Abstract
Na+-K+ ATPase is an integral component of cardiac sarcolemma and consists of three major subunits, namely the α-subunit with three isoforms (α1, α2, and α3), β-subunit with two isoforms (β1 and β2) and γ-subunit (phospholemman). This enzyme has been demonstrated to transport three Na and two K ions to generate a trans-membrane gradient, maintain cation homeostasis in cardiomyocytes and participate in regulating contractile force development. Na+-K+ ATPase serves as a receptor for both exogenous and endogenous cardiotonic glycosides and steroids, and a signal transducer for modifying myocardial metabolism as well as cellular survival and death. In addition, Na+-K+ ATPase is regulated by different hormones through the phosphorylation/dephosphorylation of phospholemman, which is tightly bound to this enzyme. The activity of Na+-K+ ATPase has been reported to be increased, unaltered and depressed in failing hearts depending upon the type and stage of heart failure as well as the association/disassociation of phospholemman and binding with endogenous cardiotonic steroids, namely endogenous ouabain and marinobufagenin. Increased Na+-K+ ATPase activity in association with a depressed level of intracellular Na+ in failing hearts is considered to decrease intracellular Ca2+ and serve as an adaptive mechanism for maintaining cardiac function. The slight to moderate depression of Na+-K+ ATPase by cardiac glycosides in association with an increased level of Na+ in cardiomyocytes is known to produce beneficial effects in failing hearts. On the other hand, markedly reduced Na+-K+ ATPase activity associated with an increased level of intracellular Na+ in failing hearts has been demonstrated to result in an intracellular Ca2+ overload, the occurrence of cardiac arrhythmias and depression in cardiac function during the development of heart failure. Furthermore, the status of Na+-K+ ATPase activity in heart failure is determined by changes in isoform subunits of the enzyme, the development of oxidative stress, intracellular Ca2+-overload, protease activation, the activity of inflammatory cytokines and sarcolemmal lipid composition. Evidence has been presented to show that marked alterations in myocardial cations cannot be explained exclusively on the basis of sarcolemma alterations, as other Ca2+ channels, cation transporters and exchangers may be involved in this event. A marked reduction in Na+-K+ ATPase activity due to a shift in its isoform subunits in association with intracellular Ca2+-overload, cardiac energy depletion, increased membrane permeability, Ca2+-handling abnormalities and damage to myocardial ultrastructure appear to be involved in the progression of heart failure.
1. Introduction
Since the discovery of Na+-K+ ATPase by JC Skou in 1957, numerous investigators have examined the physiological, biochemical and pharmacological aspects of this enzyme to establish its role in cardiac function, health and disease [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. Na+-K+ ATPase is embedded in the sarcolemmal membrane and its main function is to transport three Na and two K ions through an energy-dependent mechanism for the generation of an electrochemical gradient across the cell membrane and the maintenance of cation homeostasis in cardiomyocytes. This enzyme serves as a receptor for exogenous cardiac glycosides such as digoxin and ouabain; different cardiotonic steroids, including endogenous ouabain and marinobufagenin, are known to modulate cardiac contractile activity upon its inhibition. The regulatory role of Na+-K+ ATPase with respect to the structure–function relationship and pathological implications, as well as its therapeutic aspects, has also been studied extensively [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. Na+-K+ ATPase has been shown to act as a signal transducer, regulating different metabolic processes in cardiomyocytes as well as cellular growth, differentiation, survival and death. The activity of Na+-K+ ATPase is regulated by different hormones through the phosphorylation/dephosphorylation of phospholemman (FXYD1), which is considered to be a γ-subunit of this enzyme. Na+-K+ ATPase is known to be made up of two other subunits, namely the α-subunit and β-subunit; the α-subunit occurs in three isoforms (α1, α2 and α3) whereas the β-subunit has two isoforms (β1 and β2) in the heart. These three subunits (α, β and γ) of this enzyme have been demonstrated to play specific roles in determining the function of Na+-K+ ATPase.
While the activation of Na+-K+ ATPase is considered to be involved in cell survival as an adaptive mechanism, the marked inhibition of this enzyme was reported to induce arrhythmias, cellular damage, cell death and cardiac dysfunction. Furthermore, low doses of cardiac glycosides, which moderately inhibit Na+-K+ ATPase, are known to exert positive inotropic action and beneficial effects, whereas high doses of these agents, which produce a marked inhibition of Na+-K+ ATPase, are known to exert cardiotoxic actions during heart failure. Thus, in view of the conflicting reports regarding alterations in the Na+-K+ ATPase activities in different types of failing hearts, the exact role of this enzyme in cardiac dysfunction during the development of heart failure is not understood [31,32,33]. This article therefore aims to review the existing literature regarding the involvement of changes in the status of this enzyme in the pathogenesis of heart failure. In addition, it plans to describe the association of alterations in Na+-K+ ATPase activity with changes in different isoforms of this enzyme in failing hearts. Efforts will be made to determine the involvement of some signal transduction mechanisms in modifying the regulation of Na+-K+ ATPase in different experimental models of heart failure. The relationship among changes in Na+-K+ ATPase activity, myocardial Na+, and K+, Mg2+ and Ca2+ content, as well as contractile activity during the progression of heart failure, will also be examined to gain insight into the multifunctional and complex nature of this enzyme.
2. Status of Na+-K+ ATPase Activity and Isoenzymes
2.1. Alterations in Na+-K+ ATPase Activity in Failing Hearts
Several conflicting reports concerning changes in the activity of Na+-K+ ATPase in several types of heart failure have appeared in the literature [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. Some of the studies showing increased, unaltered and decreased Na+-K+ ATPase activities [33,34,35,36,37,38,39,40,41,42,43,44,45] in both experimental and clinical conditions of heart failure are shown in Table 1. An increase in Na+-K+ ATPase activity was seen in the BIO 14.6 strain of cardiomyopathic hamsters with a moderate degree of congestive heart failure, as well as in heart failure in dogs following mitral insufficiency or pressure overload due to aortic banding [34,35,36]. On the other hand, heart failure due to pulmonary stenosis in dogs, as well as aortic insufficiency or aortic constriction in rabbits, were not associated with any changes in Na+-K+ ATPase activity [33,37]. While the observations of no changes in Na+-K+ ATPase activity in failing hearts may be due to their being observations of early stages of heart failure, increased Na+-K+ ATPase activity has also been reported in cardiac dysfunction due to vitamin deficiency in rats [46], K+-deficiency in guinea pigs [47] or the administration of cobalt in rats [48]. An increase in the activity of Na+-K+ ATPase was suggested to reduce the intracellular concentration of Na+, decrease the entry of Ca2+ through the Na+-Ca2+ exchange system, reduce the intracellular concentration of Ca2+ and thus depress cardiac function [35,36]. In this regard, it is noteworthy that the treatment of heart failure due to pressure overload with digoxin or prazosin was found to improve cardiac function and reduce the increased Na+-K+ ATPase activity [49,50]. Furthermore, the treatment of heart failure due to mitral insufficiency with prazosin was also observed to prevent the deterioration of cardiac function as well as the increased Na+-K+ ATPase activity in the failing heart [51]. Since an increase in Na+-K+ ATPase activity can be seen to reduce the concentration of intracellular Ca2+ and suppress the occurrence of intracellular Ca2+-overload, it is suggested that the increased Na+-K+ ATPase activity in the failing heart may serve as an adaptive mechanism for the induction of beneficial effects.

Table 1.
Alterations of Na+-K+ ATPase activity in different types of failing hearts.
The involvement of Na+-K+ ATPase in the pathogenesis of heart failure was suggested on the basis of various studies showing a reduction in the activity of this enzyme in failing hearts [32,38,39,40,41,42,43,44,45]. From the information in Table 1, it may be noted that depressions in Na+-K+ ATPase activity were observed in failing hearts in rabbits due to aortic constriction, in genetic cardiomyopathy in hamsters (UM-X7.1) and in tachycardia-induced cardiomyopathy in pigs [38,39,40]. Furthermore, heart failure due to pulmonary constriction in dogs, as well as myocardial infarction in rats, showed depressed Na+-K+ ATPase activity [41,42]. Low Na+-K+ ATPase activity was also found to occur in human hearts as a consequence of end-stage dilated cardiomyopathy, congestive heart failure and idiopathic dilated cardiomyopathy [43,44,45]. Biopsies taken from failing hearts during cardiac transplantation revealed depressed levels of Na+-K+ ATPase activity [32,52]. A reduction in Na+-K+ ATPase activity was also reported in animals with hypoadrenalism, as well as in contractile failure in isolated rat hearts perfused with hypoxic or substrate-free medium [53,54,55]. It is pointed out that improvements in cardiac function following heart failure were associated with attenuation of the depressed Na+-K+ ATPase activity in heart failure in infarcted animals treated with a metabolic inhibitor, propionyl L-carnitine [56]. These observations indicate that a reduction in Na+-K+ ATPase activity in cardiomyocytes may be involved in the pathogenesis of cardiac dysfunction in heart failure.
