
Exploring a Potential Optimization Route for Peptide Ligands of the Sam Domain from the Lipid Phosphatase Ship2

 ,
,  , , ,
, , ,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
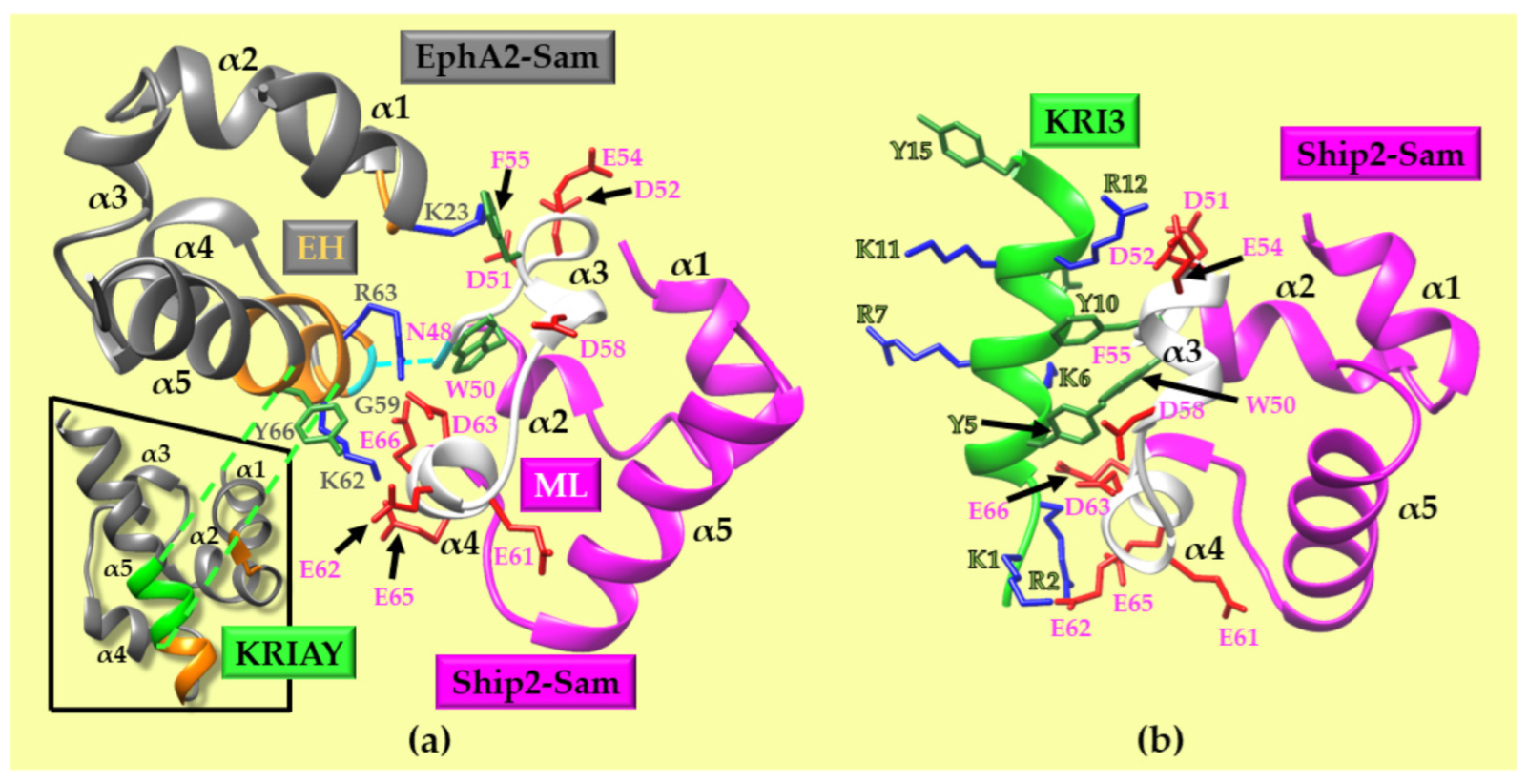
2. Results and Discussion
2.1. Computational Approach for Peptide Design and Selection
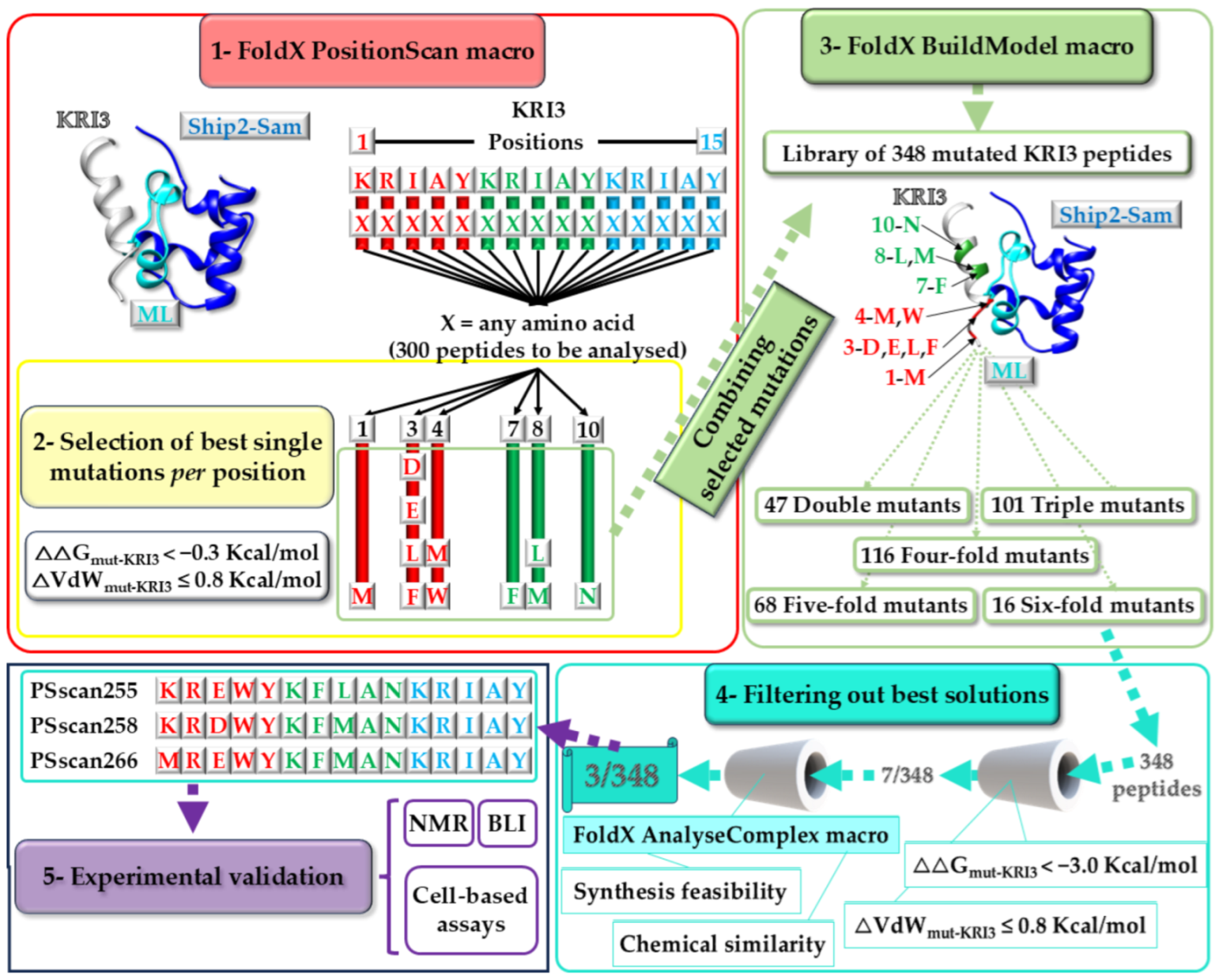
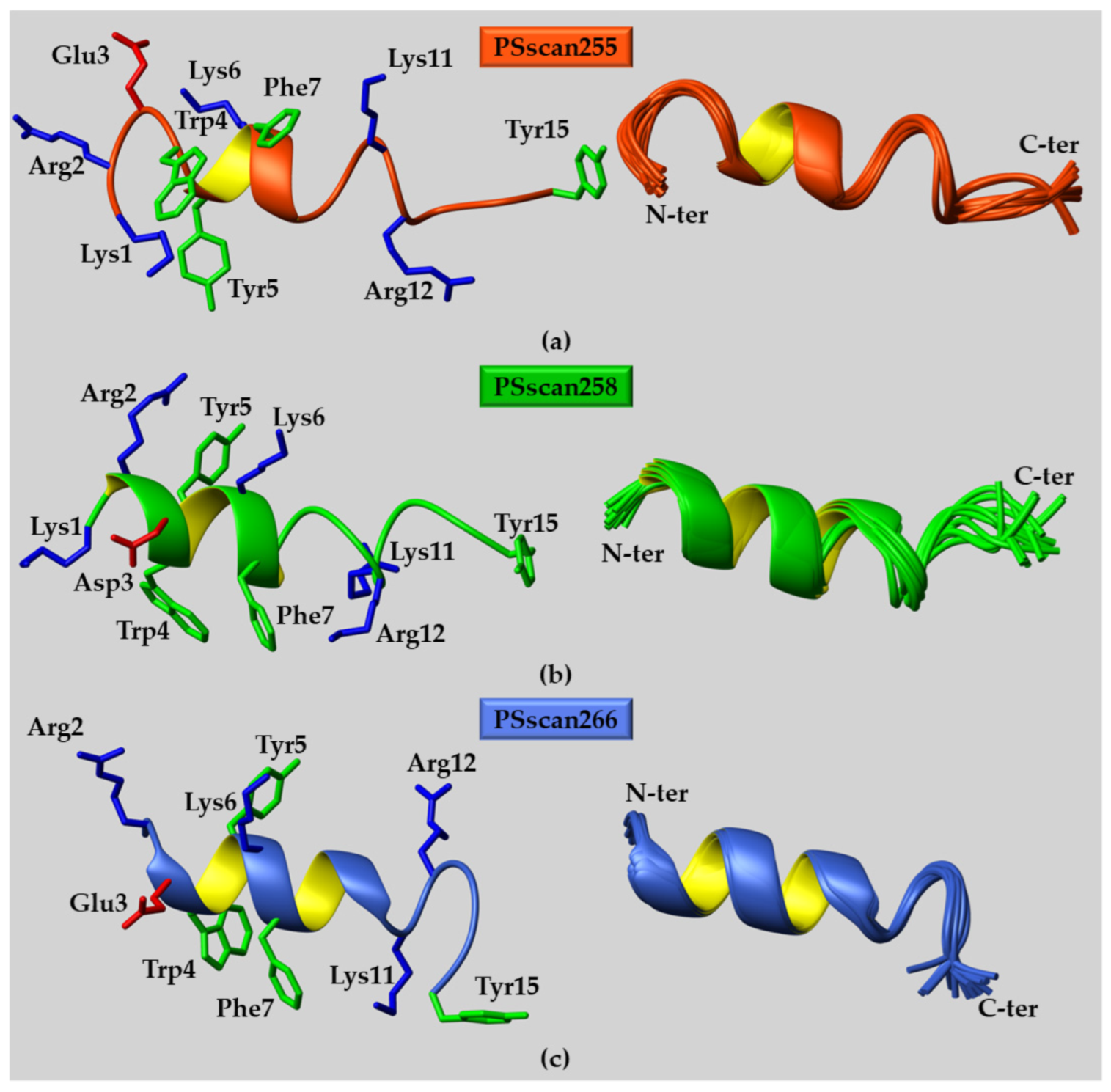
2.2. Peptide Structural Characterization
2.2.1. CD Studies
2.2.2. NMR Studies
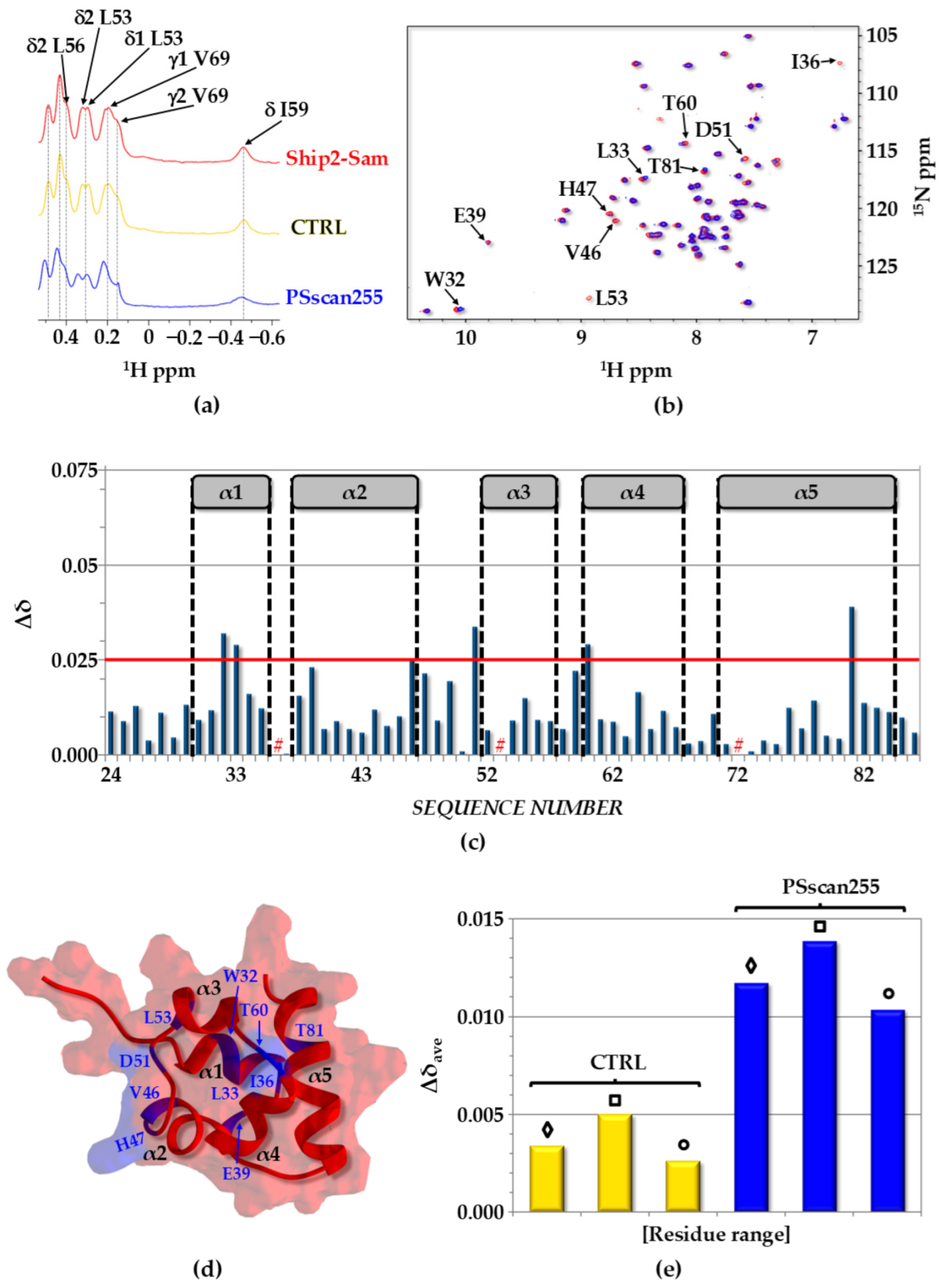
2.3. Interaction Studies: Peptides vs. Ship2-Sam
2.3.1. NMR Chemical Shift Perturbation Studies

2.3.2. Quantitative Assessment of the Binding between Ship2-Sam and PSscan255
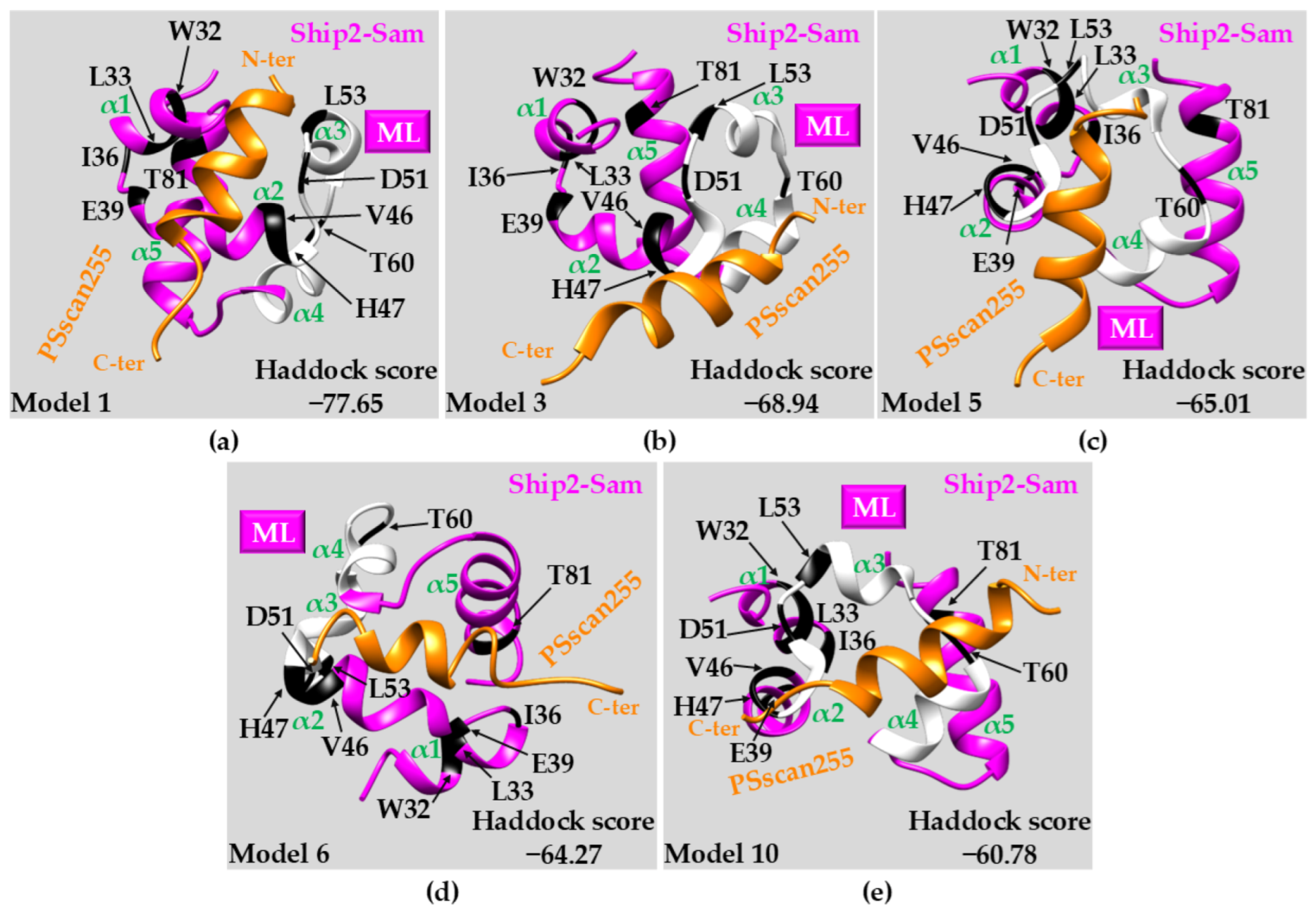
2.3.3. Docking Studies: PSscan255 vs. Ship2-Sam
2.4. Cell-Based Experiments with PSscan255
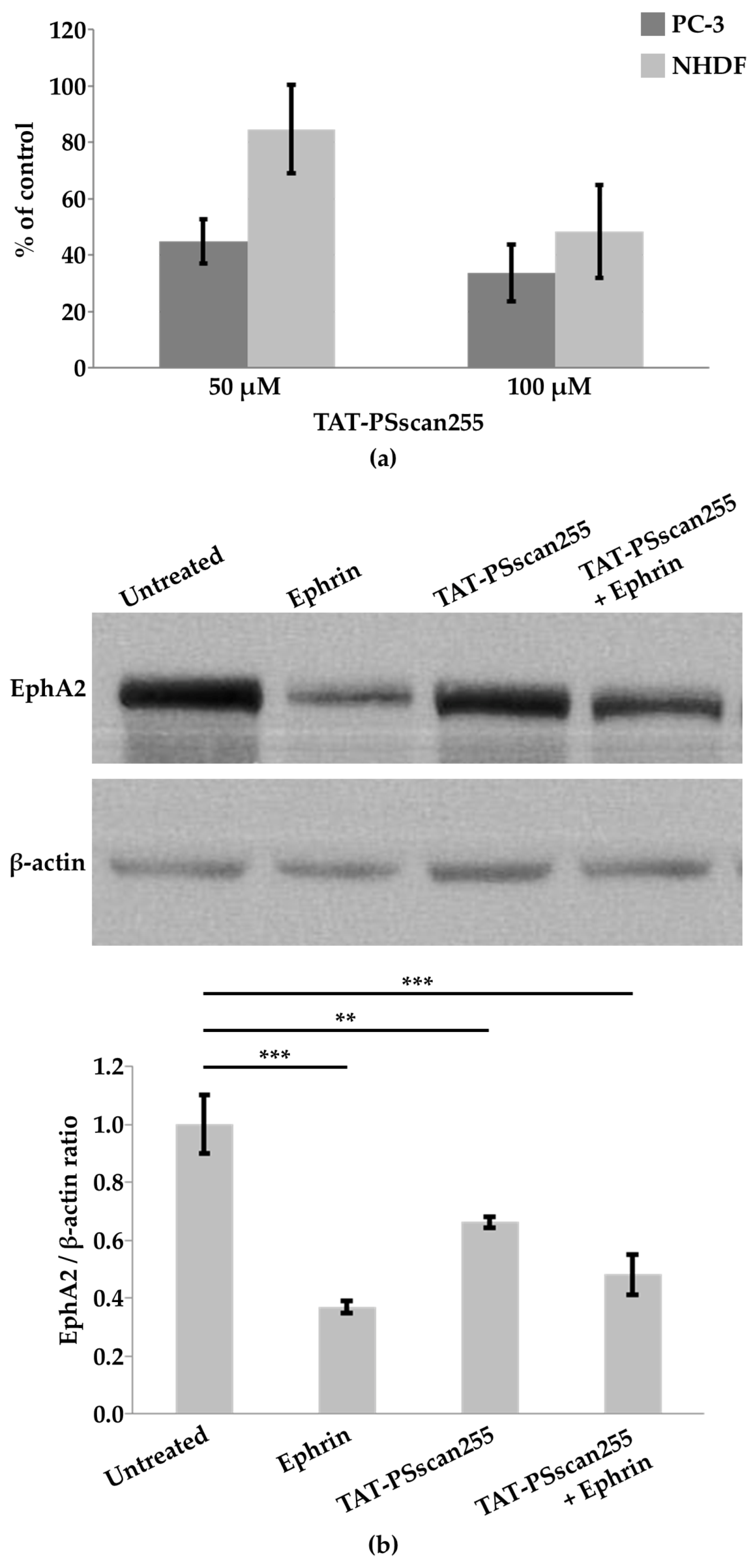
2.4.1. Assessment of Serum Stability
2.4.2. Cellular Uptake
2.4.3. Cytotoxicity
2.4.4. Western Blot Analyses to Monitor EphA2 Levels
3. Materials and Methods
3.1. FoldX Studies
3.2. Peptides
3.3. CD Spectroscopy
3.4. Protein Expression
3.5. NMR Spectroscopy
3.5.1. Conformational Studies
3.5.2. Chemical Shift Perturbation Studies
3.5.3. Solution Structure Calculations
3.6. BioLayer Interferometry
3.7. Docking Studies: Ship2-Sam/PSscan255 Complex
3.8. AlphaFold Model of the Ship2-Sam/PSscan255 Complex
3.9. Docking Studies: PSscan266 Self-Aggregation
3.10. LigPlot+ Analyses
3.11. Serum Stability
3.12. Cell Culture Conditions and Cytotoxicity
3.13. Western Blotting Analyses
