
Lignocellulolytic Potential of Microbial Consortia Isolated from a Local Biogas Plant: The Case of Thermostable Xylanases Secreted by Mesophilic Bacteria

 ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
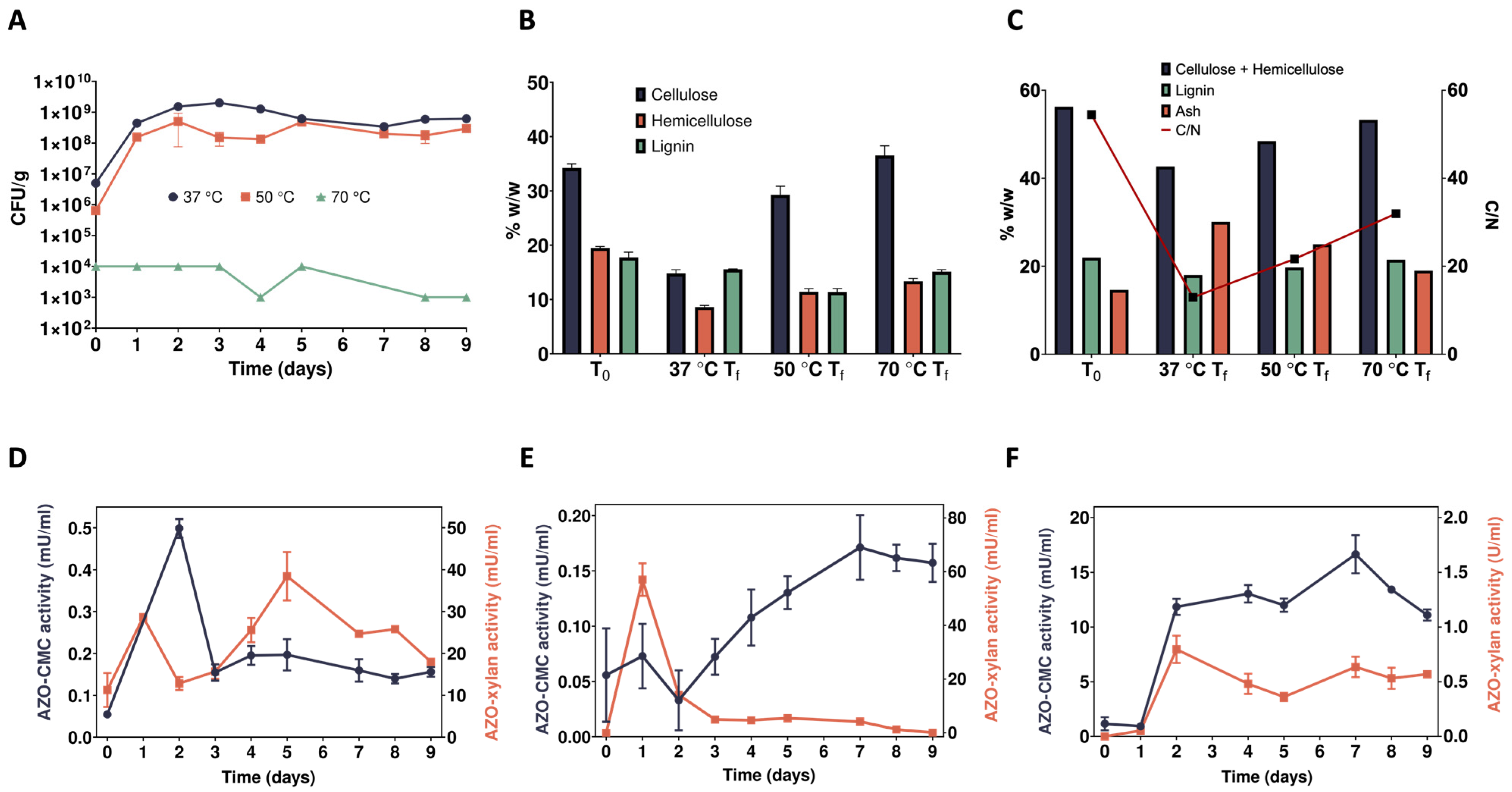
2.1. Time Course Analyses of the Enrichment Cultures
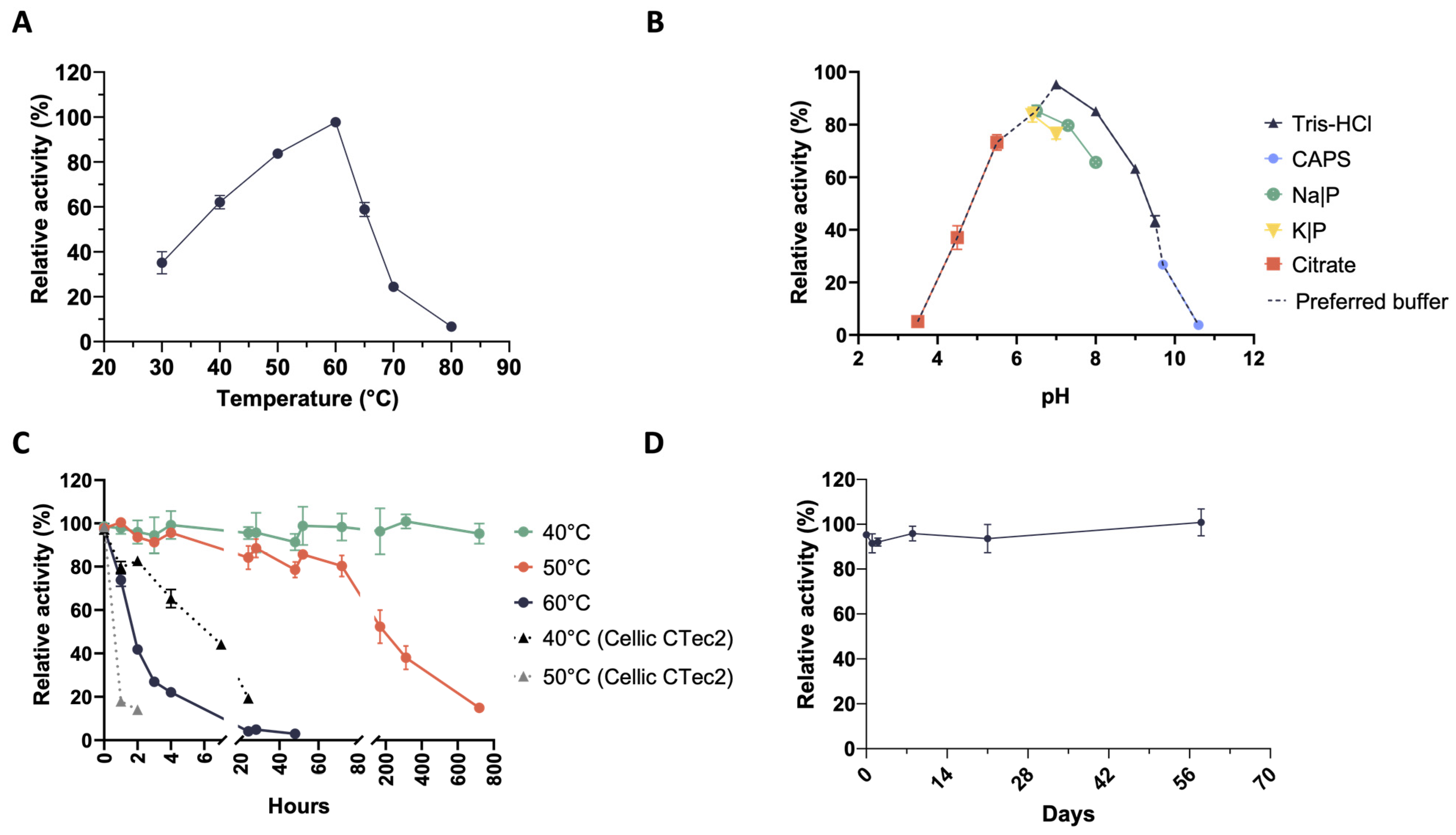
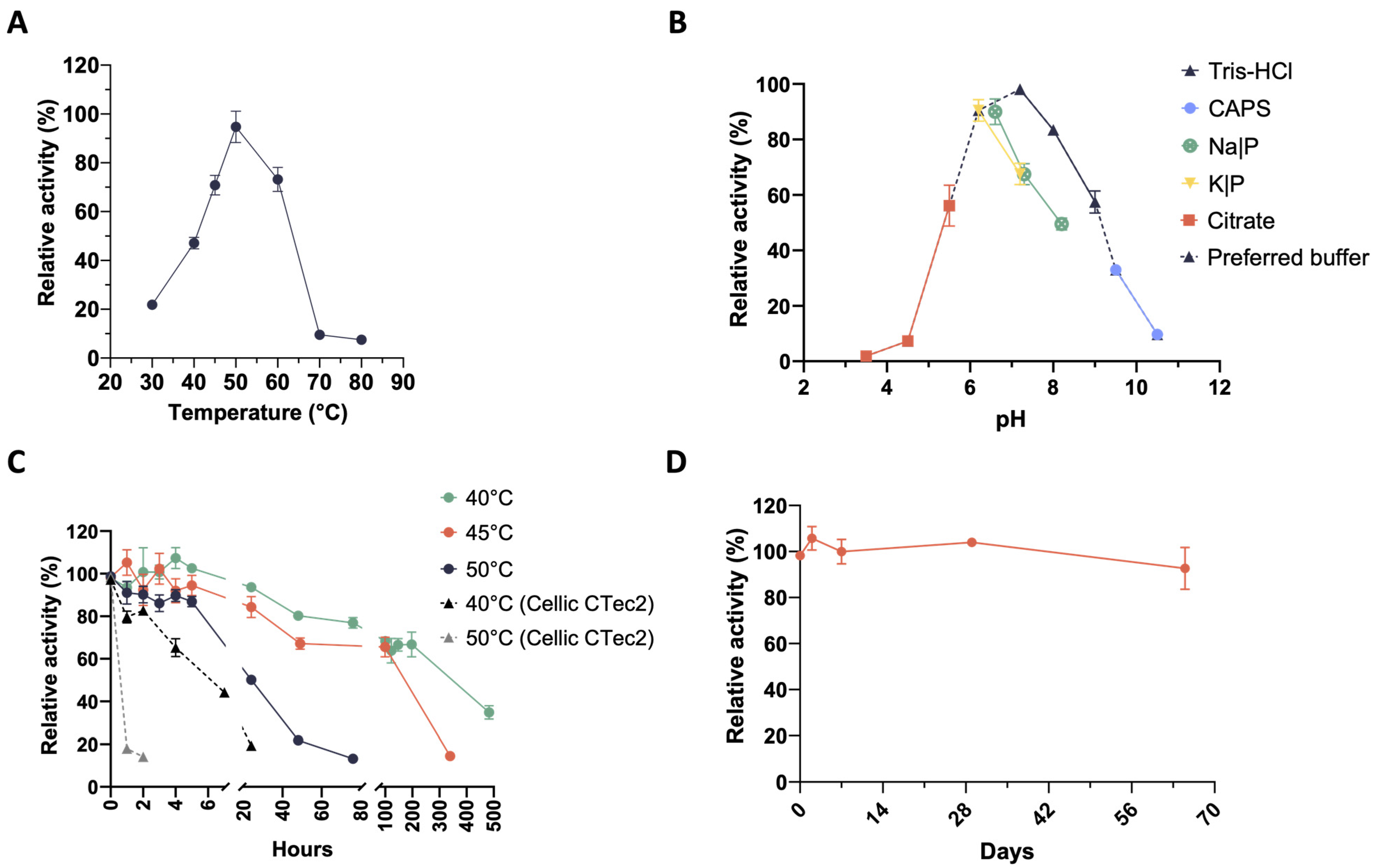
2.2. Mesophilic Microbial Consortia Secrete Thermophilic and Thermostable Xylanases
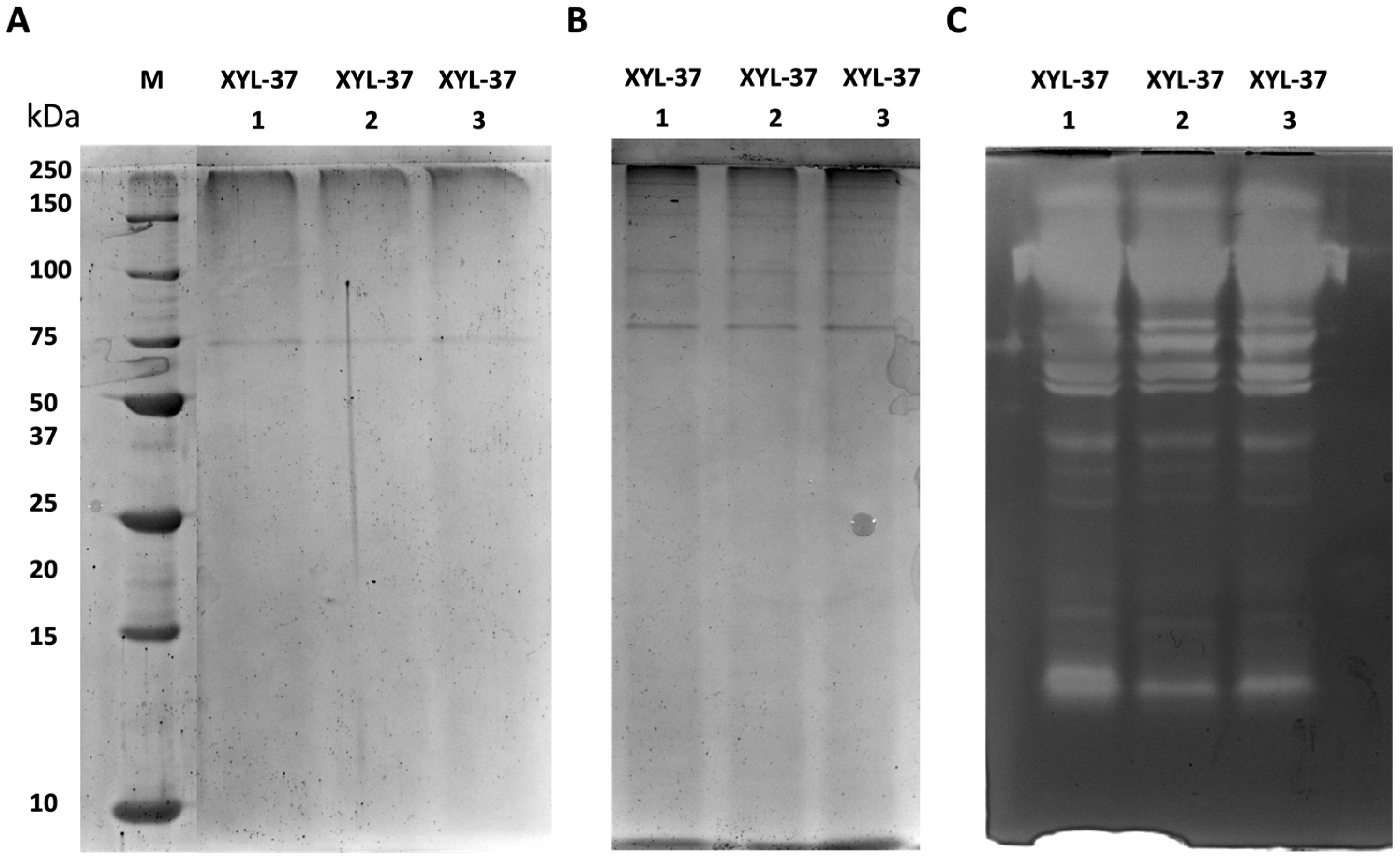
2.3. Electrophoretic and Zymographic Analyses
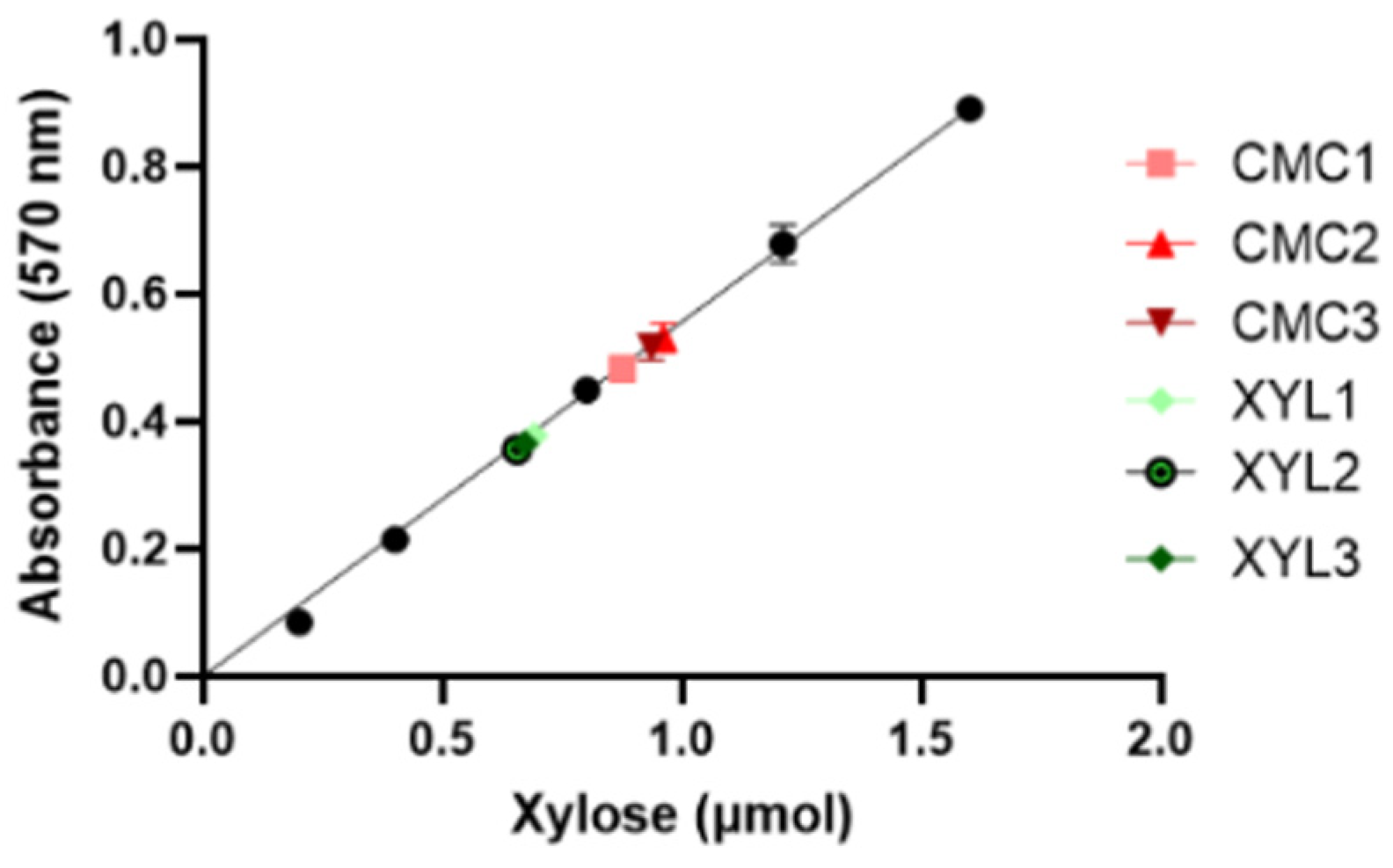
2.4. Determination of Enzyme Units
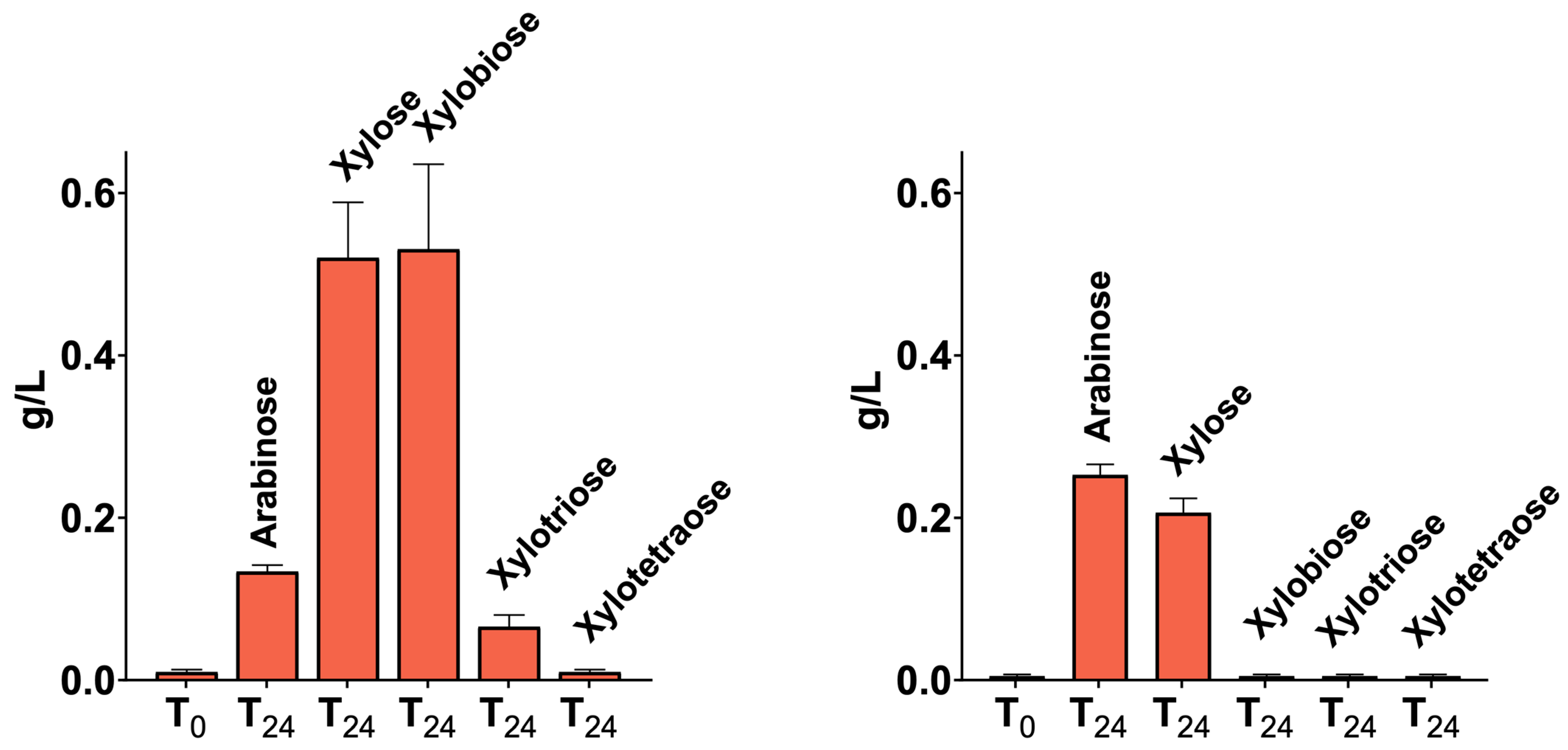
2.5. LCB-Derived Polysaccharides Hydrolysis Products Profiling
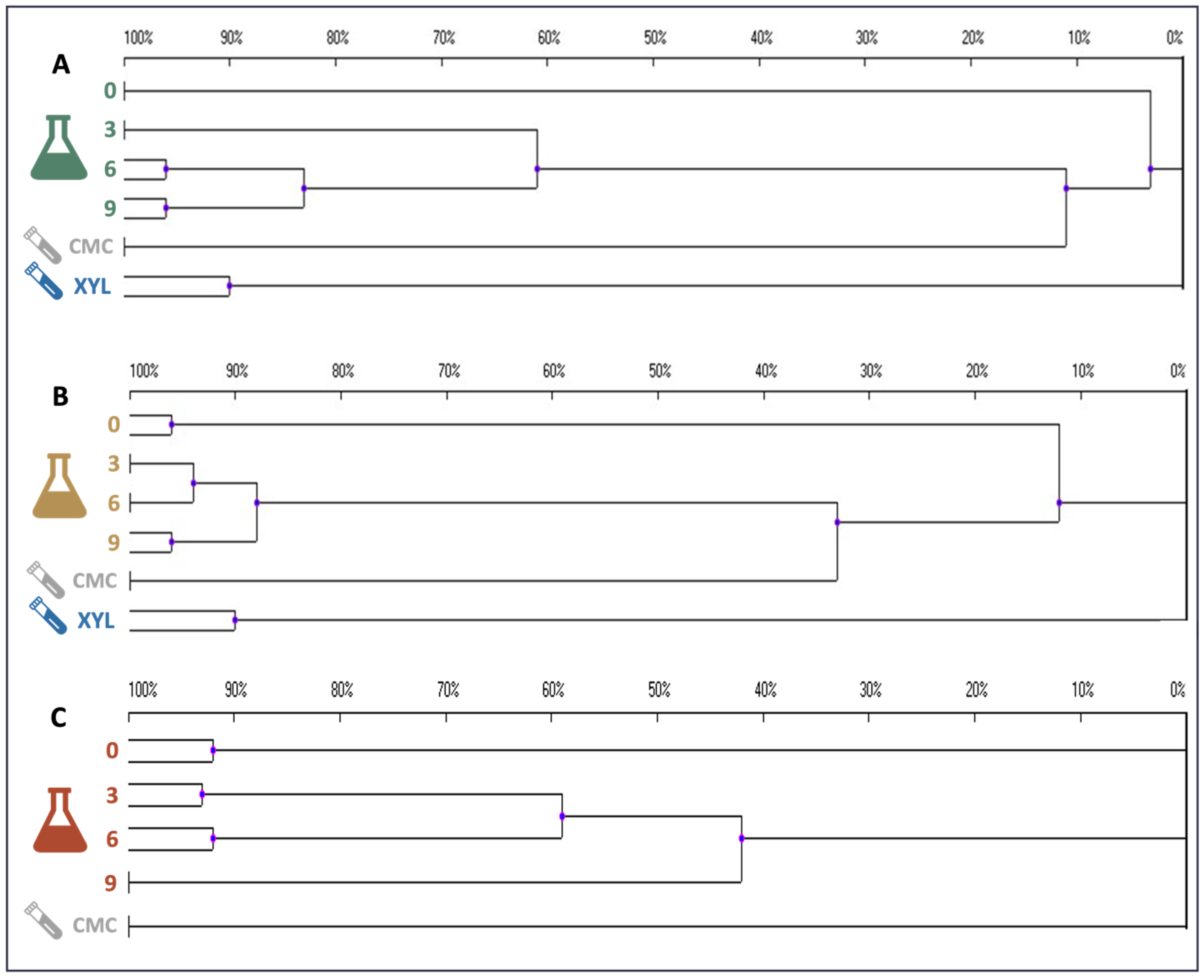
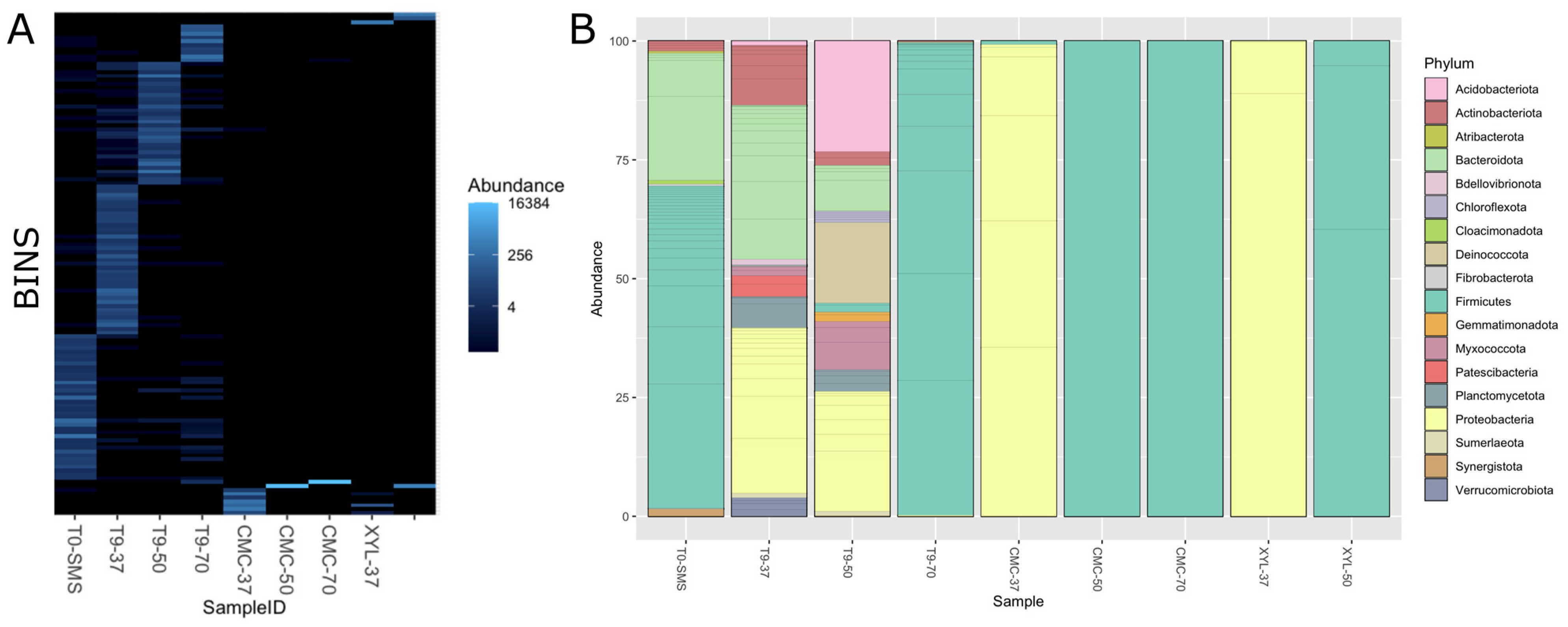
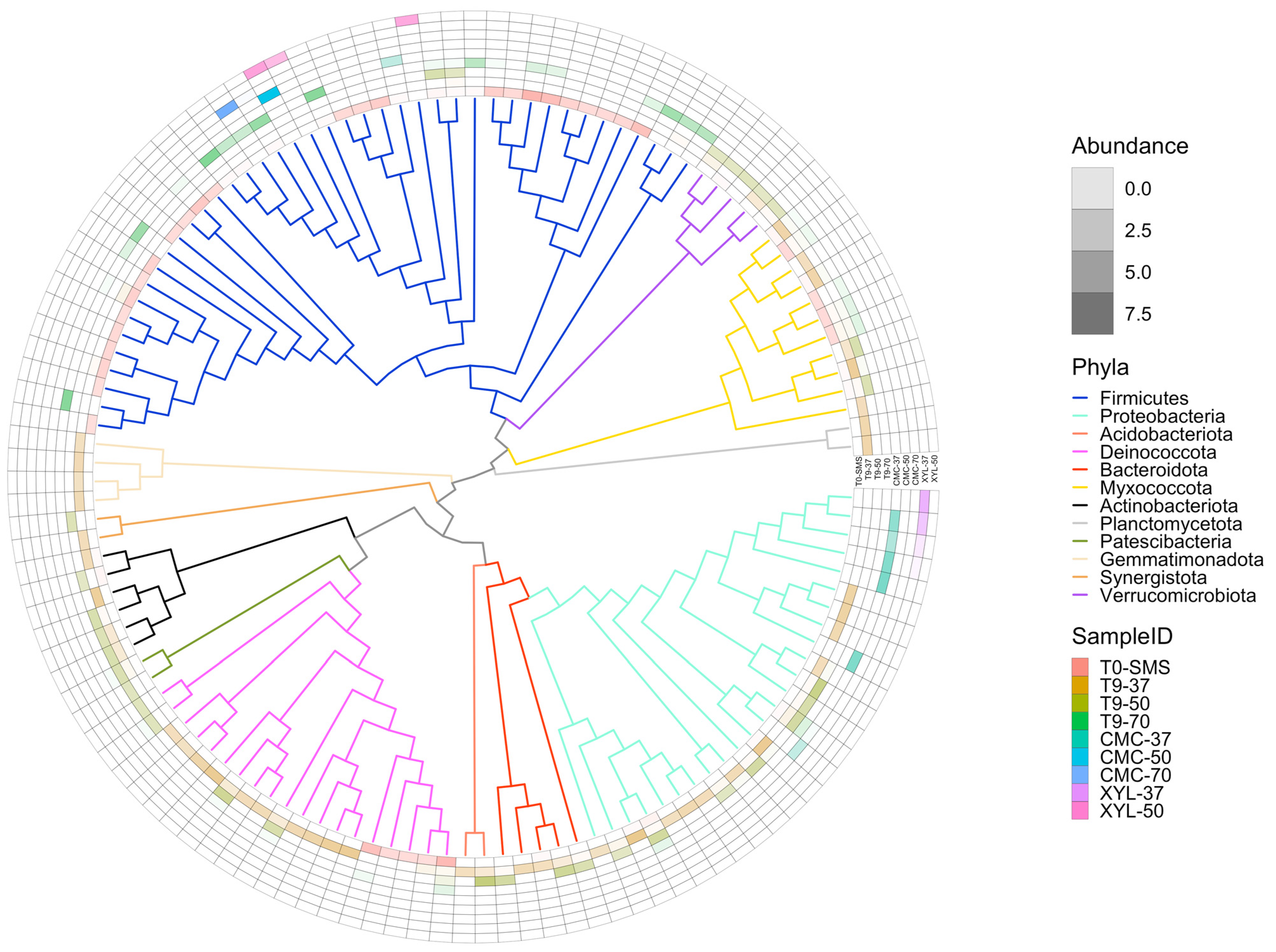
2.6. Microbial Consortia Composition through PCR-DGGE and Metagenomic Analyses
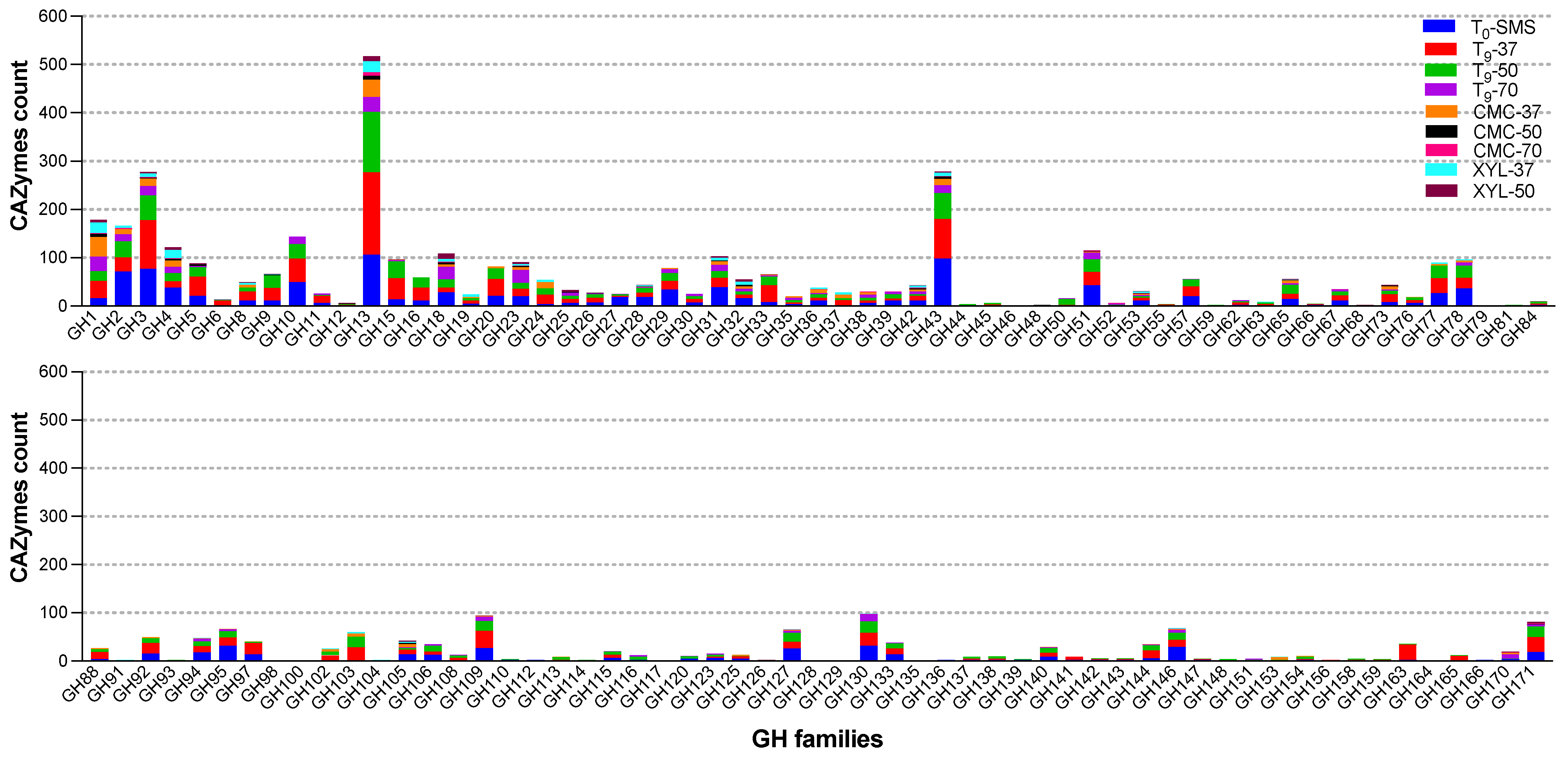
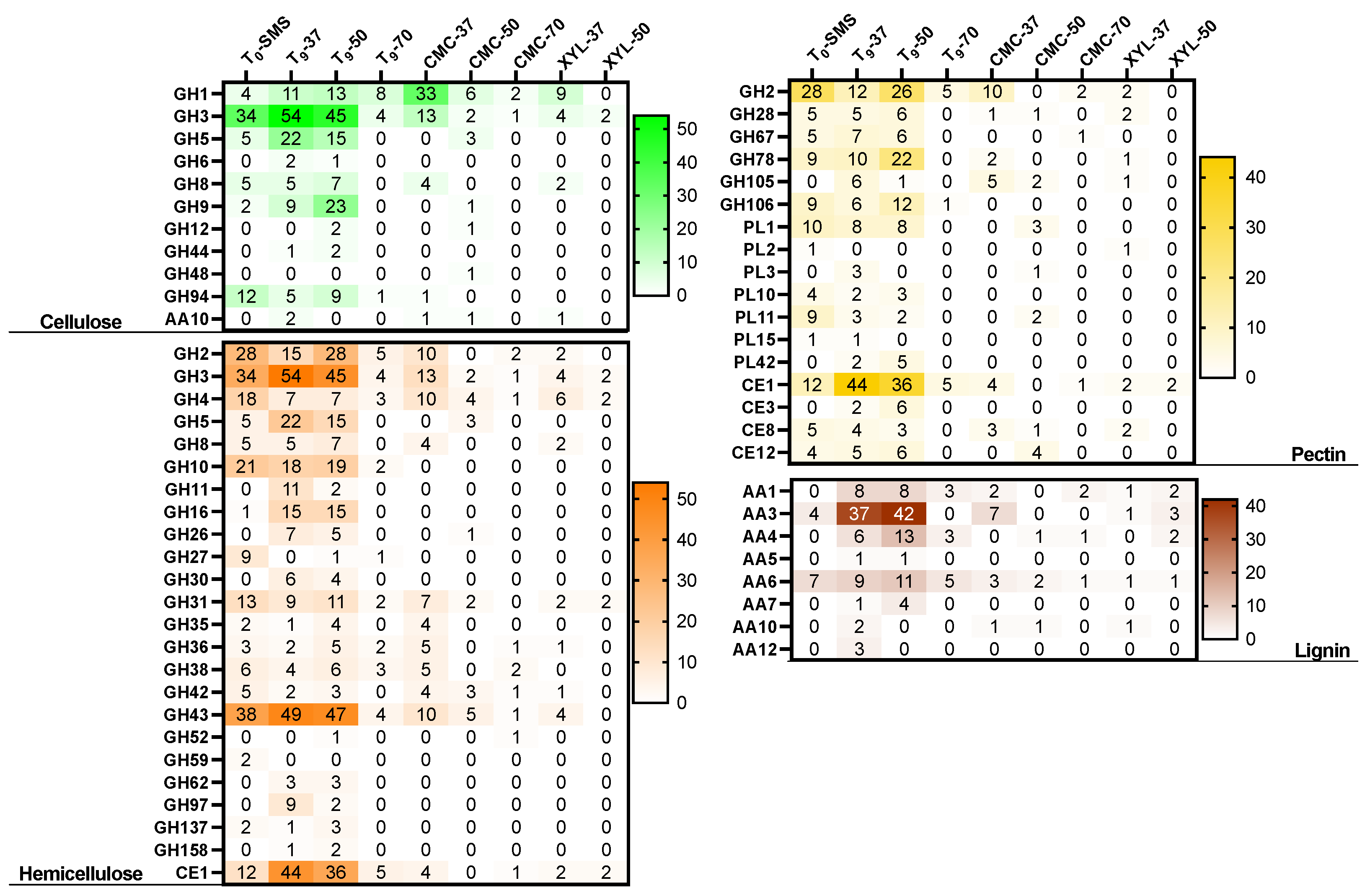
2.7. Meta-Functional Analysis of CAZymes
3. Discussion
4. Materials and Methods
4.1. Spent Mushroom Substrate and Microbial Inoculum Characterisation
4.2. Media and Chemicals
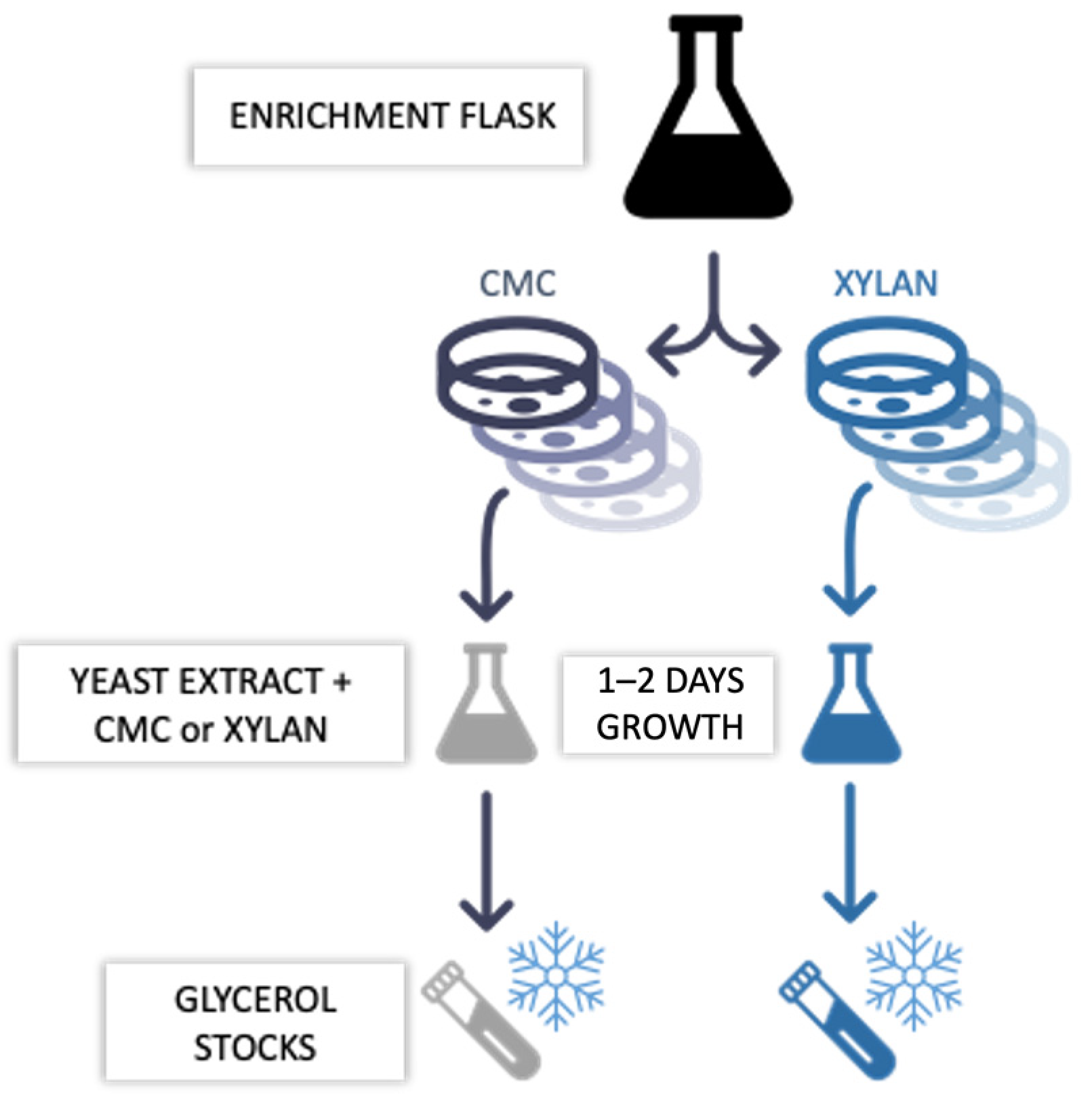
4.3. Enrichment Cultures
4.3.1. Counting of Colony-Forming Units
4.3.2. Endo-1,4-β-D-Xylanase and Endo-1,4-β-D-Glucanase Assays
4.3.3. Total Carbohydrates and Lignin Analysis
4.4. Microbial Consortia Cultivations for Enzymes Production
4.5. Reducing Sugar Assay to Measure the Enzyme Units
4.6. Hydrolysis of LCB-Derived Polysaccharides
4.7. High-Performance Anion-Exchange Chromatography (HPAEC) with Pulsed Amperometric Detection (PAD)
4.8. Electrophoretic Analysis and Zymography
4.9. Total DNA Extraction, PCR Reaction, and DGGE (Denaturing Gradient Gel Electrophoresis) Analysis
4.10. Metagenomic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Ashokkumar, V.; Venkatkarthick, R.; Jayashree, S.; Chuetor, S.; Dharmaraj, S.; Kumar, G.; Chen, W.-H.; Ngamcharussrivichai, C. Recent advances in lignocellulosic biomass for biofuels and value-added bioproducts—A critical review. Bioresour. Technol. 2022, 344, 126195. [Google Scholar] [CrossRef] [PubMed]