To gain further information regarding the role of alterations in Na+-K+ ATPase activity in cardiac dysfunction, the left-ventricle sarcolemmal ATPase activities were monitored at different stages of heart failure due to myocardial infarction in rats [57]. The results presented in Table 2 indicate that cardiac dysfunction, as reflected by a significant increase in LVEDP and depressions in both +dP/dT and −dP/dT, at early stages of heart failure was not associated with any significant alteration in Na+-K+ ATPase activity. On the other hand, cardiac dysfunction at both moderate and severe stages of heart failure was accompanied by marked depressions in Na+-K+ ATPase activity. Since sarcolemmal Mg2+-ATPase activity decreased significantly at early, moderate and severe stages of heart failure due to myocardial infarction (Table 2), it is likely that the observed reduction in Na+-K+ ATPase activity at moderate and severe stages of heart failure may be due to a generalized defect in sarcolemma. In another set of experiments [58], a marked depression of Na+-K+ ATPase activity at moderate stages of heart failure was not associated with any significant decrease in Mg2+-ATPase activity (Table 3). Furthermore, the treatment of myocardial infarcted animals with enalapril, an angiotensin-converting enzyme inhibitor, or losartan, an angiotensin II receptor antagonist, were observed to improve cardiac function and attenuate the depression in Na+-K+ ATPase activity without any changes in Mg2+-ATPase activity (Table 3). Changes in Na+-K+ ATPase activity were also examined in the UM-X7.1 strain of cardiomyopathic hamsters of different age groups [59]. On the basis of the accumulation of abdominal fluid (ascites), as well as increases in lung weight (wt), liver wt and heart to body wt ratio, these cardiomyopathic hamsters, at 90–100 days, 120–160 days, 160–200 days and 200–280 days, were considered to be at pre-failure, early failure, moderate failure and severe failure stages, respectively [59]. It can be seen from Table 4 that Na+-K+ ATPase activity was depressed at pre-failure as well as at all stages of heart failure in the UM-X7.1 strain of cardiomyopathic hamsters without any significant alterations in Mg2+-ATPase activity except in animals at the severe stage of heart failure. These observations provide further support to the view that reduced Na+-K+ ATPase activity may be specifically involved not only in the development but also in the progression of heart failure.
Table 2.
Alterations in cardiac function and sarcolemmal Na+-K+ ATPase at different stages of heart failure due to myocardial infarction in rats.
Table 3.
Improvement of cardiac dysfunction and depressed sarcolemmal Na+-K+ ATPase activity in heart failure upon treatment of infarcted rats with enalapril (10 mg/kg/day) or losartan (20 mg/kg/day).
Table 4.
General characteristics and alterations in sarcolemmal Na+-K+ ATPase activity of cardiomyopathic hamsters (UM-X 7.1) of different age groups.
2.2. Alterations in Na+-K+ ATPase Isoenzymes in Failing Hearts
Extensive studies in both humans and animals revealed that Na+-K+ ATPase in failing hearts is composed of the α-, β- and γ-subunits, which determine its overall activity and are known to have distinctly different functions from each other [7,9,17,20,31,60,61,62]. While the α-subunit primarily contains binding sites for Na+, K+ and ATP as well as cardiotonic glycosides and plays a catalytic role in the enzyme activity, the β-subunit is concerned with the transport and anchoring of the newly synthesized Na+-K+ ATPase to the sarcolemma membrane, as well as modulation of the ATPase activity. On the other hand, the γ-subunit or phospholemman regulates Na+-K+ ATPase activity upon phosphorylation/dephosphorylation because unphosphorylated and phosphorylated forms of phospholemman exert inhibitory and stimulatory actions, respectively, on Na+-K+ ATPase. Decreased Na+-K+ ATPase activity in human heart failure was found to be associated with lower protein expressions for the α1-, α3- and β1-subunits without any changes in their respective mRNA levels in cardiomyocytes [63,64]. No alterations in protein or mRNA levels for the α2-subunit of Na+-K+ ATPase were observed in human heart failure [63,64]. The downregulation of Na+-K+ ATPase density in patients with dilated cardiomyopathy was also not associated with any change in mRNA levels for the α1-, α2 and α3-subunits [65].
Since Na+-K+ ATPase α-subunit isoforms have a similar affinity for cardiac glycosides, it was suggested that the increased sensitivity that occurs during heart failure to cardiac glycosides is probably due to a depression in Na+-K+ ATPase density rather than to the selective inhibition of any α-subunit isoform of the enzyme [66]. It should also be pointed out that while reduced Na+-K+ ATPase activity in heart failure due to tachycardia, aortic valve disease, hypertension and different cardiomyopathies [67,68] is accompanied by depressed levels of the α1-, α3- and β1-subunits, the overexpression of the α2-subunit of Na+-K+ ATPase, which has been shown to be involved in Ca2+-signaling [69], was observed to attenuate cardiac hypertrophy and heart failure [70,71]. In addition, the α2-subunit has been shown to protect against cardiac remodeling and β-adrenoreceptor desensitization after myocardial infarction [72]. In contrast, β1-subunit knock-out mice were insensitive to the positive inotropic effect of ouabain [73]. Depression in the phosphorylation of phospholemman (γ-subunit) has also been reported to be associated with decreased Na+-K+ ATPase activity in heart failure [9,21,31]. Thus, different isoforms of Na+-K+ ATPase have been demonstrated to be involved in diverse pathological functions during the development of heart failure.
Because there is an increase in the plasma level of marinobufagenin and a depression in the sensitivity of Na+-K+ ATPase to this cardiotonic steroid, as well as a reduction in the α1-isoform during cardiac hypertrophy and heart failure, it was suggested that a shift in endogenous Na+-K+ ATPase ligand production is linked to a shift in myocardial Na+-K+ ATPase isoforms [74]. While the level of α1-isoform was lower in decompensated cardiac hypertrophy than that in compensated cardiac hypertrophy in guinea pigs due to aortic stenosis, increased levels of the α2-isoform were involved in decompensated hypertrophy and the development of arrhythmias [75]. Heart failure induced by tachycardia in dogs decreased Na+-K+ ATPase activity, α3-isoform protein and α3-isoform mRNA levels without any changes in α1- or α2-isoform proteins. These alterations in the failing heart were found to be similar to those seen upon chronic infusion of norepinephrine, except that the downregulation of the α3-isoform protein was not associated with a decrease in the α3-isoform mRNA levels [76,77]. The information in Table 5 indicates that the reduction in Na+-K+ ATPase activity in the UM-X7.1 strain of cardiomyopathic hamsters at a severe stage of heart failure was shown to be due to increased levels of α1- and β1-isoform proteins, but mRNA levels for the α2-subunit were decreased [78]. It can also be seen from Table 5 that the protein levels of the α3-isoform were depressed in these cardiomyopathic hamsters; however, the mRNA levels for this subunit were not detectable in either control or diseased animals [78]. The reduced Na+-K+ ATPase activity in the MS 200 strain of cardiomyopathic hamsters with dilated cardiomyopathy was accompanied by decreased levels of α1- and β1-isoform proteins without any changes in the level of the α2-subunit [79]. It was also pointed out that a reduction in Na+-K+ ATPase activity, α1-subunit protein content and α1-subunit mRNA levels was also observed in the BIO14.6 strain of hamsters with severe congestive heart failure [80,81].
Table 5.
Alterations in sarcolemmal Na+-K+ activity, isoform protein content and isoform mRNA levels in 250 days old cardiomyopathic hamsters (UM-X7.1) at severe stages of heart failure.
From Table 6, it can be observed that the reduced level of Na+-K+ ATPase activity in a rat model of heart failure due to myocardial infarction was associated with a dramatic depression in both protein and mRNA levels for the α1-, α2- and β1-subunits as well as an increase in both protein and mRNA levels for the α3-subunits; these alterations were partially prevented upon the treatment of the animals with imidapril, an angiotensin II-converting inhibitor [58]. In another study, the treatment of infarcted animals with a moderate degree of heart failure with a blocker of the renin–angiotensin system such as enalapril and losartan was observed to attenuate the decreased protein and mRNA levels in the α2-isoform, as well as the increased protein and mRNA levels in the α3-isoform in the failing heart following 8 weeks of myocardial infarction [82]. End-stage heart failure due to 40 weeks of myocardial infarction was associated with reduced Na+-K+ ATPase activity and mRNA levels in the α2-isoform, as well as increased mRNA levels in the α1- and α3-isoforms; these alterations were also partially attenuated by treatment with imidapril [83]. It should be mentioned that the reduced Na+-K+ ATPase activity in heart failure due to myocardial infarction was also reported to be associated with the decreased expression of protein and mRNA levels for the α2-subunit and increased expression of the α2-subunit, without any changes in the levels of α1- and β1-subunits of Na+-K+ ATPase [84]. These observations suggest that cardiac dysfunction in failing hearts is associated with reduced Na+-K+ ATPase activity and a wide variety of alterations in the α1-, α2-, α3- and β1-subunits of Na+-K+ ATPase depending upon the type, species and stage of heart failure. The summary of observations regarding the role of alterations in Na+-K+ ATPase isozymes in depressing the enzyme activity in heart failure is illustrated in Figure 1.
Table 6.
Alterations in sarcolemmal Na+-K+ ATPase activity, subunit protein content and subunit mRNA levels in heart failure due to myocardial infarction with or without Imidapril (1 mg/Kg/day).
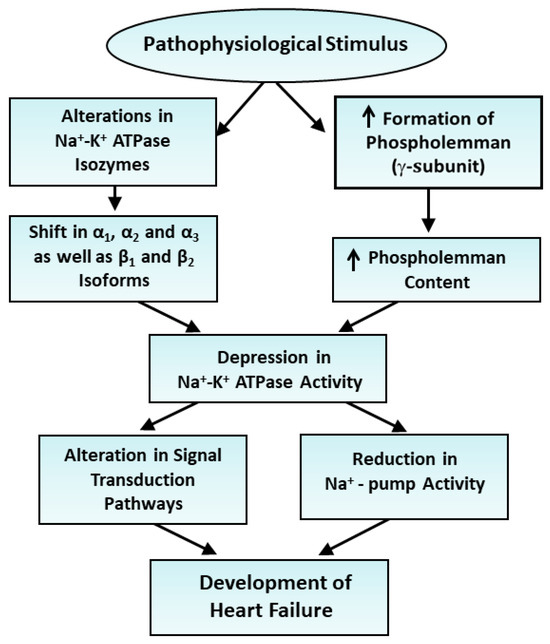
Figure 1.