3.14. Fluorescence Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vandeput, F.; Backers, K.; Villeret, V.; Pesesse, X.; Erneux, C. The influence of anionic lipids on SHIP2 phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase activity. Cell. Signal. 2006, 18, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Erneux, C.; Potter, B.V. SHIP2: Structure, Function and Inhibition. ChemBioChem 2017, 18, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.M.; Jucker, M. The Functional Roles of the Src Homology 2 Domain-Containing Inositol 5-Phosphatases SHIP1 and SHIP2 in the Pathogenesis of Human Diseases. Int. J. Mol. Sci. 2024, 25, 5254. [Google Scholar] [CrossRef] [PubMed]
- Rudge, S.A.; Wakelam, M.J. Phosphatidylinositolphosphate phosphatase activities and cancer. J. Lipid Res. 2016, 57, 176–192. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptors and ephrins in cancer progression. Nat. Rev. Cancer 2024, 24, 5–27. [Google Scholar] [CrossRef]
- Miao, H.; Li, D.Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef]
- Zhuang, G.; Hunter, S.; Hwang, Y.; Chen, J. Regulation of EphA2 receptor endocytosis by SHIP2 lipid phosphatase via phosphatidylinositol 3-Kinase-dependent Rac1 activation. J. Biol. Chem. 2007, 282, 2683–2694. [Google Scholar] [CrossRef]
- Leone, M.; Cellitti, J.; Pellecchia, M. NMR studies of a heterotypic Sam−Sam domain association: The interaction between the lipid phosphatase Ship2 and the EphA2 receptor. Biochemistry 2008, 47, 12721–12728. [Google Scholar] [CrossRef]