- Velvizhi, G.; Jacqueline, P.J.; Shetti, N.P.; Latha, K.; Mohanakrishna, G.; Aminabhavi, T.M. Emerging trends and advances in valorization of lignocellulosic biomass to biofuels. J. Environ. Manag. 2023, 345, 118527. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.u.; Qaisar, K.; Nawaz, A.; Akram, F.; Mukhtar, H.; Xu, Y.; Mumtaz, M.W.; Rashid, U.; Ghani, W.A.W.A.K.; Choong, T.S.Y. Advances in valorization of lignocellulosic biomass towards energy generation. Catalysts 2021, 11, 309. [Google Scholar] [CrossRef]
- Leong, Y.K.; Ma, T.-W.; Chang, J.-S.; Yang, F.-C. Recent advances and future directions on the valorization of spent mushroom substrate (SMS): A review. Bioresour. Technol. 2022, 344, 126157. [Google Scholar] [CrossRef] [PubMed]
- Atallah, E.; Zeaiter, J.; Ahmad, M.N.; Leahy, J.J.; Kwapinski, W. Hydrothermal carbonization of spent mushroom compost waste compared against torrefaction and pyrolysis. Fuel Process. Technol. 2021, 216, 106795. [Google Scholar] [CrossRef]
- Leong, Y.K.; Varjani, S.; Lee, D.-J.; Chang, J.-S. Valorization of spent mushroom substrate for low-carbon biofuel production: Recent advances and developments. Bioresour. Technol. 2022, 363, 128012. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, L.; Serpico, A.; De Simone, M.; Frison, N.; Fusco, S. Biorefinery gets hot: Thermophilic enzymes and microorganisms for second-generation bioethanol production. Processes 2021, 9, 1583. [Google Scholar] [CrossRef]
- Botturi, A.; Battista, F.; Andreolli, M.; Faccenda, F.; Fusco, S.; Bolzonella, D.; Lampis, S.; Frison, N. Polyhydroxyalkanoated-rich microbial cells from bio-based volatile fatty acids as potential ingredient for aquaculture feed. Energies 2021, 14, 38. [Google Scholar] [CrossRef]
- Grujić, M.; Dojnov, B.; Potočnik, I.; Duduk, B.; Vujčić, Z. Spent mushroom compost as substrate for the production of industrially important hydrolytic enzymes by fungi Trichoderma spp. and Aspergillus niger in solid state fermentation. Int. Biodeterior. Biodegrad. 2015, 104, 290–298. [Google Scholar] [CrossRef]