Role of alterations in Na+-K+ ATPase isozymes and depression in the enzyme activity in the development of heart failure. ↑—increase.
3. Signal Transduction and Regulation of Na+-K+ ATPase in Heart Failure
In addition to serving as a Na+-pump, Na+-K+ ATPase has been reported to function as a signal transducer upon inhibition by both exogenous and endogenous cardiotonic steroids [15,85,86,87,88,89,90]. The inhibition of Na+-K+ ATPase by cardiac glycosides indicates that the Ras/Raf/MEK/MAPK pathway is activated upstream of Ras by tyrosine kinase Src. Furthermore, these agents stimulate redox-activated protein kinases such as protein kinase A (PKA), protein kinase C (PKC) and calcium–calmodulin kinase II (CaMKII) [85,86,87,88,89,90]. The activated kinases are intimately involved in signal transduction mechanisms which are associated with the increased formation of oxyradicals, enhanced oxidative phosphorylation and impaired mitochondrial Ca2+ retention in both normal and failing hearts [88,89,90,91,92]. It should be emphasized that an increase in the intracellular concentration of Na+ achieved via the inhibition of this enzyme with cardiac glycosides not only promotes the elevation of intracellular Ca2+ through the Na+-Ca2+ exchange system but also promotes the activation of sarcolemmal L-type Ca2+ channels, and store-operated Ca2+-channels also help to increase the intracellular Ca2+ [93,94]. Furthermore, an increased concentration of intracellular Na+ results in the release of Ca2+ from the sarcoplasmic reticulum, involving CaMKII, PKA and inositol-3-phosphate receptor systems [95]. The signal transduction mechanisms initiated upon the inhibition of Na+-K+ ATPase have also been reported to induce cell growth, cardiac hypertrophy heart failure and arrhythmias [96,97,98]. In addition, several cardiotonic steroids, upon increases in the intracellular concentrations of both Na+ and Ca2+, have been observed to develop oxidative stress, apoptosis and cardiomyocyte injury due to the activation of diverse signal transduction systems [99,100,101,102,103].
The activity of Na+-K+ ATPase in normal and diseased hearts is regulated by different hormones, prostaglandins, growth factors and endogenous ligands through the participation of various signal transduction mechanisms [7,8,9,18,19,22,23,104]. The activation of β1-adrenergic receptors by catecholamines has been shown to affect the cyclic AMP-PKA mechanism, whereas the activation of β2- and β3-adrenergic receptors has been reported to modify Na+-K+ ATPase activity in the failing heart through some isoform-specific mechanisms [77,105,106,107,108,109,110]. On the other hand, low concentrations of angiotensin II were reported to stimulate Na+-K+ ATPase by increasing tyrosine kinase and MAP kinase activities, whereas high concentrations of this hormone are known to inhibit the enzyme via oxidative stress due to the marked activation of NADPH oxidase [111]. Estradiol and insulin-like growth factor have been shown to increase the expression of Na+-K+ ATPase isoforms due to the formation of nitric oxide [23,112]. Thromboxane β2, a product of prostaglandin, was shown to depress Na+-K+ ATPase activity [113]. Likewise, endogenous ligand marinobufagenin have been observed to decrease Na+-K+ ATPase activity by affecting the α1-subunit of the enzyme [7,8,18,114]. Thus, a wide variety of hormones and other factors produced during the development of heart failure have been reported to modify Na+-K+ ATPase activity in cardiomyocytes.
It is now well established that the regulation of Na+-K+ ATPase is mainly carried out as a consequence of the phosphorylation of phospholemman through different protein kinases as well as dephosphorylation by protein phosphatase-1 in normal and failing hearts [5,20,21,61,62,115,116,117,118,119,120]. It should be noted that unphosphorylated phospholemman exerts an inhibitory effect on Na+-K+ ATPase, whereas phosphorylated phospholemman increases the Na+-K+ ATPase activity due to changes in the protein conformation. Alterations in both the expression and phosphorylation of phospholemman have been reported to contribute to maladaptive cardiac hypertrophy, reduced Na+-K+ ATPase activity, arrhythmias and heart failure [121,122,123]. The activation of Na+-K+ ATPase by phosphorylated phospholemman is isoform-specific and has been shown to provide cardioprotection in diseased heart [124]. Stabilizing the enzyme via the antibody SSA412 at a specific site of this enzyme was shown to activate Na+-K+ ATPase and produce an inotropic effect [125]. A polyclonal antibody (DRRSAb), which was Na+-K+ ATPase DR region-specific, was also found to activate the enzyme and produce cardioprotection against ischemic injury through the stimulation of extracellular signal-regulated kinase and the phosphoinositide 3-kinase/Akt pathway [126]. Furthermore, this antibody was shown to improve cardiac function, alleviate cardiac hypertrophy and reduce oxidative stress by stabilizing membrane Na+-K+ ATPase in isoproterenol-treated mice [127]. Accordingly, it has been suggested that the activation as well as the stabilization of Na+-K+ ATPase may result in a new strategy for improving the therapy of heart failure.
A decrease in Na+-K+ ATPase activity was observed during the transition of cardiac hypertrophy to heart failure; this change was associated with an increase in the plasma level of marinobufagenin, a decrease in α1-isoform and an increase in the α3-isoform of the enzyme [7,8,18,74]. In fact, the infusion of marinobufagenin was also shown to depress Na+-K+ ATPase activity, reduce α1-isoform and induce heart failure [103]. Different mineralocorticoids such as aldosterone, as well as various inflammatory cytokines such as the tumor necrosis factor (TNF-α), the levels of which are increased in heart failure, have been reported to reduce Na+-K+ ATPase expression as well as activity and produce cardiac fibrosis [128,129,130,131,132]. Furthermore, oxidative stress, intracellular Ca2+-overload, the activation of proteases and alterations in the lipid composition of the sarcolemmal membrane have been demonstrated to depress Na+-K+ ATPase activity in heart failure [27,28,31,133]. Observations regarding the regulatory effects of diverse factors provide evidence that a reduction in Na+-K+ ATPase activity may be involved in the pathogenesis of heart failure. On the other hand, various hypertrophic stimuli, which are known to be associated with adaptive mechanisms, were shown to increase Na+-K+ ATPase activity as well as gene expression [31,134]. Thus, it appears that interventions that may induce an increase in Na+-K+ ATPase activity may prove beneficial in therapy aiming to treat heart failure.
From this discussion, it is evident that the activation of the signal transduction pathway as a consequence of a depression in Na+-K+ ATPase is a distinct function independent of its Na+-pump activity. The signal-transducing function of Na+-K+ ATPase was discovered by Zijian Xie in 2002 [89]. This view is supported by the fact that low doses of cardiac glycosides were observed to affect the signal transduction pathway without any effect on Na+ pump activity. The salient features of the involvement of signal transduction pathways in the development of heart failure are shown in Figure 2.
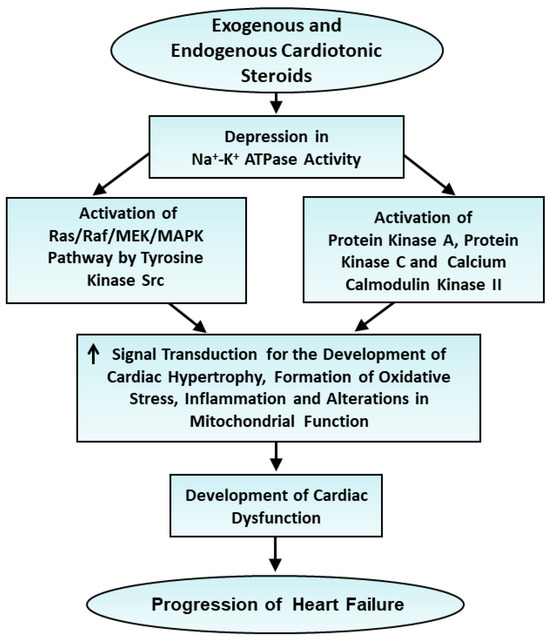
Figure 2.
Role of changes in signal transduction mechanisms following depression of Na+-K+ ATPase activity in the progression of heart failure. ↑—increase.
4. Alterations in Na+-K+ ATPase and Intracellular Cation Content in Contractile Dysfunction
In view of the important role of some cations, such as Na+, K+, Ca2+ and Mg2+, in cardiac function and metabolism, different subcellular organelles, including sarcolemma, sarcoplasmic reticulum and mitochondria, are known to be intimately involved in regulating the intracellular concentration of these cations. However, these regulatory membrane systems become defective following heart failure and lead to marked alterations in the electrolyte composition of the myocardium during the development of cardiac dysfunction in various types of failing hearts [135,136]. The high levels of intracellular Na+ and low levels of intracellular K+ in the failing heart are generally attributed to reduced Na+-K+ ATPase activity, whereas the increased levels of intracellular Ca2+ are attributed to the augmentation of Na+-Ca2+ exchange activity. However, several investigators have indicated that high levels of Na+ in the failing myocardium may also be a consequence of the increased Na+-H+ exchange systems in both the sarcolemma and mitochondria, as well as the increased activation of sarcolemmal Na+-channels [137,138,139,140,141,142,143]. Furthermore, the elevated levels of Ca2+ in the failing heart are considered to be due to the increased release of Ca2+ from the sarcoplasmic reticulum, as well as depressed Ca2+-pump activity in the sarcolemmal membrane. Thus, the involvement of the reduced activity of Na+-K+ ATPase and elevated levels of intracellular Na+ and Ca2+ cannot be considered to fully explain the pathogenesis of heart failure.Although heart failure is a complex problem and there are very many factors which may play different roles in this devastating disease, the following discussion is centered around gaining some information regarding the relationship among changes in the Na+-K+ ATPase activity, alterations in myocardial cation content and contractile dysfunction under various experimental conditions. In a rabbit model of cardiac hypertrophy and heart failure induced by a catheter with or without bacterial infection [144,145,146], it was observed that contractile dysfunction in hypertrophied failing hearts was associated with depressed Na+-K+ ATPase activity, an increased intracellular concentration of Na+ and a decreased level of intracellular K+ (Table 7). However, the intracellular Ca2+ in the hypertrophid failing heart was decreased and alterations in the intracellular Mg2+ were variable (Table 7).