- Lee, H.J.; Hota, P.K.; Chugha, P.; Guo, H.; Miao, H.; Zhang, L.Q.; Kim, S.J.; Stetzik, L.; Wang, B.C.; Buck, M. NMR Structure of a Heterodimeric SAM:SAM Complex: Characterization and Manipulation of EphA2 Binding Reveal New Cellular Functions of SHIP2. Structure 2012, 20, 41–55. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, Y.; Li, J.; Chen, W.; Li, G.; Wan, J.; Liu, W.; Zhang, M. Specific Eph receptor-cytoplasmic effector signaling mediated by SAM-SAM domain interactions. eLife 2018, 7, e35677. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Vincenzi, M.; Leone, M. Hunting for Novel Routes in Anticancer Drug Discovery: Peptides against Sam-Sam Interactions. Int. J. Mol. Sci. 2022, 23, 10397. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, Q.; Shan, H.; Liu, J.; Lv, C.; Wang, Y.; Yuan, L.; Chen, Y.; Wang, Z.; Yin, Y.; et al. SASH1: A Novel Eph Receptor Partner and Insights into SAM-SAM Interactions. J. Mol. Biol. 2023, 435, 168243. [Google Scholar] [CrossRef] [PubMed]
- Kurabi, A.; Brener, S.; Mobli, M.; Kwan, J.J.; Donaldson, L.W. A nuclear localization signal at the SAM–SAM domain interface of AIDA-1 suggests a requirement for domain uncoupling prior to nuclear import. J. Mol. Biol. 2009, 392, 1168–1177. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Vincenzi, M.; Marasco, D.; De Luca, S.; Pedone, E.M.; Leone, M. Exploring the Ability of Cyclic Peptides to Target SAM Domains: A Computational and Experimental Study. ChemBioChem 2020, 21, 702–711. [Google Scholar] [CrossRef]
- Ciura, P.; Smardz, P.; Spodzieja, M.; Sieradzan, A.K.; Krupa, P. Multilayered Computational Framework for Designing Peptide Inhibitors of HVEM-LIGHT Interaction. J. Phys. Chem. B 2024, 128, 6770–6785. [Google Scholar] [CrossRef]