- Ariff, I.N.M.; Bahrin, E.K.; Ramli, N.; Abd-Aziz, S. Direct use of spent mushroom substrate from Pleurotus pulmonarius as a readily delignified feedstock for cellulase production. Waste Biomass Valorization 2019, 10, 839–850. [Google Scholar] [CrossRef]
- Rajavat, A.S.; Rai, S.; Pandiyan, K.; Kushwaha, P.; Choudhary, P.; Kumar, M.; Chakdar, H.; Singh, A.; Karthikeyan, N.; Bagul, S.Y. Sustainable use of the spent mushroom substrate of Pleurotus florida for production of lignocellulolytic enzymes. J. Basic Microbiol. 2020, 60, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Gavande, P.V.; Goyal, A.; Fontes, C.M. Carbohydrates and Carbohydrate-Active enZymes (CAZyme): An overview. In Glycoside Hydrolases; Academic Press: Cambridge, MA, USA, 2023; pp. 1–23. [Google Scholar]
- Burlacu, A.; Cornea, C.; Israel-Roming, F. Screening of xylanase producing microorganisms. Res. J. Agric. Sci. 2016, 48, 8–15. [Google Scholar]
- Dhiman, S.; Mukherjee, G. Recent advances and industrial applications of microbial xylanases: A review. In Fungi and Their Role in Sustainable Development: Current Perspectives; Springer: Singapore, 2018; pp. 329–348. [Google Scholar]
- Gupta, G.K.; Dixit, M.; Kapoor, R.K.; Shukla, P. Xylanolytic enzymes in pulp and paper industry: New technologies and perspectives. Mol. Biotechnol. 2022, 64, 130–143. [Google Scholar] [CrossRef]
- Malhotra, G.; Chapadgaonkar, S.S. Production and applications of xylanases—An overview. BioTechnol. J. Biotechnol. Comput. Biol. Bionanotechnol. 2018, 99, 59–72. [Google Scholar] [CrossRef]
- Tyagi, D.; Sharma, D. Production and industrial applications of xylanase: A Review. Int. J. Sci. Res. Eng. Trends 2021, 7, 1866–1876. [Google Scholar]
- Burlacu, A.; Cornea, C.P.; Israel-Roming, F. Microbial xylanase: A review. Sci. Bull. Ser. F Biotechnol. 2016, 20, 335–342. [Google Scholar]
- Bajaj, P.; Mahajan, R. Cellulase and xylanase synergism in industrial biotechnology. Appl. Microbiol. Biotechnol. 2019, 103, 8711–8724. [Google Scholar] [CrossRef]
