
Mesenchymal Stem/Stromal Cells Induce Myeloid-Derived Suppressor Cells in the Bone Marrow via the Activation of the c-Jun N-Terminal Kinase Signaling Pathway

Abstract
:1. Introduction
2. Results
2.1. MAPK-Related Genes Are Upregulated in MSC-Induced MDSCs
2.2. MSCs Activate the JNK Pathway in BM Cells
2.3. JNK Inhibition Abrogates MSC Effects on MDSC Induction
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Sequencing
4.3. Western Blot Assays
4.4. Quantitative Real-Time Reverse-Transcription PCR (qRT-PCR)
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galipeau, J. Macrophages at the nexus of mesenchymal stromal cell potency: The emerging role of chemokine cooperativity. Stem Cells 2021, 39, 1145–1154. [Google Scholar] [CrossRef]
- Cheung, T.S.; Dazzi, F. Mesenchymal-myeloid interaction in the regulation of immunity. Semin Immunol. 2018, 35, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Chen, H.Y.; Wang, L.T.; Wang, F.H.; Fang, L.W.; Lai, H.Y.; Chen, H.H.; Lu, J.; Hung, M.S.; Cheng, Y.; et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190, 5065–5077. [Google Scholar] [CrossRef]
- Yen, B.L.; Yen, M.L.; Hsu, P.J.; Liu, K.J.; Wang, C.J.; Bai, C.H.; Sytwu, H.K. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Rep. 2013, 1, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ko, J.H.; Jeong, H.J.; Ko, A.Y.; Kim, M.K.; Wee, W.R.; Yoon, S.O.; Oh, J.Y. Mesenchymal stem/stromal cells protect against autoimmunity via CCL2-dependent recruitment of myeloid-derived suppressor cells. J. Immunol. 2015, 194, 3634–3645. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, X.; Yang, S.; Xiao, Y.; Jia, Y.; Zhong, J.; Gao, Q.; Zhang, X. Umbilical cord-derived mesenchymal stem cells promote myeloid-derived suppressor cell enrichment by secreting CXCL1 to prevent graft-versus-host disease after hematopoietic stem cell transplantation. Cytotherapy 2021, 23, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Queckbörner, S.; Jylhä, C.E.; Andrén, A.T.; Forshell, T.Z.P.; Blanc, K.L. Lysis and phenotypic modulation of mesenchymal stromal cells upon blood contact triggers anti-inflammatory skewing of the peripheral innate immune repertoire. Cytotherapy 2023, 25, 956–966. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Trikha, P.; Carson, W.E., 3rd. Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta 2014, 1846, 55–65. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Lee, H.J.; Ko, J.H.; Kim, H.J.; Jeong, H.J.; Oh, J.Y. Mesenchymal stromal cells induce distinct myeloid-derived suppressor cells in inflammation. JCI Insight 2020, 18, e136059. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Ko, J.H.; Oh, J.Y. Mesenchymal stromal cells regulate THP-1-differentiated macrophage cytokine production by activating Akt/mammalian target of rapamycin complex 1 pathway. Cytotherapy 2023, 25, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef]
- Dobreva, Z.G.; Miteva, L.D.; Stanilova, S.A. The inhibition of JNK and p38 MAPKs downregulates IL-10 and differentially affects c-Jun gene expression in human monocytes. Immunopharmacol. Immunotoxicol. 2009, 31, 195–201. [Google Scholar] [CrossRef]