- Wu, X.; Lin, H.; Bai, R.; Duan, H. Deep learning for advancing peptide drug development: Tools and methods in structure prediction and design. Eur. J. Med. Chem. 2024, 268, 116262. [Google Scholar] [CrossRef]
- Guo, H.B.; Perminov, A.; Bekele, S.; Kedziora, G.; Farajollahi, S.; Varaljay, V.; Hinkle, K.; Molinero, V.; Meister, K.; Hung, C.; et al. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 2022, 12, 10696. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Design of Cyclic Peptides Targeting Protein-Protein Interactions Using AlphaFold. Int. J. Mol. Sci. 2023, 24, 13257. [Google Scholar] [CrossRef] [PubMed]
- Rettie, S.A.; Campbell, K.V.; Bera, A.K.; Kang, A.; Kozlov, S.; De La Cruz, J.; Adebomi, V.; Zhou, G.; DiMaio, F.; Ovchinnikov, S.; et al. Cyclic peptide structure prediction and design using AlphaFold. bioRxiv 2023. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Marasco, D.; Calce, E.; Vincenzi, M.; Pedone, E.M.; De Luca, S.; Leone, M. Design and analysis of EphA2-SAM peptide ligands: A multi-disciplinary screening approach. Bioorg. Chem. 2019, 84, 434–443. [Google Scholar] [CrossRef]
- Vincenzi, M.; Mercurio, F.A.; Palumbo, R.; La Manna, S.; Pirone, L.; Marasco, D.; Pedone, E.M.; Leone, M. Inhibition of the EphA2-Sam/Ship2-Sam Association through Peptide Ligands: Studying the Combined Effect of Charge and Aromatic Character. J. Med. Chem. 2024, 67, 16649–16663. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Iannitti, R.; Marasco, D.; Pedone, E.; Palumbo, R.; Leone, M. The Sam-Sam interaction between Ship2 and the EphA2 receptor: Design and analysis of peptide inhibitors. Sci. Rep. 2017, 7, 17474. [Google Scholar] [CrossRef]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef]
