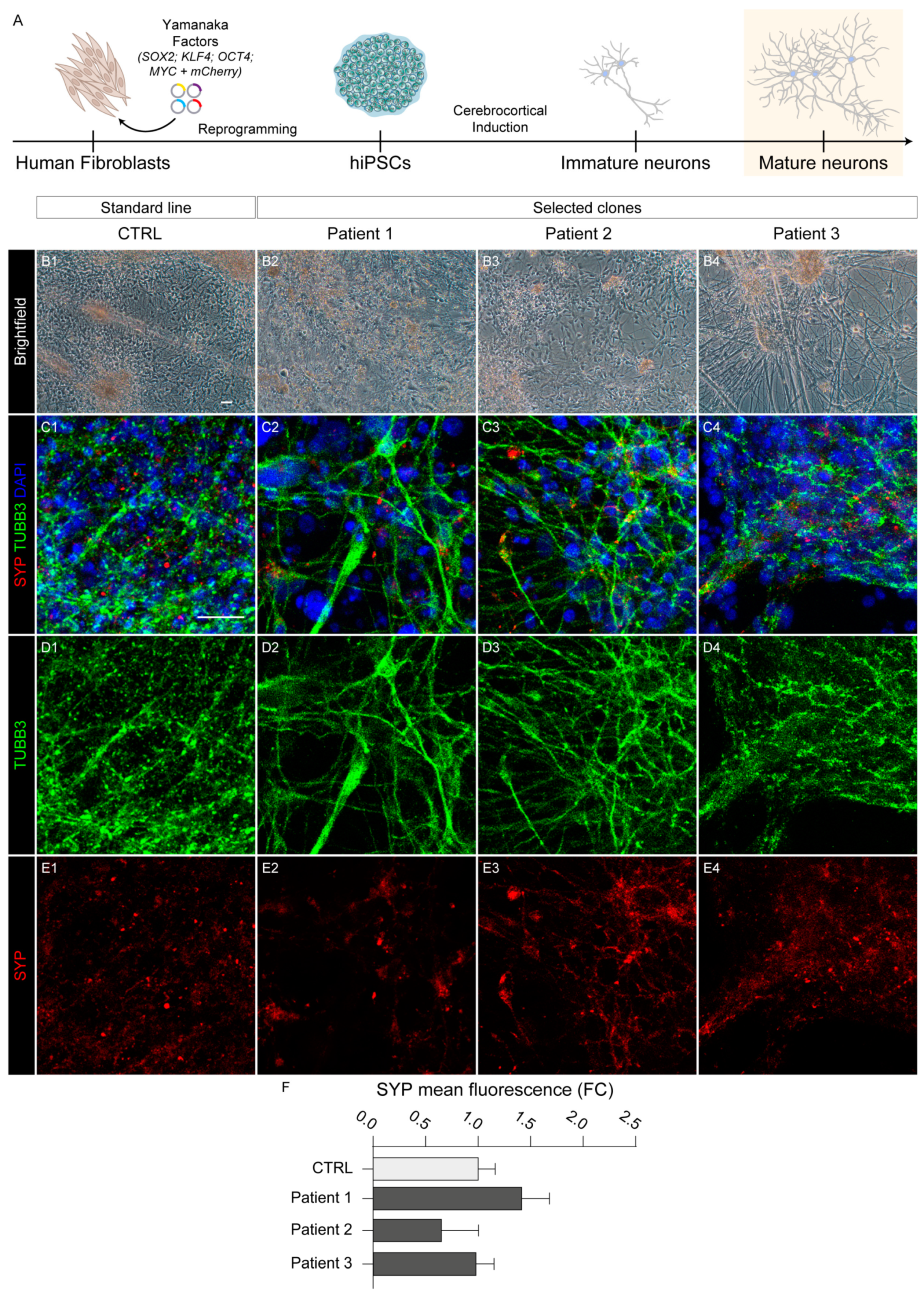
2.5. Neocortical Differentiation
3. Discussion
4. Materials and Methods
4.1. Reprogramming of Skin Fibroblasts
4.2. Culture of hiPS Cells
4.3. Expression of Pluripotency Markers and Mutation Analysis
4.4. Image Analysis
4.5. Trilineage Differentiation
4.6. STR Analysis
4.7. Genomic Analysis by Array-CGH
4.8. Mycoplasma Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fink, J.K. Hereditary Spastic Paraplegia: Clinico-Pathologic Features and Emerging Molecular Mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the Complexity of Hereditary Spastic Paraplegias: How Unexpected Phenotypes and Inheritance Modes Are Revolutionizing Their Nosology. Hum. Genet. 2015, 134, 511–538. [Google Scholar] [CrossRef] [PubMed]
- Damiani, D.; Baggiani, M.; Della Vecchia, S.; Naef, V.; Santorelli, F.M. Pluripotent Stem Cells as a Preclinical Cellular Model for Studying Hereditary Spastic Paraplegias. Int. J. Mol. Sci. 2024, 25, 2615. [Google Scholar] [CrossRef] [PubMed]
- Murala, S.; Nagarajan, E.; Bollu, P.C. Hereditary Spastic Paraplegia. Neurol. Sci. 2021, 42, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Husain, R.A.; Grimmel, M.; Wagner, M.; Hennings, J.C.; Marx, C.; Feichtinger, R.G.; Saadi, A.; Rostásy, K.; Radelfahr, F.; Bevot, A.; et al. Bi-Allelic HPDL Variants Cause a Neurodegenerative Disease Ranging from Neonatal Encephalopathy to Adolescent-Onset Spastic Paraplegia. Am. J. Hum. Genet. 2020, 107, 364–373. [Google Scholar] [CrossRef]
- Ghosh, S.G.; Lee, S.; Fabunan, R.; Chai, G.; Zaki, M.S.; Abdel-Salam, G.; Sultan, T.; Ben-Omran, T.; Alvi, J.R.; McEvoy-Venneri, J.; et al. Biallelic Variants in HPDL, Encoding 4-Hydroxyphenylpyruvate Dioxygenase-like Protein, Lead to an Infantile Neurodegenerative Condition. Genet. Med. 2021, 23, 524–533. [Google Scholar] [CrossRef]
- Morgan, N.V.; Yngvadottir, B.; O’Driscoll, M.; Clark, G.R.; Walsh, D.; Martin, E.; Tee, L.; Reid, E.; Titheradge, H.L.; Maher, E.R. Evidence That Autosomal Recessive Spastic Cerebral Palsy-1 (CPSQ1) Is Caused by a Missense Variant in HPDL. Brain Commun. 2021, 3, fcab002. [Google Scholar] [CrossRef]
- Numata-Uematsu, Y.; Uematsu, M.; Yamamoto, T.; Saitsu, H.; Katata, Y.; Oikawa, Y.; Saijyo, N.; Inui, T.; Murayama, K.; Ohtake, A.; et al. Leigh Syndrome-like MRI Changes in a Patient with Biallelic HPDL Variants Treated with Ketogenic Diet. Mol. Genet. Metab. Rep. 2021, 29, 100800. [Google Scholar] [CrossRef]