- Bayik, D.; Tross, D.; Haile, L.A.; Verthelyi, D.; Klinman, D.M. Regulation of the maturation of human monocytes into immunosuppressive macrophages. Blood Adv. 2017, 1, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, H.; Zhang, Z.; Lin, W.; Wei, X.; Shao, B. Targeting the MDSCs of Tumors In Situ With Inhibitors of the MAPK Signaling Pathway to Promote Tumor Regression. Front. Oncol. 2021, 11, 647312. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yang, J.; Qian, J.; Liu, R.; Huang, E.; Wang, Y.; Luo, F.; Chu, Y. TLR1/TLR2 signaling blocks the suppression of monocytic myeloid-derived suppressor cell by promoting its differentiation into M1-type macrophage. Mol. Immunol. 2019, 112, 266–273. [Google Scholar] [CrossRef]
- Liu, L.; Song, H.; Duan, H.; Chai, J.; Yang, J.; Li, X.; Yu, Y.; Zhang, X.; Hu, X.; Xiao, M.; et al. TSG-6 secreted by human umbilical cord-MSCs attenuates severe burn-induced excessive inflammation via inhibiting activations of P38 and JNK signaling. Sci. Rep. 2016, 6, 30121. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Sánchez-Tilló, E.; Arpa, L.; Park, J.M.; Caelles, C.; Lloberas, J.; Celada, A. Selective roles of MAPKs during the macrophage response to IFN-gamma. J. Immunol. 2008, 180, 4523–4529. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensébé, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef]
- Lee, M.J.; Park, S.Y.; Ko, J.H.; Lee, H.J.; Ryu, J.S.; Park, J.W.; Khwarg, S.I.; Yoon, S.O.; Oh, J.Y. Mesenchymal stromal cells promote B-cell lymphoma in lacrimal glands by inducing immunosuppressive microenvironment. Oncotarget 2017, 8, 66281–66292. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar] [CrossRef]
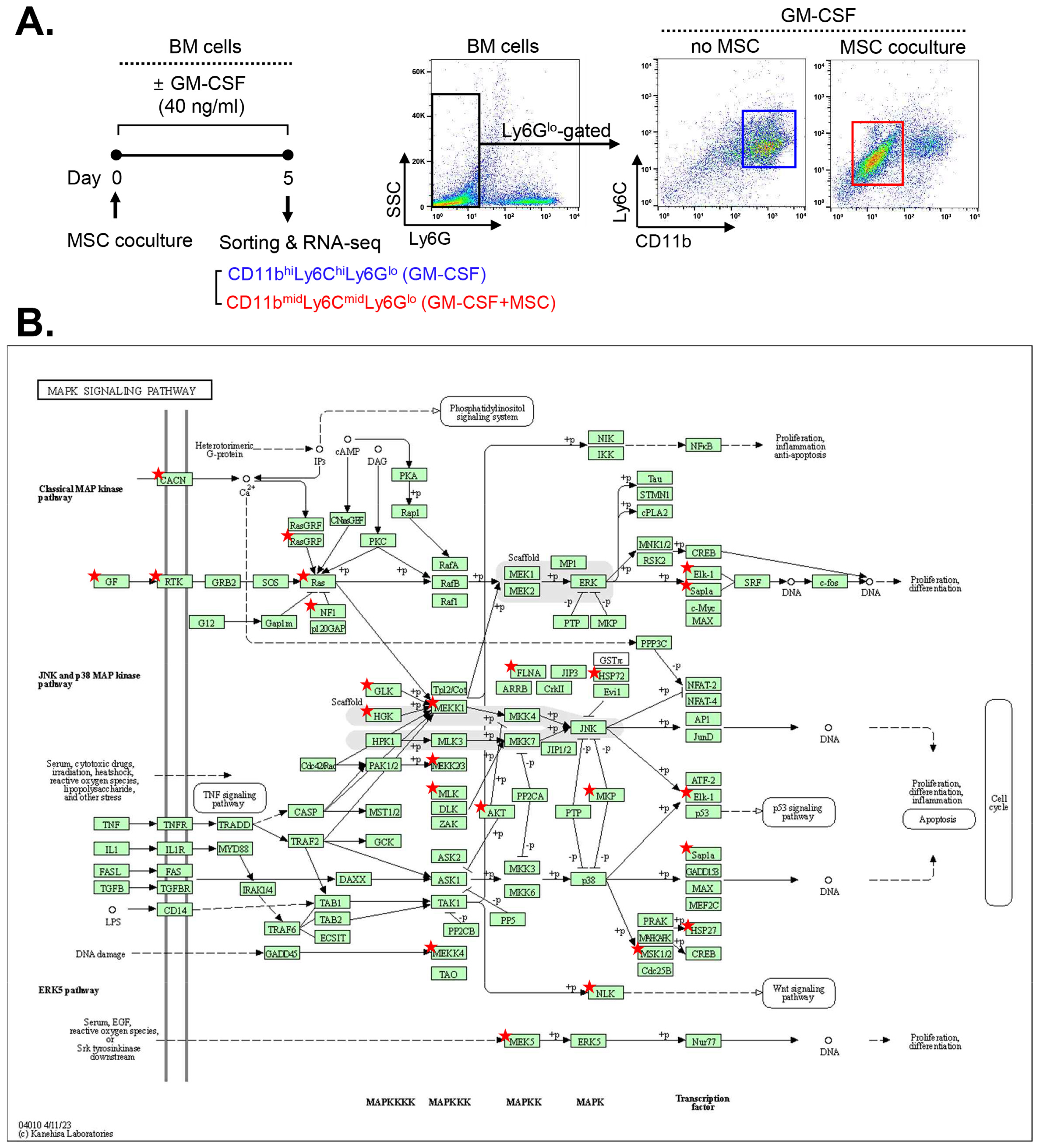
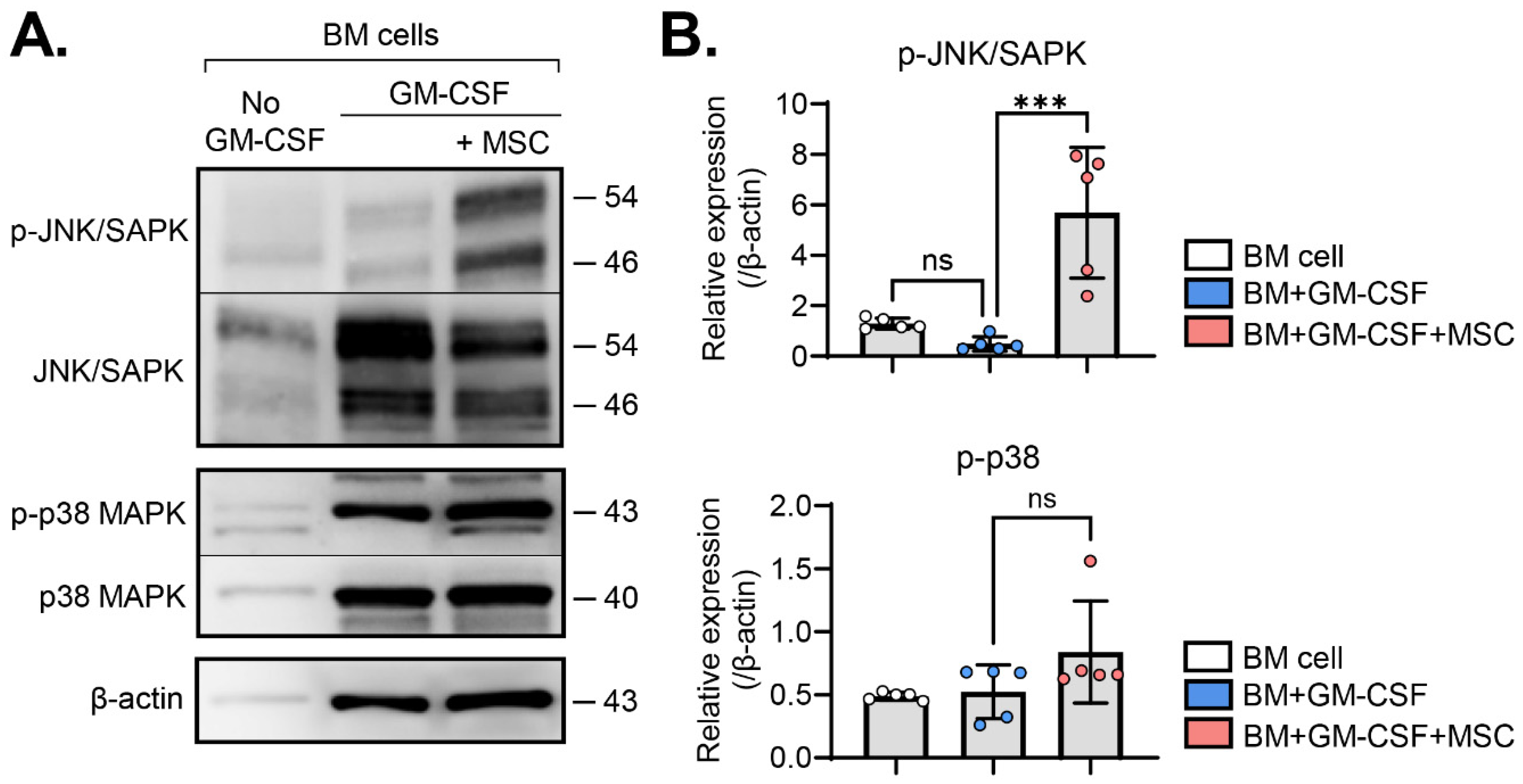
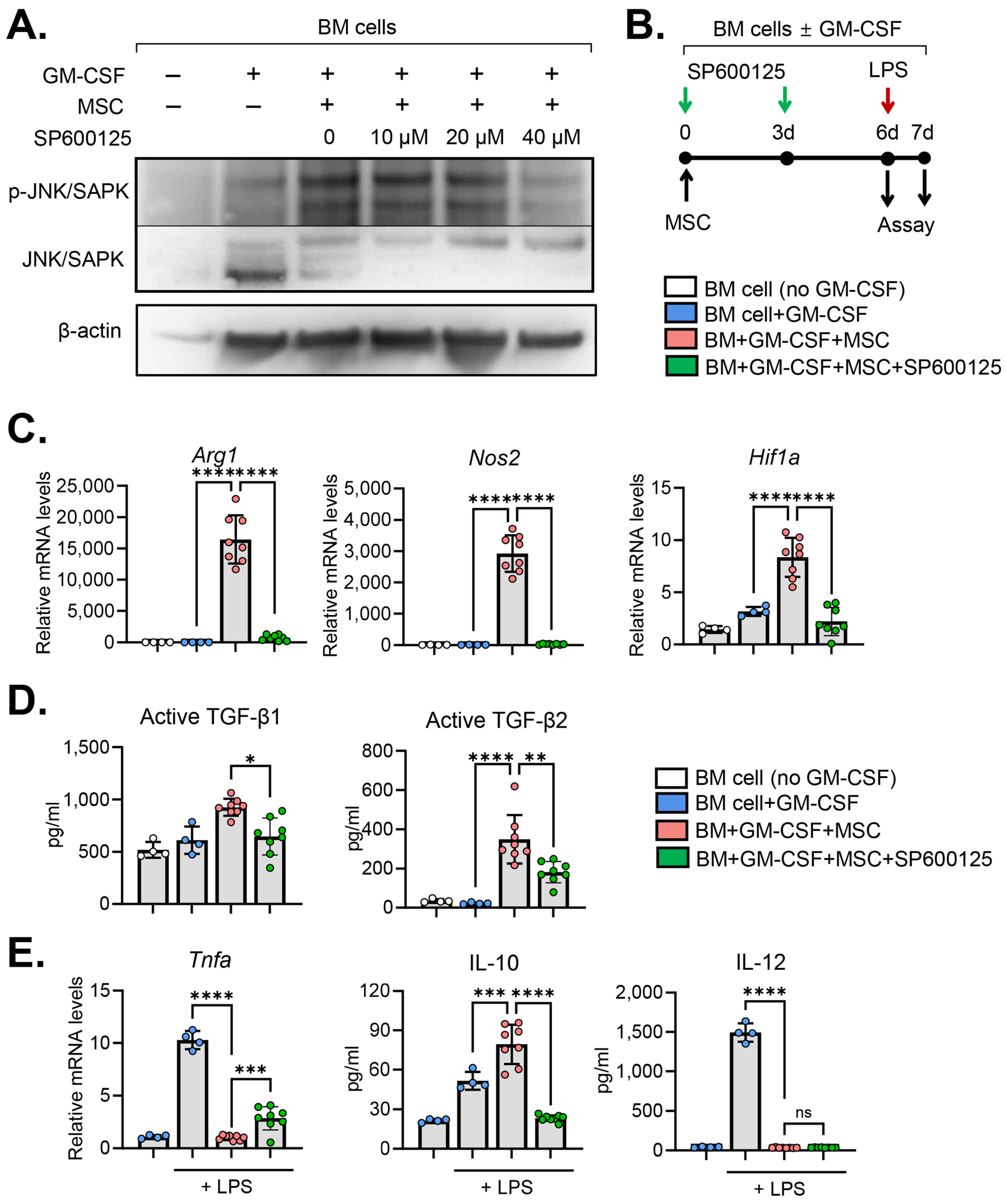

{kind=link}
{kind=link}
{kind=link}
| Gene Description | Gene Symbol | Fold Change * |
|---|---|---|