- Hu, J.; Arantes, V.; Saddler, J.N. The enhancement of enzymatic hydrolysis of lignocellulosic substrates by the addition of accessory enzymes such as xylanase: Is it an additive or synergistic effect? Biotechnol. Biofuels 2011, 4, 36. [Google Scholar] [CrossRef]
- Song, H.-T.; Gao, Y.; Yang, Y.-M.; Xiao, W.-J.; Liu, S.-H.; Xia, W.-C.; Liu, Z.-L.; Yi, L.; Jiang, Z.-B. Synergistic effect of cellulase and xylanase during hydrolysis of natural lignocellulosic substrates. Bioresour. Technol. 2016, 219, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, J.; Saritha, M.; Nain, L.; Arora, A. Enhanced saccharification of steam-pretreated rice straw by commercial cellulases supplemented with xylanase. J. Bioprocess. Biotech. 2014, 4, 1–6. [Google Scholar] [CrossRef]
- Strazzulli, A.; Fusco, S.; Cobucci-Ponzano, B.; Moracci, M.; Contursi, P. Metagenomics of microbial and viral life in terrestrial geothermal environments. Rev. Environ. Sci. Bio/Technol. 2017, 16, 425–454. [Google Scholar] [CrossRef]
- Aulitto, M.; Fusco, S.; Franzén, C.J.; Strazzulli, A.; Moracci, M.; Bartolucci, S.; Contursi, P. Draft Genome Sequence of Bacillus coagulans MA-13, a Thermophilic Lactic Acid Producer from Lignocellulose. Microbiol. Resour. Announc. 2019, 8, e00341-19. [Google Scholar] [CrossRef]
- Battista, F.; Zuliani, L.; Rizzioli, F.; Fusco, S.; Bolzonella, D. Biodiesel, biogas and fermentable sugars production from Spent coffee Grounds: A cascade biorefinery approach. Bioresour. Technol. 2021, 342, 125952. [Google Scholar] [CrossRef]
- Lambertz, C.; Garvey, M.; Klinger, J.; Heesel, D.; Klose, H.; Fischer, R.; Commandeur, U. Challenges and advances in the heterologous expression of cellulolytic enzymes: A review. Biotechnol. Biofuels 2014, 7, 135. [Google Scholar] [CrossRef]
- Zhao, X.-Q.; Liu, C.-G.; Bai, F.-W. Making the biochemical conversion of lignocellulose more robust. Trends Biotechnol. 2023; in press. [Google Scholar]
- Ferremi Leali, N.; Binati, R.L.; Martelli, F.; Gatto, V.; Luzzini, G.; Salini, A.; Slaghenaufi, D.; Fusco, S.; Ugliano, M.; Torriani, S. Reconstruction of simplified microbial consortia to modulate sensory quality of kombucha tea. Foods 2022, 11, 3045. [Google Scholar] [CrossRef]