- Sun, Y.; Wei, X.; Fang, F.; Shen, Y.; Wei, H.; Li, J.; Ye, X.; Zhan, Y.; Ye, X.; Liu, X.; et al. HPDL Deficiency Causes a Neuromuscular Disease by Impairing the Mitochondrial Respiration. J. Genet. Genom. 2021, 48, 727–736. [Google Scholar] [CrossRef]
- Wiessner, M.; Maroofian, R.; Ni, M.-Y.; Pedroni, A.; Müller, J.S.; Stucka, R.; Beetz, C.; Efthymiou, S.; Santorelli, F.M.; Alfares, A.A.; et al. Biallelic Variants in HPDL Cause Pure and Complicated Hereditary Spastic Paraplegia. Brain 2021, 144, 1422–1434. [Google Scholar] [CrossRef]
- Yu, H.; Wei, Q.; Luo, W.-J.; Wu, Z.-Y. Novel Bi-Allelic HPDL Variants Cause Hereditary Spastic Paraplegia in a Chinese Patient. Clin. Genet. 2021, 100, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wei, X.; Liao, J.; Chen, P.; Li, X.; Chen, Y.; Yang, Y.; Zhao, Q.; Sun, H.; Pan, L.; et al. 4-Hydroxyphenylpyruvate Dioxygenase-Like Protein Promotes Pancreatic Cancer Cell Progression and Is Associated with Glutamine-Mediated Redox Balance. Front. Oncol. 2021, 10, 617190. [Google Scholar] [CrossRef]
- Banh, R.S.; Kim, E.S.; Spillier, Q.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Shi, G.; Jones, D.R.; Kimmelman, A.C.; Pacold, M.E. The Polar Oxy-Metabolome Reveals the 4-Hydroxymandelate CoQ10 Synthesis Pathway. Nature 2021, 597, 420–425. [Google Scholar] [CrossRef]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of Pluripotent Stem Cells from Fibroblast Cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic Instability of IPSCs: Challenges towards Their Clinical Applications. Stem Cell Rev. Rep. 2017, 13, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly Efficient Neural Conversion of Human ES and IPS Cells by Dual Inhibition of SMAD Signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An Efficient Nonviral Method to Generate Integration-Free Human-Induced Pluripotent Stem Cells from Cord Blood and Peripheral Blood Cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Baggiani, M.; Santorelli, F.M.; Mero, S.; Privitera, F.; Damiani, D.; Tessa, A. Generation of a Human Induced Pluripotent Stem Cell Line (FSMi001-A) from Fibroblasts of a Patient Carrying Heterozygous Mutation in the REEP1 Gene. Stem Cell Res. 2024, 79, 103472. [Google Scholar] [CrossRef]
- Schmitt, C.E.; Morales, B.M.; Schmitz, E.M.H.; Hawkins, J.S.; Lizama, C.O.; Zape, J.P.; Hsiao, E.C.; Zovein, A.C. Fluorescent Tagged Episomals for Stoichiometric Induced Pluripotent Stem Cell Reprogramming. Stem Cell Res. Ther. 2017, 8, 132. [Google Scholar] [CrossRef]