Table 7.
Alterations in cardiac function, cation content and Na+-K+ ATPase activity at different times of inducing bacterial endocarditis in rabbits.
Although the observed changes in the concentrations of Na+, K+ and Mg2+ in this model of cardiac hypertrophy and failure are in agreement with the results for other types of failing hearts [38,39,40,41,42,43,44,45,147], the decrease in the concentration of intracellular Ca2+ in these experimental hearts was different from the other results. In this regard, it is pointed out that heart failure has been shown to be associated with either Ca2+ overload or Ca2+ deficiency [136]. We also examined the relationship between changes in cation content and Na+-K+ ATPase activity using isolated rat hearts perfused with different interventions known to induce contractile dysfunction [135]. The perfusion of hearts with Ca2+-free medium, which is known to generate no contractile activity [135], was found to decrease all cation content as well as Na+-K+ ATPase activity (Table 8). In contrast, reperfusion of the Ca2+-free perfused hearts with Ca2+-containing medium [135] was observed to increase Na+ and Ca2+ content in addition to depressing Na+-K+ ATPase activity, as well as K+ and Mg2+ content (Table 8).
Table 8.
Changes in sarcolemmal Na+-K+ ATPase activity and cation content in isolated rat hearts subjected to perfusion with Ca2+-free medium for 20 min or reperfusion for 10 min with medium containing 1.25 mM Ca2+.
The inability of the heart to generate contractile activity upon perfusion for 20 min with Na+-free or K+-free medium [135] was associated with a depression in Na+-K+ ATPase activity (Table 9). However, myocardial Na+, K+ and Mg2+ contents were decreased and Ca2+ level was increased upon perfusion with the Na+-free medium, whereas Na+ and Ca2+ contents were increased and K+ as well as Mg2+ contents were decreased upon perfusion with the K+-free medium (Table 9). The perfusion of hearts with substrate-free medium for 2 h resulted in loss of contractile function [135], a marked reduction in Na+-K+ ATPase activity and a decrease in K+ and Mg2+ content, in addition to an increase in Na+ content, without any changes in Ca2+ levels (Table 10). Although hearts perfused with hypoxic medium for 30 min showed no contractile function [135] and a depression in Na+-K+ ATPase activity, myocardial Na+ content was increased whereas K+, Ca2+ and Mg2+ contents were decreased (Table 10). These observations provide evidence that this reduced Na+-K+ ATPase activity may be associated with the development of cardiac dysfunction but showed no relationship with changes in myocardial cation content. It appears that alterations in cation content in the failing heart are determined by diverse mechanisms including changes in Na+-K+ ATPase activity, sarcolemmal and mitochondrial Na+-H+ exchange activities, sarcolemmal and sarcoplasmic reticulum cation channels’ activities, and sarcolemmal permeability. Thus, alterations in Na+ and Ca2+ contents in the failing heart cannot be considered to be exclusively due to the reduction in Na+-K+ ATPase expression or activity in the failing heart. The ways in which Na+-K+ ATPase induces alterations in cardiac cation content and subsequent changes in metabolic processes, as well as changes in the pathogenesis of heart failure and associated arrhythmias and cardiac dysfunction, are depicted in Figure 3.
Table 9.
Changes in sarcolemmal Na+-K+ ATPase activity and cation content of isolated rat hearts perfused with sodium-free or potassium-free medium for 20 min.
Table 10.
Changes in sarcolemmal Na+-K+ ATPase activity and cation content of isolated rat hearts upon perfusion with substrate-free or hypoxia medium.
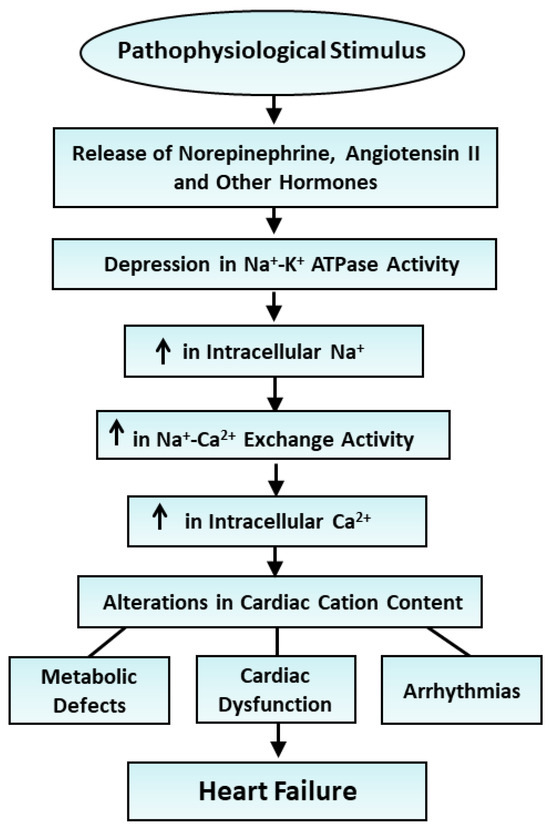
Figure 3.
Role of various hormones in the depression Na+-K+ ATPase activity and changes in cardiac cation contents in the development of heart failure. ↑—increase.
5. Conclusions
Extensive studies have indicated that Na+-K+ ATPase not only acts as a Na+-pump for maintaining myocardial cation composition but also serves as a signal transducer, modifying cardiac function and metabolism, when its activity is inhibited by different cardiotonic glycosides and steroids. Different isoforms of the α- and β-subunits of Na+-K+ ATPase are involved in the catalytic function and in anchoring this enzyme to the sarcolemmal membrane, respectively, whereas the γ-subunit (phospholemman) of Na+-K+ ATPase, by virtue of its phosphorylation/dephosphorylation ability, is known to regulate the enzyme activity. Several hormones and growth factors have been shown to regulate Na+-K+ ATPase activity by affecting different redox-sensitive or redox-insensitive tyrosine kinases in an isoform-specific manner in both normal and failing hearts. Some endogenous ligands, such as marinobufagenin, the levels of which are increased in heart failure, are known to have an inhibitory affect on Na+-K+ ATPase activity. Some interventions, such as oxidative stress and inflammatory cytokines, the levels of which are increased in heart failure, have been shown to adversely affect Na+-K+ ATPase activity as well as inducing the development of apoptosis and fibrosis. However, in spite of the numerous associations between alterations in Na+-K+ ATPase activity and a wide variety of defects in metabolic, cation transport and functional processes, as well as Ca2+-handling abnormalities, the exact role that changes in Na+-K+ ATPase activity play in the pathogenesis of heart failure remains far from clear.
In this article, we reviewed the literature regarding alterations in Na+-K+ ATPase activity during the development of heart failure. It was observed that Na+-K+ ATPase activity is increased, decreased or unaltered in the failing heart in both experimental and clinical situations. These variable changes in Na+-K+ ATPase activity appear to depend upon the stage and type of pathological stimuli responsible for inducing heart failure. Nonetheless, some studies have shown that increased Na+-K+ ATPase activity in the failing heart may promote the efflux of Na+, depress the intracellular concentration of Na+ and thereby reduce the intracellular level of Ca2+ due to its lack of effect on the Na+-Ca2+ exchange system. Accordingly, the increased Na+-K+ ATPase activity due to heart failure may serve as an adaptive mechanism. On the other hand, the reduced activity of Na+-K+ ATPase in the failing heart has been reported to depress the removal of Na+ from cardiomyocytes, increase the intracellular concentration of Na+ and thus increase the intracellular concentration of Ca2+ due to the participation of the Na+-Ca2+ exchange system. In view of the harmful effects of an excessive amount of intracellular Ca2+ on cardiac function and metabolism, the reduced Na+-K+ ATPase activity in the failing heart is considered to be a pathogenic factor for the development and/or progression of heart failure. It is therefore of critical importance that some novel therapies are developed to increase Na+-K+ ATPase activity in the failing heart to improve the treatment of heart failure. This view is supported by the observation that some specific Na+-K+ ATPase antibody preparations, which stabilize and activate Na+-K+ ATPase, have been reported to improve cardiac function and prevent arrhythmias in heart failure.
Author Contributions
The literature search and the writing of the first draft were conducted by V.E. and A.D.A.; the data were tabulated by V.E. and A.D.A. The conceptualization of the project, interpretation of results and preparation of final manuscript were conducted by N.S.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The infrastructure support for the preparation of this article was provided by the St. Boniface Hospital Research Foundation, Winnipeg, Canada. Thanks are due to Khushman Kaur for helping to prepare this manuscript.
Conflicts of Interest
The authors have no conflicts of interest.