- Duncker, K.E.; Holmes, Z.A.; You, L. Engineered microbial consortia: Strategies and applications. Microb. Cell Factories 2021, 20, 211. [Google Scholar] [CrossRef]
- Liang, Y.; Ma, A.; Zhuang, G. Construction of environmental synthetic microbial consortia: Based on engineering and ecological principles. Front. Microbiol. 2022, 13, 829717. [Google Scholar] [CrossRef]
- Tom, L.M.; Aulitto, M.; Wu, Y.-W.; Deng, K.; Gao, Y.; Xiao, N.; Rodriguez, B.G.; Louime, C.; Northen, T.R.; Eudes, A. Low-abundance populations distinguish microbiome performance in plant cell wall deconstruction. Microbiome 2022, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Gladkov, G.V.; Kimeklis, A.K.; Afonin, A.M.; Lisina, T.O.; Orlova, O.V.; Aksenova, T.S.; Kichko, A.A.; Pinaev, A.G.; Andronov, E.E. The Structure of Stable Cellulolytic Consortia Isolated from Natural Lignocellulosic Substrates. Int. J. Mol. Sci. 2022, 23, 10779. [Google Scholar] [CrossRef] [PubMed]
- Gavande, P.; Basak, A.; Sen, S.; Lepcha, K.; Murmu, N.; Rai, V.; Mazumdar, D.; Saha, S.; Das, V.; Ghosh, S. Functional characterization of thermotolerant microbial consortium for lignocellulolytic enzymes with central role of Firmicutes in rice straw depolymerization. Sci. Rep. 2021, 11, 3032. [Google Scholar] [CrossRef]
- Sheng, P.; Huang, J.; Zhang, Z.; Wang, D.; Tian, X.; Ding, J. Construction and characterization of a cellulolytic consortium enriched from the hindgut of Holotrichia parallela larvae. Int. J. Mol. Sci. 2016, 17, 1646. [Google Scholar] [CrossRef] [PubMed]
- Levesque, M.; Dinel, H. Applicability of thermal methods for characterization of peats and plants. Geoderma 1978, 20, 201–213. [Google Scholar] [CrossRef]
- Provenzano, M.R.; Cavallo, O.; Malerba, A.D.; Fabbri, C.; Zaccone, C. Unravelling (maize silage) digestate features throughout a full-scale plant: A spectroscopic and thermal approach. J. Clean. Prod. 2018, 193, 372–378. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Z.; Hu, S.; Yu, J. On the molecular mechanism of GC content variation among eubacterial genomes. Biol. Direct 2012, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.-S.; Christiansen, C.; Abou Hachem, M.; Nielsen, M.; Fukuda, K.; Bozonnet, S.; Blennow, A.; Aghajari, N.; Haser, R.; Svensson, B. An enzyme family reunion—Similarities, differences and eccentricities in actions on α-glucans. Biologia 2008, 63, 967–979. [Google Scholar] [CrossRef]