| filamin, beta | Flnb | 559.322 |
| FMS-like tyrosine kinase 1 | Flt1 | 223.11 |
| neurofibromin 1 | Nf1 | 179.681 |
| ephrin A2 | Efna2 | 135.17 |
| calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | Cacna1a | 123.802 |
| dual-specificity phosphatase 10 | Dusp10 | 119.544 |
| thymoma viral proto-oncogene 3 | Akt3 | 112.279 |
| mitogen-activated protein kinase kinase kinase 9 | Map3k9 | 69.346 |
| platelet-derived growth factor receptor, beta polypeptide | Pdgfc | 37.249 |
| fibroblast growth factor receptor 1 | Fgfr1 | 29.61 |
| calcium channel, voltage-dependent, N type, alpha 1B subunit | Cacna1b | 27.199 |
| related RAS viral (r-ras) oncogene 2 | Rras2 | 21.369 |
| mitogen-activated protein kinase kinase kinase 4 | Map3k4 | 21.317 |
| mitogen-activated protein kinase kinase kinase kinase 3 | Map4k3 | 12.556 |
| mitogen-activated protein kinase kinase kinase kinase 4 | Map4k4 | 7.147 |
| ELK1, member of ETS oncogene family | Elk1 | 12.104 |
| nemo-like kinase | Nlk | 6.99 |
| calcium channel, voltage-dependent, L type, alpha 1D subunit | Cacna1d | 5.488 |
| hepatocyte growth factor | Hgf | 3.193 |
| mitogen-activated protein kinase kinase kinase 1 | Map3k1 | 3.146 |
| platelet-derived growth factor receptor, beta polypeptide | Pdgfrb | 3.010 |
| heat shock protein 1 | Hspb1 | 2.873 |
| mitogen-activated protein kinase kinase 5 | Map2k5 | 2.75 |
| heat shock protein 1B | Hspa1b | 2.626 |
| RAS, guanyl-releasing protein 3 | Rasgrp3 | 2.563 |
| fibroblast growth factor 7 | Fgf7 | 2.471 |
| mitogen-activated protein kinase kinase kinase 3 | Map3k3 | 2.372 |
| ribosomal protein S6 kinase, polypeptide 5 | Rps6ka5 | 2.286 |
| dual-specificity phosphatase 4 | Dusp4 | 2.278 |