- Maria Turco, E.; Maria Giada Giovenale, A.; Rotundo, G.; Mazzoni, M.; Zanfardino, P.; Frezza, K.; Torrente, I.; Mary Carletti, R.; Damiani, D.; Santorelli, F.M.; et al. Generation and Characterization of CSSi016-A (9938) Human Pluripotent Stem Cell Line Carrying Two Biallelic Variants in MTMR5/SBF1 Gene Resulting in a Case of Severe CMT4B3. Stem Cell Res. 2022, 65, 102946. [Google Scholar] [CrossRef]
- Lam, A.Q.; Freedman, B.S.; Morizane, R.; Lerou, P.H.; Valerius, M.T.; Bonventre, J.V. Rapid and Efficient Differentiation of Human Pluripotent Stem Cells into Intermediate Mesoderm That Forms Tubules Expressing Kidney Proximal Tubular Markers. J. Am. Soc. Nephrol. 2014, 25, 1211–1225. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| HPDL Variants | |||
| Mutations | NM_032756.2: c.147C>A(p.(Ser49Arg)) | NM_032756.2: c.256delG(p.(Ala86Glyfs*45)) c.797T>C(p.(Ile266Thr)) | NM_032756.2: c.91T>C(p.(Phe31Leu)) c.832G>A(p.(Gly278Ser)) |
| Allele frequency (gnomAD) | 0.001527% | 0.0004896%/0.0008506% | Not reported |
| Clinical Features and Age at Skin Biopsy | |||
| First manifestations | Developmental delay, neonatal seizures, spasticity in the lower limb | Motor delay and gait problem | Spastic gait |
| Age at onset (years) | Neonatal | 1 | 4 |
| Age at skin biopsy (years) | 12 | 14 | 7 |
| Disease severity/course | Intermediate phenotype | Intermediate phenotype | Mild phenotype |
| Target | Size of Band | Forward/Reverse Primer (5′-3′) |
|---|---|---|
| OCT4 | 143 bp | Fwd: CCC CAG GGC CCC ATT TTG GTA CC |
| Rev: ACC TCA GTT TGA ATG CAT GGG AGA GC | ||
| L-MYC | 143 bp | Fwd: GCG AAC CCA AGA CCC AGG CCT GCT CC |
| Rev: CAG GGG GTC TGC TCG CAC CGT GAT G | ||
| SOX2 | 80 bp | Fwd: TTC ACA TGT CCC AGC ACT ACC AGA |
| Rev: TCA CAT GTG TGA GAG GGG CAG TGT GC | ||
| GAPDH | 110 bp | Fwd: GGA AGG ACT CAT GAC CAC AGT |
| Rev: GGA TGA TGT TCT GGA GAG CCC | ||
| eOCT4 | 124 bp | Fwd: CAT TCA AAC TGA GGT AAG GG |
| Rev: TAG CGT AAA AGG AGC AAC ATA G | ||
| eLIN28 | 251 bp | Fwd: AGC CAT ATG GTA GCC TCA TGT CCG C |
| Rev: TAG CGT AAA AGG AGC AAC ATA G | ||
| eSOX2 | 111 bp | Fwd: TTC ACA TGT CCC AGC ACT ACC AGA |
| Rev: TTT GTT TGA CAG GAG CGA CAA T | ||
| eKLF4 | 156 bp | Fwd: CCA CCT CGC CTT ACA CAT GAA GA |
| Rev: TAG CGT AAA AGG AGC AAC ATA G | ||
| HPDL A | 785 bp | Fwd: CTTTCCGGAAGAAAGCGAGGAA |
| (Genotyping) | Rev: CCTCAGTCCCCCAAGCCCAA | |
| HPDL B | 661 bp | Fwd: TGCGCTGGTTCCACGACTGC |