- Lahiri, D.; Nag, M.; Banerjee, R.; Mukherjee, D.; Garai, S.; Sarkar, T.; Dey, A.; Sheikh, H.I.; Pathak, S.K.; Edinur, H.A. Amylases: Biofilm inducer or biofilm inhibitor? Front. Cell. Infect. Microbiol. 2021, 11, 660048. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.-T.; Lin, C.-W.; Lai, C.-Y.; Wu, C.-H. Ethanol production from lignocelluloses by native strain Klebsiella oxytoca THLC0409. Waste Biomass Valorization 2011, 2, 389–396. [Google Scholar] [CrossRef]
- Zaccone, C.; Plaza, C.; Ciavatta, C.; Miano, T.M.; Shotyk, W. Advances in the determination of humification degree in peat since: Applications in geochemical and paleoenvironmental studies. Earth-Sci. Rev. 2018, 185, 163–178. [Google Scholar] [CrossRef]
- Association, A.P.H. Standard Methods for the Examination of Water and Wastewater; American Public Health Association: Washington, DC, USA, 1926; Volume 6. [Google Scholar]
- Istituto di Ricerca sulle Acque-CNR, A. Analytical Methods for Waters; Manual and Guidline; IRSA/CNR: Brugherio, Italy, 2003; Volume 3. [Google Scholar]
- Verduyn, C.; Postma, E.; Scheffers, W.A.; Van Dijken, J.P. Effect of benzoic acid on metabolic fluxes in yeasts: A continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 1992, 8, 501–517. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass: Laboratory Analytical Procedure; National Renewable Energy Laboratory: Golden, CO, USA, 2008; Volume 1617, pp. 1–16. [Google Scholar]
- Deshavath, N.N.; Mukherjee, G.; Goud, V.V.; Veeranki, V.D.; Sastri, C.V. Pitfalls in the 3, 5-dinitrosalicylic acid (DNS) assay for the reducing sugars: Interference of furfural and 5-hydroxymethylfurfural. Int. J. Biol. Macromol. 2020, 156, 180–185. [Google Scholar] [CrossRef]
- Ghose, T. Measurement of cellulase activities. Pure Appl. Chem. 1987, 59, 257–268. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680. [Google Scholar] [CrossRef] [PubMed]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; De Waal, E.C.; Uitterlinden, A. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef]