- Gerasimavicius, L.; Liu, X.; Marsh, J.A. Identification of pathogenic missense mutations using protein stability predictors. Sci. Rep. 2020, 10, 15387. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Mercurio, F.A.; Di Natale, C.; Palumbo, R.; Pirone, L.; La Manna, S.; Marasco, D.; Pedone, E.M.; Leone, M. Targeting Ship2-Sam with peptide ligands: Novel insights from a multidisciplinary approach. Bioorg. Chem. 2022, 122, 105680. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Chen, Z.; Wu, X.; Yan, W.; Lu, F.; Liu, F. Improving the thermal stability and catalytic activity of ulvan lyase by the combination of FoldX and KnowVolution campaign. Int. J. Biol. Macromol. 2024, 257, 128577. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, C.; Li, D.; Wang, L. Enhancing the thermostability of Streptomyces cyaneofuscatus strain Ms1 tyrosinase by multi-factors rational design and molecular dynamics simulations. PLoS ONE 2023, 18, e0288929. [Google Scholar] [CrossRef]
- Fan, S.; Lu, X.; Wei, X.; Lu, R.; Feng, C.; Jin, Y.; Yan, M.; Yang, Z. Computational design of α-amylase from Bacillus licheniformis to increase its activity and stability at high temperatures. Comput. Struct. Biotechnol. J. 2024, 23, 982–989. [Google Scholar] [CrossRef]
- Miller, C.R.; Johnson, E.L.; Burke, A.Z.; Martin, K.P.; Miura, T.A.; Wichman, H.A.; Brown, C.J.; Ytreberg, F.M. Initiating a watch list for Ebola virus antibody escape mutations. PeerJ 2016, 4, e1674. [Google Scholar] [CrossRef]