| (Genotyping) | Rev: GCAGATGTTCCTCAGTTCTGTG |
| Antibody | Dilution | Company, Cat # |
|---|---|---|
| Rabbit anti-OCT4 | 1:500 | Abcam (Cambridge, UK), Cat #ab19857 |
| Rabbit anti-Nanog (D73G4) | 1:200 | Cell Signaling Technology (Danvers, MA, USA), Cat #4903 |
| Mouse anti-TRA-1-60 | 1:500 | Cell Signaling Technology, Cat #4746 |
| Mouse anti-SSEA4 (MC813) | 1:500 | Cell Signaling Technology, Cat #4755 |
| Rabbit anti-FOXG1 | 1:500 | Abcam, Cat #ab18259 |
| Rabbit anti-synaptophisin | 1:100 | Cell Signaling Technology, Cat #36406 |
| Mouse anti-TUBB3 | 1:1000 | Abcam, Cat #ab7751 |
| Rabbit anti-brachyury (D2Z3J) | 1:500 | Cell Signaling Technology, Cat #81694 |
| Rabbit anti-SOX17 (D1T8M) | 1:500 | Cell Signaling Technology, Cat #81778 |
| Mouse anti-MAP2 | 1:200 | Sigma (St. Louis, MO, USA), Cat #ZMS1013 |
| Rabbit anti-RBFOX3/NeuN | 1:500 | Millipore (Burlington, MA, USA), Cat #ABN78 |
| Goat Anti-Mouse Alexa Fluor 488 | 1:500 | Thermo Fisher Scientific, Cat #a11029 |
| Goat Anti-Rabbit Alexa Fluor 555 | 1:500 | Thermo Fisher Scientific, Cat #a21429 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baggiani, M.; Damiani, D.; Privitera, F.; Della Vecchia, S.; Tessa, A.; Santorelli, F.M. Generation and Characterization of hiPS Lines from Three Patients Affected by Different Forms of HPDL-Related Neurological Disorders. Int. J. Mol. Sci. 2024, 25, 10614. https://doi.org/10.3390/ijms251910614
Baggiani M, Damiani D, Privitera F, Della Vecchia S, Tessa A, Santorelli FM. Generation and Characterization of hiPS Lines from Three Patients Affected by Different Forms of HPDL-Related Neurological Disorders. International Journal of Molecular Sciences. 2024; 25(19):10614. https://doi.org/10.3390/ijms251910614
Chicago/Turabian StyleBaggiani, Matteo, Devid Damiani, Flavia Privitera, Stefania Della Vecchia, Alessandra Tessa, and Filippo Maria Santorelli. 2024. "Generation and Characterization of hiPS Lines from Three Patients Affected by Different Forms of HPDL-Related Neurological Disorders" International Journal of Molecular Sciences 25, no. 19: 10614. https://doi.org/10.3390/ijms251910614
APA StyleBaggiani, M., Damiani, D., Privitera, F., Della Vecchia, S., Tessa, A., & Santorelli, F. M. (2024). Generation and Characterization of hiPS Lines from Three Patients Affected by Different Forms of HPDL-Related Neurological Disorders. International Journal of Molecular Sciences, 25(19), 10614. https://doi.org/10.3390/ijms251910614