References
- Skou, J.C. Enzymatic basis for active transport of Na+ and K+ across cell membrane. Physiol. Rev. 1965, 45, 596–617. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H. Biochemistry of Na, K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Ziegelhoffer, A.; Kjeldsen, K.; Bundgaard, H.; Breier, A.; Vrbjarm, N.; Dzurba, A. Na, K-ATPase in the myocardium: Molecular principles, functional and clinical aspects. Gen. Physiol. Biophys. 2000, 19, 9–47. [Google Scholar]
- Jorgensen, P.L.; Hakansson, K.O.; Karlish, S.J. Structure and mechanism of Na, K-ATPase: Functional sites and their interactions. Annu. Rev. Physiol. 2003, 65, 817–849. [Google Scholar] [CrossRef]
- Garty, H.; Karlish, S.J. Role of FXYD proteins in ion transport. Annu. Rev. Physiol. 2006, 68, 431–459. [Google Scholar] [CrossRef]
- Shinoda, T.; Ogawa, H.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 2009, 459, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef]
- Lingrel, J.B. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na, K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef]
- Fuller, W.; Tulloch, L.B.; Shattock, M.J.; Calaghan, S.C.; Howie, J.; Wypijewski, K.J. Regulation of the cardiac sodium pump. Cell Mol. Life Sci. 2013, 70, 1357–1380. [Google Scholar] [CrossRef]
- Schuurmans-Stekhoven, F.; Banting, S.L. Transport adenosine triphosphatase properties and functions. Physiol. Rev. 1981, 61, 2–76. [Google Scholar] [CrossRef]
- Schwartz, A.; Lindenmayer, G.E.; Allen, J.C. The sodium-potassium adenosine triphosphatase: Pharmacological, physiological and biochemical aspects. Pharmacol. Rev. 1975, 27, 3–134. [Google Scholar]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Horisberger, J.D.; Lemas, V.; Kraehenbuhl, J.P.; Rossier, B.C. Structure-function relationship of Na, K-ATPase. Annu. Rev. Physiol. 1991, 53, 565–584. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J. Isozymes of the Na+/K+ -ATPase. Biochim. Biophys. Acta 1989, 988, 185–220. [Google Scholar] [CrossRef]
- Chakraborti, S.; Dhalla, N.S. (Eds.) Regulation of Membrane Na+-K+ ATPase; Advances in Biochemistry in Health and Disease; Springer International Publishing: Cham, Switzerland, 2016; Volume 15, pp. 1–436. [Google Scholar] [CrossRef]
- McDonough, A.A.; Velotta, J.B.; Schwinger, R.H.; Philipson, K.D.; Farley, R.A. The cardiac sodium pump: Structure and function. Basic. Res. Cardiol. 2002, 97, I19–I24. [Google Scholar] [CrossRef] [PubMed]
- Müller-Ehmsen, J.; McDonough, A.A.; Farley, R.A.; Schwinger, R.H. Sodium pump isoform expression in heart failure: Implication for treatment. Basic. Res. Cardiol. 2002, 97, I25–I30. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef]
- Galougahi, K.K.; Liu, C.C.; Bundgaard, H.; Rasmussen, H.H. β-Adrenergic regulation of the cardiac Na+-K+ ATPase mediated by oxidative signaling. Trends Cardiovasc. Med. 2012, 22, 83–87. [Google Scholar] [CrossRef]
- Cheung, J.Y.; Zhang, X.Q.; Song, J.; Gao, E.; Chan, T.O.; Rabinowitz, J.E.; Koch, W.J.; Feldman, A.M.; Wang, J. Coordinated regulation of cardiac Na+/Ca2+ exchanger and Na+-K+-ATPase by phospholemman (FXYD1). Adv. Exp. Med. Biol. 2013, 961, 175–190. [Google Scholar]
- Shattock, M.J. Phospholemman: Its role in normal cardiac physiology and potential as a druggable target in disease. Curr. Opin. Pharmacol. 2009, 9, 160–166. [Google Scholar] [CrossRef]
- Silva, E.; Soares-da-Silva, P. New insights into the regulation of Na+,K+-ATPase by ouabain. Int. Rev. Cell Mol. Biol. 2012, 294, 99–132. [Google Scholar] [PubMed]
- Obradovic, M.; Stanimirovic, J.; Panic, A.; Bogdanovic, N.; Sudar-Milovanovic, E.; Cenic-Milosevic, D.; Isenovic, E.R. Regulation of Na+/K+-ATPase by estradiol and IGF-1 in cardio-metabolic diseases. Curr. Pharm. Des. 2017, 23, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Bundgaard, H.; Müller-Ehmsen, J.; Kjeldsen, K. The Na, K-ATPase in the failing human heart. Cardiovasc. Res. 2003, 57, 913–920. [Google Scholar] [CrossRef]
- Bartlett, D.E.; Miller, R.B.; Thiesfeldt, S.; Lakhani, H.V.; Shapiro, J.I.; Sodhi, K. The role of Na/K-ATPase signaling in oxidative stress related to aging: Implications in obesity and cardiovascular disease. Int. J. Mol. Sci. 2018, 19, 2139. [Google Scholar] [CrossRef]
- Baartscheer, A.; van Borren, M.M. Sodium ion transporters as new therapeutic targets in heart failure. Cardiovasc. Hematol. Agents Med. Chem. 2008, 6, 229–236. [Google Scholar] [CrossRef]
- Figtree, G.A.; Keyvan Karimi, G.; Liu, C.C.; Rasmussen, H.H. Oxidative regulation of the Na+-K+ pump in the cardiovascular system. Free Radic. Biol. Med. 2012, 53, 2263–2268. [Google Scholar] [CrossRef]
- Liu, C.C.; Fry, N.A.; Hamilton, E.J.; Chia, K.K.; Garcia, A.; Karimi Galougahi, K.; Figtree, G.A.; Clarke, R.J.; Bundgaard, H.; Rasmussen, H.H. Redox-dependent regulation of the Na⁺-K⁺ pump: New twists to an old target for treatment of heart failure. J. Mol. Cell Cardiol. 2013, 61, 94–101. [Google Scholar] [CrossRef]
- Khan, H.; Metra, M.; Blair, J.E.; Vogel, M.; Harinstein, M.E.; Filippatos, G.S.; Sabbah, H.N.; Porchet, H.; Valentini, G.; Gheorghiade, M. Istaroxime, a first in class new chemical entity exhibiting SERCA-2 activation and Na-K-ATPase inhibition: A new promising treatment for acute heart failure syndromes? Heart Fail. Rev. 2009, 14, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Ambrosy, A.P.; Ferrandi, M.; Ferrari, P. Combining SERCA2a activation and Na-K ATPase inhibition: A promising new approach to managing acute heart failure syndromes with low cardiac output. Discov. Med. 2011, 12, 141–151. [Google Scholar]
- Elimban, V.; Bartekova, M.; Xu, Y.-J.; Dhalla, N.S. Regulation of membrane Na+-K+ ATPase in health and disease. In Regulation of Membrane Na+-K+ ATPase; Advances in Biochemistry in Health and Disease; Chakraborti, S., Dhalla, N.S., Eds.; Springer Int Pub.: Cham, Switzerland, 2016; Volume 15, pp. 311–322. [Google Scholar] [CrossRef]
- Bundgaard, H.; Kjeldsen, K. Human myocardial Na, K-ATPase concentration in heart failure. Mol. Cell Biochem. 1996, 163–164, 277–283. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Sulakhe, P.V.; Dhalla, N.S. Alterations in the activity of cardiac Na+ K+-stimulated ATPase in congestive heart failure. Exp. Mol. Path. 1973, 18, 100–111. [Google Scholar] [CrossRef]
- Prasad, K.; Khatter, J.C.; Bharadwaj, B. Intra- and extracellular electrolytes and sarcolemmal ATPase in the failing heart due to pressure overload in dogs. Cardiovasc. Res. 1979, 13, 95–104. [Google Scholar] [CrossRef]
- Khatter, J.C.; Prasad, K. Myocardial sarcolemmal ATPase in dogs with induced mitral insufficiency. Cardiovasc. Res. 1976, 10, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Peterson, M.B.; Welty, J.D. Sarcolemmal and sarcoplasmic reticular ATPase activities in the failing canine heart. Circ. Res. 1971, 29, 14–20. [Google Scholar] [CrossRef]
- Yazaki, Y.; Fujii, J. Depressed Na+-K+-ATPase activity in the failing rabbit heart. Jap. Heart J. 1972, 13, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Singh, J.N.; Bajusz, E.; Jasmin, G. Comparison of heart sarcolemmal enzyme activities in normal and cardiomyopathic (UM-X7.1) hamsters. Clin. Sci. Mol. Med. 1976, 51, 233–242. [Google Scholar] [CrossRef]
- Spinale, F.G.; Clayton, C.; Tanaka, R.; Fulbright, B.M.; Mukherjee, R.; Schulte, B.A.; Crawford, F.A.; Zile, M.R. Myocardial Na+, K+-ATPase in tachycardia induced cardiomyopathy. J. Mol. Cell Cardiol. 1992, 24, 277–294. [Google Scholar] [CrossRef]
- Fan, T.H.; Frantz, R.P.; Elam, H.; Sakamoto, S.; Imai, N.; Liang, C.S. Reductions of myocardial Na-K-ATPase activity and ouabain binding sites in heart failure: Prevention by nadolol. Am. J. Physiol. 1993, 265, H2086–H2093. [Google Scholar] [CrossRef]
- Dixon, I.M.; Hata, T.; Dhalla, N.S. Sarcolemmal Na+-K+-ATPase activity in congestive heart failure due to myocardial infarction. Am. J. Physiol. 1992, 262, C664–C671. [Google Scholar] [CrossRef]