- Malbranke, C.; Rostain, W.; Depardieu, F.; Cocco, S.; Monasson, R.; Bikard, D. Computational design of novel Cas9 PAM-interacting domains using evolution-based modelling and structural quality assessment. PLoS Comput. Biol. 2023, 19, e1011621. [Google Scholar] [CrossRef]
- Chi, L.A.; Barnes, J.E.; Suresh Patel, J.; Ytreberg, F.M. Exploring the ability of the MD+FoldX method to predict SARS-CoV-2 antibody escape mutations using large-scale data. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.R.; Wuthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. [Google Scholar] [CrossRef]
- Wuthrich, K. NMR of Proteins and Nucleic Acids; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Kjaergaard, M.; Brander, S.; Poulsen, F.M. Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 2011, 49, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, T.; Guntert, P.; Wuthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Pellecchia, M.; Bertini, I.; Cowburn, D.; Dalvit, C.; Giralt, E.; Jahnke, W.; James, T.L.; Homans, S.W.; Kessler, H.; Luchinat, C.; et al. Perspectives on NMR in drug discovery: A technique comes of age. Nat. Rev. Drug Discov. 2008, 7, 738–745. [Google Scholar] [CrossRef]
- Pellecchia, M.; Sem, D.S.; Wuthrich, K. NMR in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 211–219. [Google Scholar] [CrossRef]
- Joshi, R.; Qin, L.; Cao, X.; Zhong, S.; Voss, C.; Min, W.; Li, S.S.C. DLC1 SAM domain-binding peptides inhibit cancer cell growth and migration by inactivating RhoA. J. Biol. Chem. 2020, 295, 645–656. [Google Scholar] [CrossRef]