- Andreolli, M.; Lampis, S.; Bernardi, P.; Calò, S.; Vallini, G. Bacteria from black crusts on stone monuments can precipitate CaCO3 allowing the development of a new bio-consolidation protocol for ornamental stone. Int. Biodeterior. Biodegrad. 2020, 153, 105031. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]

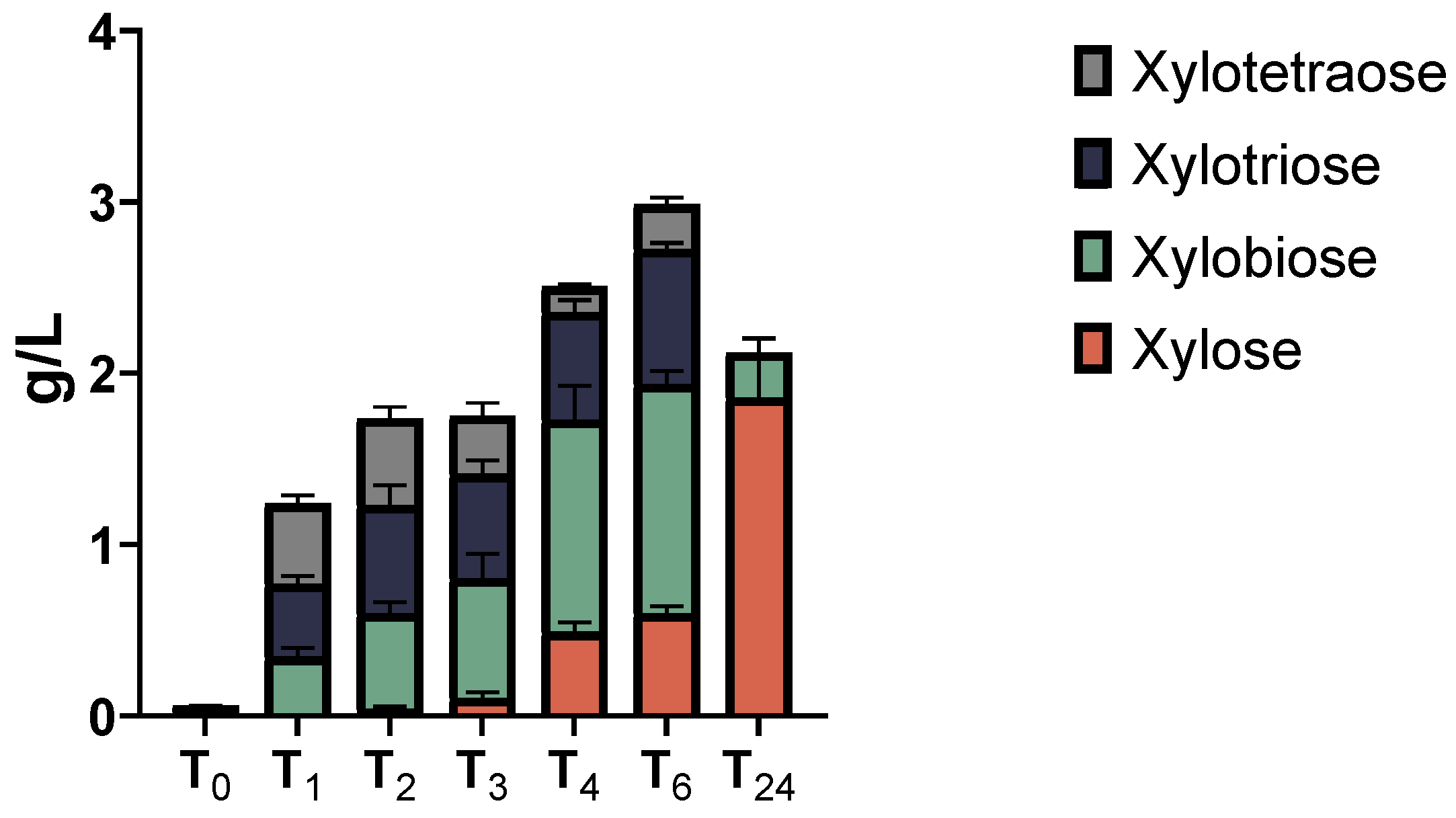

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secretome Name | Units/mL |
|---|---|
| CMC-37 | 2.29 ± 0.20 |
| XYL-37 | 0.47 ± 0.02 |
| Contigs | Contigs (≥5 Kb) | N50 | L50 | |
|---|---|---|---|---|
| T0-SMS | 52,084 | 5182 | 3538 | 8157 |
| T9-37 | 65,657 | 7924 | 6133 | 6137 |
| T9-50 | 39,024 | 5777 | 10,431 | 2652 |
| T9-70 | 13,240 | 1959 | 8148 | 1135 |
| CMC-37 | 6257 | 1174 | 12,179 | 422 |
| CMC-50 | 247 | 50 | 365,423 | 5 |
| CMC-70 | 37 | 29 | 360,411 | 4 |
| XYL-37 | 2790 | 697 | 10,645 | 230 |
| XYL-5050 | 2274 | 630,630 | 2,374,023,740 | 155,155 |
| # MAGs | # Contigs in MAGs | # Total Contigs | % Contigs in MAGs | |
|---|---|---|---|---|
| T0-SMS | 38 | 12,744 | 52,084 | 24.5 |
| T9-37 | 39 | 14,065 | 65,657 | 21.4 |
| T9-50 | 32 | 14,983 | 39,024 | 38.4 |
| T9-70 | 10 | 3115 | 13,240 | 23.5 |
| CMC-37 | 7 | 4024 | 6257 | 64.3 |
| CMC-50 | 1 | 100 | 247 | 40.5 |
| CMC-70 | 1 | 21 | 37 | 56.8 |
| XYL-37 | 1 | 109 | 2790 | 3.9 |
| YXYL-50 | 3 | 788 | 2274 | 34.7 |
| Metagenomes | CAZyme Classes | |||||
|---|---|---|---|---|---|---|
| GH | GT | CE | AA | PL | CBM | |
| T0-SMS | 1335 | 612 | 246 | 29 | 52 | 53 |
| T9-37 | 1477 | 1109 | 352 | 151 | 53 | 76 |
| T9-50 | 1099 | 879 | 205 | 132 | 55 | 53 |
| T9-70 | 390 | 288 | 92 | 21 | 5 | 25 |
| CMC-37 | 280 | 152 | 32 | 22 | 8 | 3 |
| CMC-50 | 59 | 28 | 11 | 4 | 8 | 4 |
| CMC-70 | 32 | 16 | 6 | 4 | 0 | 2 |
| XYL-37 | 157 | 77 | 15 | 10 | 12 | 2 |
| XYL-50 | 85 | 57 | 29 | 10 | 7 | 4 |
| No. Sequences | 4914 | 3218 | 988 | 383 | 200 | 222 |
| Relative abundance (%) | 49.5 | 32.4 | 10.0 | 3.9 | 2.0 | 2.2 |
| a SMS | Digestate | |
|---|---|---|