- Nørgaard, A.; Bjerregaard, P.; Baandrup, U.; Kjeldsen, K.; Reske-Nielsen, E.; Thomsen, P.E. The concentration of the Na, K-pump in skeletal and heart muscle in congestive heart failure. Int. J. Cardiol. 1990, 26, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, O.; Holthe, M.R.; Svindland, A.; Aksnes, G.; Sejersted, O.M.; Ilebekk, A. Na, K-pump concentration in hypertrophied human hearts. Eur. Heart J. 1994, 15, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Ishino, K.; Bøtker, H.E.; Clausen, T.; Hetzer, R.; Sehested, J. Myocardial adenine nucleotides, glycogen, and Na, K-ATPase in patients with idiopathic dilated cardiomyopathy requiring mechanical circulatory support. Am. J. Cardiol. 1999, 83, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Fedelesova, M.; Sulakhe, P.V.; Yates, J.C.; Dhalla, N.S. Biochemical basis of heart function. IV. Energy metabolism and calcium transport in hearts of vitamin E deficient rats. Can. J. Physiol. Pharmacol. 1971, 49, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.; Bolte, H.D.; Lüderitz, B. The (Na+ K+)-ATPase activity of guinea pig heart muscle in potassium deficiency. Arch. Biochem. Biophys. 1971, 145, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Dransfeld, H.; Lipinski, J.; Borsch-Galetke, E. Die Na+ + K+- aktivierte transport-ATPase bei experimenteller herzinsuffizienz durch kobaltchlorid. Naunyn-Schmiedebergs Arch. Pharmak. 1971, 270, 335–342. [Google Scholar] [CrossRef]
- O’Neil, C.L.; Bharadwaj, B.; Prasad, K. Effect of chronic digoxin treatment on cardiac function, electrolytes and ATPase in failing heart due to pressure overload. Cardiovasc. Res. 1984, 18, 502–510. [Google Scholar] [CrossRef]
- Prasad, K.; O’Neil, C.L.; Bharadwaj, B. Effect of prolonged prazosin treatment on hemodynamic and biochemical changes in the dog heart due to chronic pressure overload. Jpn. Heart J. 1984, 25, 461–476. [Google Scholar] [CrossRef]
- Prasad, K.; O’Neil, C.L.; Bharadwaj, B. Effect of prazosin treatment on the cardiac sarcolemmal ATPase in failing heart due to mitral insufficiency in dogs. Cardiovasc. Res. 1985, 19, 406–410. [Google Scholar] [CrossRef]
- Lindenmayer, G.E.; Sordahl, L.A.; Harigaya, S.; Allen, J.C.; Besch, H.R., Jr.; Schwartz, A. Some biochemical studies on subcellular systems isolated from fresh recipient human cardiac tissue obtained during transplantation. Am. J. Cardiol. 1971, 27, 277–283. [Google Scholar] [CrossRef]
- Borsch-Galetke, E.; Dransfeld, H.; Greeff, K. Specific activity and sensitivity to strophanthin of the Na+ + K+-activated ATPase in rats and guinea-pigs with hypoadrenalism. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1972, 274, 74–80. [Google Scholar] [CrossRef]
- Balasubramanian, V.; McNamara, D.B.; Singh, J.N.; Dhalla, N.S. Biochemical basis of heart function. X. Reduction in the Na+ -K+-stimulated ATPase activity in failing rat heart due to hypoxia. Can. J. Physiol. Pharmacol. 1973, 51, 504–510. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Singh, J.N.; Fedelesova, M.; Balasubramanian, V.; McNamara, D.B. Biochemical basis of heart function. XII. Sodium-potassium stimulated adenosine triphosphatase activity in the perfused rat heart made to fail by substrate-lack. Cardiovasc. Res. 1974, 8, 227–236. [Google Scholar] [CrossRef]
- Sethi, R.; Dhalla, K.S.; Ganguly, P.K.; Ferrari, R.; Dhalla, N.S. Beneficial effects of propionyl L-carnitine on sarcolemmal changes in congestive heart failure due to myocardial infarction. Cardiovasc. Res. 1999, 42, 607–615. [Google Scholar] [CrossRef]
- Dixon, I.M.; Afzal, N.; Takeda, N.; Magano, M.; Dhalla, N.S. Remodeling of cardiac membranes during the development of congestive heart failure due to myocardial infarction. In The Failing Heart; Dhalla, N.S., Beamish, R., Takeda, N., Nagano, M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1995; pp. 217–230. [Google Scholar]
- Shao, Q.; Ren, B.; Elimban, V.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2637–H2646. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Lee, S.L.; Shah, K.R.; Elimban, V.; Suzuki, S.; Jasmine, G. Behavior of subcellular organelles during the development of congestive heart failure in cardiomyopathic hamsters (UM-X7.1). In The Cardiomyopathic Heart; Nagano, M., Takeda, N., Dhalla, N.S., Eds.; Raven Press, Ltd.: New York, NY, USA, 1994; pp. 1–14. [Google Scholar]
- Geering, K. Functional roles of Na, K-ATPase subunits. Curr. Opin. Nephrol. Hypertens. 2008, 17, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Crambert, G.; Fuzesi, M.; Garty, H.; Karlish, S.; Geering, K. Phospholemman (FXYD1) associates with Na, K-ATPase and regulates its transport properties. Proc. Natl. Acad. Sci. USA 2002, 99, 11476–11481. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Roy, S.; Schaer, D.; Horisberger, J.D.; Geering, K. Phosphorylation of phospholemman (FXYD1) by protein kinases A and C modulates distinct Na, K-ATPase isozymes. J. Biol. Chem. 2008, 283, 476–486. [Google Scholar] [CrossRef]
- Shamraj, O.I.; Grupp, I.L.; Grupp, G.; Melvin, D.; Gradoux, N.; Kremers, W.; Lingrel, J.B.; Pover, A.D. Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 1993, 27, 2229–2237. [Google Scholar] [CrossRef]
- Schwinger, R.H.; Wang, J.; Frank, K.; Müller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef]
- Sylvén, C.; Jansson, E.; Sotonyi, P.; Waagstein, F.; Brönnegård, M. Na, K-ATPase receptor subunits alpha 1, alpha 2 and alpha 3 mRNA in dilated cardiomyopathy. Biol. Pharm. Bull. 1995, 18, 907–909. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Velotta, J.B.; McDonough, A.A.; Farley, R.A. All human Na+-K+-ATPase α-subunit isoforms have a similar affinity for cardiac glycosides. Am. J. Physiol. Cell Physiol. 2001, 50, C1336–C1343. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.A.; Larsen, J.S.; Shannon, R.P.; Komamura, K.; Vatner, D.E.; Kjeldsen, K. Reduced 3H-ouabain binding site (Na, K-ATPase) concentration in ventricular myocardium of dogs with tachycardia induced heart failure. Basic. Res. Cardiol. 1993, 88, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.S.; Schmidt, T.A.; Bundgaard, H.; Kjeldsen, K. Reduced concentration of myocardial Na+,K+-ATPase in human aortic valve disease as well as of Na+,K+- and Ca2+-ATPase in rodents with hypertrophy. Mol. Cell Biochem. 1997, 169, 85–93. [Google Scholar] [CrossRef] [PubMed]
- James, P.F.; Grupp, I.L.; Grupp, G.; Woo, A.L.; Askew, G.R.; Croyle, M.L.; Walsh, R.A.; Lingrel, J.B. Identification of a specific role for the Na, K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell. 1999, 3, 555–563. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Chen, L.; Hamlyn, J.M.; Leenen, F.H.; Lingrel, J.B.; Wier, W.G.; Zhang, J. Pivotal role of α2 Na+ pumps and their high affinity ouabain binding site in cardiovascular health and disease. J. Physiol. 2016, 594, 6079–6103. [Google Scholar] [CrossRef]
- Correll, R.N.; Eder, P.; Burr, A.R.; Despa, S.; Davis, J.; Bers, D.M.; Molkentin, J.D. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 2014, 114, 249–256. [Google Scholar] [CrossRef]
- Cellini, A.; Höfler, D.; Arias-Loza, P.A.; Bandleon, S.; Langsenlehner, T.; Kohlhaas, M.; Maack, C.; Bauer, W.R.; Eder-Negrin, P. The α2-isoform of the Na+/K+-ATPase protects against pathological remodeling and β-adrenergic desensitization after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H650–H662. [Google Scholar] [CrossRef]
- Barwe, S.P.; Jordan, M.C.; Skay, A.; Inge, L.; Rajasekaran, S.A.; Wolle, D.; Johnson, C.L.; Neco, P.; Fang, K.; Rozengurt, N.; et al. Dysfunction of ouabain-induced cardiac contractility in mice with heart-specific ablation of Na, K-ATPase beta1-subunit. J. Mol. Cell Cardiol. 2009, 47, 552–560. [Google Scholar] [CrossRef][Green Version]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Coordinated shifts in Na/K-ATPase isoforms and their endogenous ligands during cardiac hypertrophy and failure in NaCl-sensitive hypertension. J. Hypertens. 2004, 22, 389–397. [Google Scholar] [CrossRef]