- Farmer, B.T., 2nd; Constantine, K.L.; Goldfarb, V.; Friedrichs, M.S.; Wittekind, M.; Yanchunas, J., Jr.; Robertson, J.G.; Mueller, L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat. Struct. Biol. 1996, 3, 995–997. [Google Scholar] [CrossRef]
- Honorato, R.V.; Trellet, M.E.; Jimenez-Garcia, B.; Schaarschmidt, J.J.; Giulini, M.; Reys, V.; Koukos, P.I.; Rodrigues, J.; Karaca, E.; van Zundert, G.C.P.; et al. The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat. Protoc. 2024. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Versini, R.; Sritharan, S.; Aykac Fas, B.; Tubiana, T.; Aimeur, S.Z.; Henri, J.; Erard, M.; Nusse, O.; Andreani, J.; Baaden, M.; et al. A Perspective on the Prospective Use of AI in Protein Structure Prediction. J. Chem. Inf. Model. 2024, 64, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 2022, 31, e4379. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Magana, P.; Kovalevskiy, O. pLDDT: Understanding Local Confidence. Available online: https://www.ebi.ac.uk/training/online/courses/alphafold/inputs-and-outputs/evaluating-alphafolds-predicted-structures-using-confidence-scores/plddt-understanding-local-confidence/ (accessed on 11 September 2024).
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Magana, P.; Kovalevskiy, O. Confidence Scores in AlphaFold-Multimer. Available online: https://www.ebi.ac.uk/training/online/courses/alphafold/inputs-and-outputs/evaluating-alphafolds-predicted-structures-using-confidence-scores/confidence-scores-in-alphafold-multimer/ (accessed on 11 September 2024).