| TS (% w/w f.m.) | 94.99 ± 2.18 | 7.04 ± 0.01 |
| TVS (% w/w f.m.) | 81.17 ± 1.97 | 4.11 ± 0.03 |
| C (% w/w d.w.) | 38.64 ± 0.11 | 29.55 ± 0.19 |
| N (% w/w d.w.) | 0.71 ± 0.01 | 3.00 ± 0.08 |
| S (% w/w d.w.) | 0.51 ± 0.01 | 1.13 ± 0.01 |
| H (% w/w d.w.) | 5.72 ± 0.05 | 4.11 ± 0.05 |
| C/N | 54.43 ± 1.11 | 9.85 ± 0.31 |
| Cellulose (% w/w) | 34.24 ± 0.71 | – |
| Hemicellulose (% w/w) | 19.45 ± 0.31 | – |
| Lignin (% w/w) | 17.73 ± 0.98 | – |
| Ash (% w/w f.w.) | 13.81 ± 0.21 | 2.93 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bombardi, L.; Salini, A.; Aulitto, M.; Zuliani, L.; Andreolli, M.; Bordoli, P.; Coltro, A.; Vitulo, N.; Zaccone, C.; Lampis, S.; et al. Lignocellulolytic Potential of Microbial Consortia Isolated from a Local Biogas Plant: The Case of Thermostable Xylanases Secreted by Mesophilic Bacteria. Int. J. Mol. Sci. 2024, 25, 1090. https://doi.org/10.3390/ijms25021090
Bombardi L, Salini A, Aulitto M, Zuliani L, Andreolli M, Bordoli P, Coltro A, Vitulo N, Zaccone C, Lampis S, et al. Lignocellulolytic Potential of Microbial Consortia Isolated from a Local Biogas Plant: The Case of Thermostable Xylanases Secreted by Mesophilic Bacteria. International Journal of Molecular Sciences. 2024; 25(2):1090. https://doi.org/10.3390/ijms25021090
Chicago/Turabian StyleBombardi, Luca, Andrea Salini, Martina Aulitto, Luca Zuliani, Marco Andreolli, Paola Bordoli, Annalaura Coltro, Nicola Vitulo, Claudio Zaccone, Silvia Lampis, and et al. 2024. "Lignocellulolytic Potential of Microbial Consortia Isolated from a Local Biogas Plant: The Case of Thermostable Xylanases Secreted by Mesophilic Bacteria" International Journal of Molecular Sciences 25, no. 2: 1090. https://doi.org/10.3390/ijms25021090
APA StyleBombardi, L., Salini, A., Aulitto, M., Zuliani, L., Andreolli, M., Bordoli, P., Coltro, A., Vitulo, N., Zaccone, C., Lampis, S., & Fusco, S. (2024). Lignocellulolytic Potential of Microbial Consortia Isolated from a Local Biogas Plant: The Case of Thermostable Xylanases Secreted by Mesophilic Bacteria. International Journal of Molecular Sciences, 25(2), 1090. https://doi.org/10.3390/ijms25021090