- Trouve, P.; Carre, F.; Belikova, I.; Leclercq, C.; Dakhli, T.; Soufir, L.; Coquard, I.; Ramirez-Gil, J.; Charlemagne, D. Na+-K+ ATPase alpha(2)-isoform expression in guinea pig hearts during transition from compensation to decompensation. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1972–H1981. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Fan, T.H.; Kelly, P.F.; Himura, Y.; Delehanty, J.M.; Hang, C.L.; Liang, C.S. Isoform-specific regulation of myocardial Na, K-ATPase alpha-subunit in congestive heart failure. Role of norepinephrine. Circulation 1994, 89, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.P.; Fan, T.H.; Delehanty, J.M.; Yatani, A.; Liang, C.S. Elevated myocardial interstitial norepinephrine concentration contributes to the regulation of Na+,K+-ATPase in heart failure. Eur. J. Pharmacol. 1996, 309, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Lukas, A.; Chapman, D.C.; Dhalla, N.S. Changes in the expression of cardiac Na+ -K+ ATPase subunits in the UM-X7.1 cardiomyopathic hamster. Life Sci. 2000, 67, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, F.; Mougenot, R.; Maixentt, J.M. Defective activity and isoform of the Na, K-ATPase in the dilated cardiomyopathic hamster. Cell Mol. Biol. 2001, 47, 255–260. [Google Scholar] [PubMed]
- Maixent, J.M.; Pierre, S.V.; Sadrin, S.; Guieu, R.; Paganelli, F. Effects of long-term anti-ischemic drug treatment on Na, K-ATPase isoforms in cardiomyopathic hamsters. Cell Mol. Biol. 2021, 67, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Tsuruya, Y.; Ikeda, U.; Ohta, T.; Yamamoto, K.; Seino, Y.; Ebata, H. Na, K-ATPase gene expression in cardiomyopathic hearts. In The Cardiomyopathic Heart; Nagano, M., Takeda, N., Dhalla, Eds.; Raven Press Ltd.: New York, NY, USA, 1994; pp. 15–21. [Google Scholar]
- Guo, X.; Wang, J.; Elimban, V.; Dhalla, N.S. Both enalapril and losartan attenuate sarcolemmal Na+-K+-ATPase remodeling in failing rat heart due to myocardial infarction. Can. J. Physiol. Pharmacol. 2008, 86, 139–147. [Google Scholar] [CrossRef]
- Ren, B.; Shao, Q.; Ganguly, P.K.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Influence of long-term treatment of imidapril on mortality, cardiac function, and gene expression in congestive heart failure due to myocardial infarction. Can. J. Physiol. Pharmacol. 2004, 82, 1118–1127. [Google Scholar] [CrossRef]
- Semb, S.O.; Lunde, P.K.; Holt, E.; Tønnessen, T.; Christensen, G.; Sejersted, O.M. Reduced myocardial Na+, K(+)-pump capacity in congestive heart failure following myocardial infarction in rats. J. Mol. Cell Cardiol. 1998, 30, 1311–1328. [Google Scholar] [CrossRef]
- Chen, W.; Dando, R. Membrane potential hyperpolarization in mammalian cardiac cells by synchronization modulation of Na/K pumps. J. Membr. Biol. 2008, 221, 165–173. [Google Scholar] [CrossRef]
- Wang, W.; Gao, J.; Entcheva, E.; Cohen, I.S.; Gordon, C.; Mathias, R.T. A transmural gradient in the cardiac Na/K pump generates a transmural gradient in Na/Ca exchange. J. Membr. Biol. 2010, 233, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Horie, M.; Haruna, T.; Ai, T.; Nishimoto, T.; Fujiwara, H.; Sasayama, S. Functional Communication Between Cardiac ATPSensitive K+ Channel and Na/K ATPase. J. Cardiovasc. Electrophysiol. 2010, 9, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.Y. Activation of KATP channels by Na/K pump in isolated cardiac myocytes and giant membrane patches. Biophys. J. 1998, 75, 2858–2867. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, Z. Ouabain interaction with cardiac Na/K -ATPase reveals that the enzyme can act as a pump and as a signal transducer. Cell Mol. Biol. 2002, 47, 383–390. [Google Scholar]
- Xie, Z.; Xie, J. The Na/K-ATPase-mediated signal transduction as a target for new drug development. Front. Biosci. 2005, 10, 3100–3109. [Google Scholar] [CrossRef]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+/K+ ATPase can impair mitochondrial energetics and induce abnormall Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar]
- Sag, C.M.; Wagner, S.; Maier, L.S. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic. Biol. Med. 2013, 63, 338–349. [Google Scholar] [CrossRef]
- Netticadan, T.; Kato, K.; Tappia, P.; Elimban, V.; Dhalla, N.S. Phosphorylation of cardiac Na+-K+ ATPase by Ca2+/calmodulin dependent protein kinase. Biochem. Biophys. Res. Commun. 1997, 238, 544–548. [Google Scholar] [CrossRef]
- Saini, H.K.; Dhalla, N.S. Sarcolemmal cation channels and exchangers modify the increase in intracellular calcium in cardiomyocytes on inhibiting Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H169–H181. [Google Scholar] [CrossRef]
- Saini-Chohan, H.K.; Goyal, R.K.; Dhalla, N.S. Involvement of sarcoplasmic reticulum in changing intracellular calcium due to Na+/K+-ATPase inhibition in cardiomyocytes. Can. J. Physiol. Pharmacol. 2010, 88, 702–715. [Google Scholar] [CrossRef]
- Askari, A. The sodium pump and digitalis drugs: Dogmas and fallacies. Pharmacol. Res. Perspect. 2019, 7, e00505. [Google Scholar] [CrossRef]
- Wansapura, A.N.; Lasko, V.M.; Lingrel, J.B.; Lorenz, J.N. Mice expressing ouabain-sensitive α1-Na,K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H347–H355. [Google Scholar] [CrossRef] [PubMed]
- Gonano, L.A.; Petroff, M.V. Subcellular mechanisms underlying digitalis-induced arrhythmias: Role of calcium/calmodulin-dependent kinase II (CaMKII) in the transition from an inotropic to an arrhythmogenic effect. Heart Lung Circ. 2014, 23, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Kumar, S.P.; Modi, K.M.; Soni, M.N.; Modi, N.R.; Pandya, H.A. Cardiotonic steroids as potential Na+/K+-ATPase inhibitors—A computational study. J. Recept. Signal Transduct. Res. 2019, 39, 226–234. [Google Scholar] [CrossRef]
- Sehirli, A.O.; Aykac, A.; Tetik, S.; Yiginer, O.; Cetinel, S.; Ozkan, N.; Akkiprik, M.; Kaya, Z.; Yegen, B.C.; Tezcan, M.; et al. The effects of resveratrol treatment on caveolin-3 expression and Na+/K+ ATPase activity in rats with isoproterenol-induced myocardial injury. North. Clin. Istanb. 2020, 7, 313–320. [Google Scholar]
- Şehirli, A.Ö.; Koyun, D.; Tetik, Ş.; Özsavcı, D.; Yiğiner, Ö.; Çetinel, Ş.; Tok, O.E.; Kaya, Z.; Akkiprik, M.; Kılıç, E.; et al. Melatonin protects against ischemic heart failure in rats. J. Pineal Res. 2013, 55, 138–148. [Google Scholar] [CrossRef]
- Sapia, L.; Palomeque, J.; Mattiazzi, A.; Petroff, M.V. Na+/K+-ATPase inhibition by ouabain induces CaMKII-dependent apoptosis in adult rat cardiac myocytes. J. Mol. Cell Cardiol. 2010, 49, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bai, Y.; Chen, Y.; Wang, Y.; Sottejeau, Y.; Liu, L.; Li, X.; Lingrel, J.B.; Malhotra, D.; Cooper, C.J.; et al. Reduction of Na/K-ATPase potentiates marinobufagenin-induced cardiac dysfunction and myocyte apoptosis. J. Biol. Chem. 2012, 287, 16390–16398. [Google Scholar] [CrossRef]
- Liao, Q.Q.; Dong, Q.Q.; Zhang, H.; Shu, H.P.; Tu, Y.C.; Yao, L.J. Contributions of SGK3 to transporter-related diseases. Front. Cell Dev. Biol. 2022, 10, 1007924. [Google Scholar] [CrossRef]
- Galougahi, K.K.; Liu, C.C.; Garcia, A.; Fry, N.A.; Hamilton, E.J.; Rasmussen, H.H.; Figtree, G.A. Protein kinase-dependent oxidative regulation of the cardiac Na+-K+ pump: Evidence from in vivo and in vitro modulation of cell signalling. J. Physiol. 2013, 591, 2999–3015. [Google Scholar] [CrossRef]
- Yan, X.; Li, M.; Lan, P.; Xun, M.; Zhang, Y.; Shi, J.; Wang, R.; Zheng, J. Regulation of Na+-K+-ATPase leads to disturbances of isoproterenol-induced cardiac dysfunction via interference of Ca2+-dependent cardiac metabolism. Clin. Sci. 2024, 138, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Bastug-Özel, Z.; Wright, P.T.; Kraft, A.E.; Pavlovic, D.; Howie, J.; Froese, A.; Fuller, W.; Gorelik, J.; Shattock, M.J.; Nikolaev, V.O. Heart failure leads to altered β2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain. Cardiovasc. Res. 2019, 115, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Fry, N.A.S.; Liu, C.C.; Garcia, A.; Hamilton, E.J.; Karimi Galougahi, K.; Kim, Y.J.; Whalley, D.W.; Bundgaard, H.; Rasmussen, H.H. Targeting Cardiac Myocyte Na+ -K+ Pump Function With β3 Adrenergic Agonist in Rabbit Model of Severe Congestive Heart Failure. Circ. Heart Fail. 2020, 13, e006753. [Google Scholar] [CrossRef] [PubMed]
- Stimers, J.R.; Dobretsov, M. Adrenergic stimulation of Na/K pump current in adult rat cardiac myocytes in short-term culture. J. Membr. Biol. 1998, 163, 205–216. [Google Scholar] [CrossRef]
- Bers, D.M.; Despa, S. Na/K-ATPase--an integral player in the adrenergic fight-or-flight response. Trends Cardiovasc. Med. 2009, 19, 111–118. [Google Scholar] [CrossRef]
- Banday, A.A.; Lokhandwala, M.F. Loss of biphasic effect on Na/K-ATPase activity by Angiotensin II involves defective angiotensin type 1 receptor–nitric oxide signaling. Hypertension 1979, 52, 1099–1105. [Google Scholar] [CrossRef]