- Barquilla, A.; Lamberto, I.; Noberini, R.; Heynen-Genel, S.; Brill, L.M.; Pasquale, E.B. Protein kinase A can block EphA2 receptor–mediated cell repulsion by increasing EphA2 S897 phosphorylation. Mol. Biol. Cell 2016, 27, 2757–2770. [Google Scholar] [CrossRef]
- Arias, M.; Piga, K.B.; Hyndman, M.E.; Vogel, H.J. Improving the Activity of Trp-Rich Antimicrobial Peptides by Arg/Lys Substitutions and Changing the Length of Cationic Residues. Biomolecules 2018, 8, 19. [Google Scholar] [CrossRef]
- Ma, Y.X.; Yao, A.F.; Chen, X.L.; Wang, L.; Ma, C.B.; Xi, X.P.; Chen, T.B.; Shaw, C.; Zhou, M. Generation of truncated derivatives through in silico enzymatic digest of peptide GV30 target MRSA both in vitro and in vivo. Comput. Struct. Biotechnol. J. 2021, 19, 4984–4996. [Google Scholar] [CrossRef]
- Walker-Daniels, J.; Riese, D.J., 2nd; Kinch, M.S. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol. Cancer Res. 2002, 1, 79–87. [Google Scholar]
- Locard-Paulet, M.; Lim, L.; Veluscek, G.; McMahon, K.; Sinclair, J.; van Weverwijk, A.; Worboys, J.D.; Yuan, Y.; Isacke, C.M.; Jorgensen, C. Phosphoproteomic analysis of interacting tumor and endothelial cells identifies regulatory mechanisms of transendothelial migration. Sci. Signal. 2016, 9, ra15. [Google Scholar] [CrossRef] [PubMed]
- Alves, D.S.; Westerfield, J.M.; Shi, X.; Nguyen, V.P.; Stefanski, K.M.; Booth, K.R.; Kim, S.; Morrell-Falvey, J.; Wang, B.C.; Abel, S.M.; et al. A novel pH-dependent membrane peptide that binds to EphA2 and inhibits cell migration. eLife 2018, 7, e36645. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R.; Kanvinde, P.; King, C.; Pasquale, E.B.; Hristova, K. The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun. Biol. 2018, 1, 15. [Google Scholar] [CrossRef]
- Baggio, C.; Udompholkul, P.; Gambini, L.; Pellecchia, M. Targefrin: A Potent Agent Targeting the Ligand Binding Domain of EphA2. J. Med. Chem. 2022, 65, 15443–15456. [Google Scholar] [CrossRef]
- Gomez-Soler, M.; Gehring, M.P.; Lechtenberg, B.C.; Zapata-Mercado, E.; Ruelos, A.; Matsumoto, M.W.; Hristova, K.; Pasquale, E.B. Ligands with different dimeric configurations potently activate the EphA2 receptor and reveal its potential for biased signaling. iScience 2022, 25, 103870. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.; Borg, J.; Vanhee, P.; Verschueren, E.; Baeten, L.; Delgado, J.; Schymkowitz, J.; Rousseau, F.; Stricher, F.; et al. BUILDMODEL. Available online: https://foldxsuite.crg.eu/command/BuildModel (accessed on 3 June 2024).
- Micsonai, A.; Moussong, E.; Wien, F.; Boros, E.; Vadaszi, H.; Murvai, N.; Lee, Y.H.; Molnar, T.; Refregiers, M.; Goto, Y.; et al. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]
- Leone, M.; Cellitti, J.; Pellecchia, M. The Sam domain of the lipid phosphatase Ship2 adopts a common model to interact with Arap3-Sam and EphA2-Sam. BMC Struct. Biol. 2009, 9, 59. [Google Scholar] [CrossRef]
- Griesinger, C.; Otting, G.; Wuethrich, K.; Ernst, R.R. Clean TOCSY for proton spin system identification in macromolecules. J. Am. Chem. Soc. 1988, 110, 7870–7872. [Google Scholar] [CrossRef]
- Piantini, U.; Sorensen, O.W.; Ernst, R.R. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A.J. Water suppression that works. Excitation sculpting using arbitrary waveforms and pulsed field gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Bartels, C.; Xia, T.; Billeter, M.; Güntert, P.; Wüthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M.; Poulsen, F.M. Sequence correction of random coil chemical shifts: Correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR 2011, 50, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Bonvin Lab. HADDOCK v2.4 Settings. Available online: https://wenmr.science.uu.nl/haddock2.4/settings (accessed on 11 June 2024).
- Bonvin Lab. HADDOCK2.2 Manual. Ambiguous Interaction Restraints. Available online: https://www.bonvinlab.org/software/haddock2.2/generate_air_help/ (accessed on 11 June 2024).
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef]
- Beach, S.S.; Hull, M.A.; Ytreberg, F.M.; Patel, J.S.; Miura, T.A. Molecular Modeling Predicts Novel Antibody Escape Mutations in the Respiratory Syncytial Virus Fusion Glycoprotein. J. Virol. 2022, 96, e0035322. [Google Scholar] [CrossRef]
- Kiel, C.; Serrano, L. Structure-energy-based predictions and network modelling of RASopathy and cancer missense mutations. Mol. Syst. Biol. 2014, 10, 727. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincenzi, M.; Mercurio, F.A.; La Manna, S.; Palumbo, R.; Pirone, L.; Marasco, D.; Pedone, E.M.; Leone, M. Exploring a Potential Optimization Route for Peptide Ligands of the Sam Domain from the Lipid Phosphatase Ship2. Int. J. Mol. Sci. 2024, 25, 10616. https://doi.org/10.3390/ijms251910616
Vincenzi M, Mercurio FA, La Manna S, Palumbo R, Pirone L, Marasco D, Pedone EM, Leone M. Exploring a Potential Optimization Route for Peptide Ligands of the Sam Domain from the Lipid Phosphatase Ship2. International Journal of Molecular Sciences. 2024; 25(19):10616. https://doi.org/10.3390/ijms251910616
Chicago/Turabian StyleVincenzi, Marian, Flavia Anna Mercurio, Sara La Manna, Rosanna Palumbo, Luciano Pirone, Daniela Marasco, Emilia Maria Pedone, and Marilisa Leone. 2024. "Exploring a Potential Optimization Route for Peptide Ligands of the Sam Domain from the Lipid Phosphatase Ship2" International Journal of Molecular Sciences 25, no. 19: 10616. https://doi.org/10.3390/ijms251910616
APA StyleVincenzi, M., Mercurio, F. A., La Manna, S., Palumbo, R., Pirone, L., Marasco, D., Pedone, E. M., & Leone, M. (2024). Exploring a Potential Optimization Route for Peptide Ligands of the Sam Domain from the Lipid Phosphatase Ship2. International Journal of Molecular Sciences, 25(19), 10616. https://doi.org/10.3390/ijms251910616