- Palacios, J.; Marusic, E.T.; Lopez, N.C.; Gonzalez, M.; Michea, L. Estradiol-induced expression of N+-K+-ATPase catalytic isoforms in rat arteries: Gender differences in activity mediated by nitric oxide donors. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1793–H1800. [Google Scholar] [CrossRef]
- Karmazyn, M.; Dhalla, N.S. Thromboxane B2: A cardiodepressant of isolated rat hearts and inhibitor of sarcolemma Na+—K+ stimulated ATPase activity. Prostaglandins Med. 1979, 3, 81–93. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Droy-Lefaix, M.T.; Lakatta, E.G.; Bagrov, A.Y. Myocardial PKC beta2 and the sensitivity of Na/K-ATPase to marinobufagenin are reduced by cicletanine in Dahl hypertension. Hypertension 2003, 41, 505–511. [Google Scholar] [CrossRef]
- Silverman, B.; Fuller, W.; Eaton, P.; Deng, J.; Moorman, J.R.; Cheung, J.Y.; James, A.F.; Shattock, M.J. Serine 68 phosphorylation of phospholemman: Acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc. Res. 2005, 65, 93–103. [Google Scholar] [CrossRef]
- Despa, S.; Tucker, A.L.; Bers, D.M. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation 2008, 117, 1849–1855. [Google Scholar] [CrossRef]
- Bossuyt, J.; Despa, S.; Han, F.; Hou, Z.; Robia, S.L.; Lingrel, J.B.; Bers, D.M. Isoform specificity of the Na/K-ATPase association and regulation by phospholemman. J. Biol. Chem. 2009, 284, 26749–26757. [Google Scholar] [CrossRef] [PubMed]
- El-Armouche, A.; Wittköpper, K.; Fuller, W.; Howie, J.; Shattock, M.J.; Pavlovic, D. Phospholemman-dependent regulation of the cardiac Na/K-ATPase activity is modulated by inhibitor-1 sensitive type-1 phosphatase. FASEB J. 2011, 25, 4467–4475. [Google Scholar] [CrossRef]
- Teriete, P.; Thai, K.; Choi, J.; Marassi, F.M. Effects of PKA phosphorylation on the conformation of the Na,K-ATPase regulatory protein FXYD1. Biochim. Biophys. Acta 2009, 1788, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, D.; Fuller, W.; Shattock, M.J. The intracellular region of FXYD1 is sufficient to regulate cardiac Na/K ATPase. FASEB J. 2007, 21, 1539–1546. [Google Scholar] [CrossRef]
- Mirza, M.A.; Lane, S.; Yang, Z.; Karaoli, T.; Akosah, K.; Hossack, J.; McDuffie, M.; Wang, J.; Zhang, X.; Song, J.; et al. Phospholemman deficiency in postinfarct hearts: Enhanced contractility but increased mortality. Clin. Transl. Sci. 2012, 5, 235–242. [Google Scholar] [CrossRef]
- Bossuyt, J.; Ai, X.; Moorman, J.R.; Pogwizd, S.M.; Bers, D.M. Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 2005, 97, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Boguslavskyi, A.; Pavlovic, D.; Aughton, K.; Clark, J.E.; Howie, J.; Fuller, W.; Shattock, M.J. Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc. Res. 2014, 104, 72–82. [Google Scholar] [CrossRef]
- Li, W.; Wang, X.; He, M.; Wang, C.; Qiao, Z.; Wang, Q.; Ren, S.; Yu, Q. Activating Na+-K+ ATPase: A potential cardioprotective therapy during early hemorrhagic shock. Med. Hypotheses. 2014, 83, 685–687. [Google Scholar] [CrossRef]
- Xu, K.Y.; Takimoto, E.; Fedarko, N.S. Activation of (Na+ + K+)-ATPase induces positive inotropy in intact mouse heart in vivo. Biochem. Biophys. Res. Commun. 2006, 349, 582–587. [Google Scholar] [CrossRef]
- Zheng, J.; Koh, X.; Hua, F.; Li, G.; Larrick, J.W.; Bian, J.S. Cardioprotection induced by Na+/K+-ATPase activation involves extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/Akt pathway. Cardiovasc. Res. 2011, 89, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Wu, Z.; Yan, X.; Zheng, J.; Sun, H.; Cao, X.; Bian, J.-S. DR region of Na+-K+-ATPase is a new target to protect heart against oxidative injury. Sci. Rep. 2018, 8, 13100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lu, X.; Li, J.; Chidiac, P.; Sims, S.M.; Feng, Q. Inhibition of Na/K ATPase promotes myocardial tumor necrosis factor-alpha protein expression and cardiac dysfunction via calcium/m TOR signaling in endotoxemia. Basic. Res. Cardiol. 2012, 107, 254. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Schiffrin, E.L. Role of mineralocorticoid receptor antagonist in cardiovascular disease. Circ. Res. 2015, 116, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Xie, J.; Tian, J. Reducing cardiac fibrosis: Na/K-ATPase signaling complex as a novel target. Cardiovasc. Pharm. 2017, 6, 204. [Google Scholar]
- Skayian, Y.; Kreydiyyeh, S.I. Tumor necrosis factor alpha alters Na+-K+ ATPase activity in rat cardiac myocytes: Involvement of NF-kappaB, AP-1 and PGE2. Life Sci. 2006, 80, 173–180. [Google Scholar] [CrossRef]
- Xie, Z.J.; Novograd, J.; Itzkowitz, Y.; Sher, A.; Buchen, Y.D.; Sodhi, K.; Abraham, N.G.; Shapiro, J.I. The pivotal role of adipocyte-Na K peptide in reversing systemic inflammation in obesity and covid-19 in the development of heart failure. Antioxidants 2020, 9, 1129. [Google Scholar] [CrossRef]
- Buhagiar, K.A.; Hansen, P.S.; Kong, B.Y.; Clarke, R.J.; Fernandes, C.; Rasmussen, H.H. Dietary cholesterol alters Na+/K+ selectivity at intracellular Na+/K+ pump sites in cardiac myocytes. Am. J. Physiol. Cell Physiol. 2004, 286, C398–C405. [Google Scholar] [CrossRef]
- Kometiani, P.; Tian, J.; Li, J.; Nabih, Z.; Gick, G.; Xie, Z. Regulation of Na/K- ATPase beta1-subunit gene expression by ouabain and other hypertrophic stimuli in neonatal stimuli in neonatal rat cardiac myocytes. Mol. Cell Biochem. 2000, 215, 65–72. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Tomlinson, C.W.; Singh, J.N.; Lee, S.L.; McNamara, D.B.; Harrow, J.A.; Yates, J.C. Role of sarcolemmal changes in cardiac pathophysiology. Recent. Adv. Stud. Cardiac Struct. Metab. 1976, 9, 377–394. [Google Scholar]
- Dhalla, N.S.; Das, P.K.; Sharma, G.P. Subcellular basis of cardiac contractile failure. J. Mol. Cell Cardiol. 1978, 10, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Bers, D.M. Na+ transport in the normal and failing heart- remember the balance. J. Mol. Cell Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Van Borren, M.M.G.J.; Belterman, C.N.W.; Coronel, R.; Fiolet, J.W.T. Increased Na+/H+- exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc. Res. 2003, 57, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Hoare, Z.; Baark, F.; Yu, C.S.; Guo, J.; Fuller, W.; Southworth, R.; Katschinski, D.M.; Murphy, M.P.; Eykyn, T.R.; et al. Elevated Na is a dynamic and reversible modulator of mitochondrial metabolism in the heart. Nat. Commun. 2024, 15, 4277. [Google Scholar] [CrossRef] [PubMed]
- Morotti, S.; Ni, H.; Peters, C.H.; Rickert, C.; Asgari-Targi, A.; Sato, D.; Glukhov, A.V.; Proenza, C.; Grandi, E. Intracellular Na+ modulates pacemaking activity in murine sinoastrial node myocytes: An in silico analysis. Int. J. Mol. Sci. 2021, 22, 5645. [Google Scholar] [CrossRef] [PubMed]
- Trenor, B.; Cardona, K.; Gomez, J.F.; Rajamani, S.; Ferrero, J.M., Jr.; Belardinelli, L.; Saiz, J. Simulation and mechanistic investigation of the arrhythmogenic role of the late sodium current in human heart failure. PLoS ONE 2012, 7, e32659. [Google Scholar] [CrossRef]
- Wasserstrom, J.A. Changes in intracellular Na+ in heart failure following SERCA knockout-more of a solution or more of a problem? J. Physiol. 2012, 588 Pt 7, 1027. [Google Scholar] [CrossRef] [PubMed]
- Despa, S. Myocyte [Na+]I Dysregulation in heart failure and diabetic cardiomyopathy. Front. Physiol. 2018, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.W.; Dhalla, N.S. Alterations in myocardial function during bacterial infective cardiomyopathy. Am. J. Cardiol. 1976, 37, 373–381. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Ziegelhoffer, A.; Singal, P.K.; Panagia, V.; Dhillon, K.S. Subcellular changes during cardiac hypertrophy and heart failure due to bacterial endocarditis. Basic. Res. Cardiol. 1980, 75, 81–91. [Google Scholar] [CrossRef]
- Tomlinson, C.W.; Dhalla, N.S. Alterations in calcium metabolism in cardiac hypertrophy and failure caused by bacterial infection. Recent. Adv. Stud. Cardiac Struct. Metab. 1978, 12, 191–202. [Google Scholar]
- Lossnitzer, K.; Bajusz, E. Water and electrolyte alterations during the life course of the BIO 14.6 Syrian golden hamsters. A disease model of hereditary cardiomyopathy. J. Mol. Cell Cardiol. 1974, 6, 163–177. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).