| ELK4, member of ETS oncogene family (Elk4) | Elk4 | 2.264 |
| dual-specificity phosphatase 8 | Dusp8 | 2.192 |
| fibroblast growth factor receptor 2 | Fgfr2 | 2.189 |
| nerve growth factor | Ngf | 2.1 |
| Gene Description | Gene Symbol | Fold Change * |
|---|---|---|
| toll-like receptor 4 | Tlr4 | 0.009 |
| thioredoxin 2 | Txn2 | 0.012 |
| BCL2-like 1 | Bcl2l1 | 0.013 |
| C-C motif chemokine ligand 5 | Ccl5 | 0.014 |
| gasdermin D | Gsdmd | 0.014 |
| interleukin 1 beta | Il1b | 0.014 |
| NLR family, apoptosis inhibitory protein 5 | Naip5 | 0.016 |
| v-rel reticuloendotheliosis viral oncogene homolog A | Rela | 0.016 |
| caspase 8 | Casp8 | 0.018 |
| C-X-C motif chemokine ligand 3 | Cxcl3 | 0.02 |
| mitochondrial antiviral-signaling protein | Mavs | 0.021 |
| nucleotide-binding oligomerization domain-containing 1 | Nod1 | 0.023 |
| TNF receptor-associated factor 6 | Traf6 | 0.024 |
| autophagy related 12 | Atg12 | 0.032 |
| signal transducer and activator of transcription 1 | Stat1 | 0.032 |
| NLR family, apoptosis inhibitory protein 2 | Naip2 | 0.039 |
| inhibitor of kappaB kinase beta | Ikbkb | 0.044 |
| TGF-beta-activated kinase 1/MAP3K7 binding protein 1 | Tab1 | 0.048 |
| TNF receptor-associated factor 3 | Traf3 | 0.051 |
| interleukin 18 | Il18 | 0.053 |
| inositol 1,4,5-triphosphate receptor 2 | Itpr2 | 0.054 |
| dynamin 1-like | Dnm1l | 0.055 |
| tumor necrosis factor | Tnf | 0.06 |
| RanBP-type and C3HC4-type zinc finger containing 1 | Rbck1 | 0.074 |
| nuclear factor of kappa light polypeptide gene enhancer in B cells 1, p105 | Nfkb1 | 0.085 |
| thioredoxin 1 | Txn1 | 0.086 |
| transient receptor potential cation channel, subfamily M, member 2 | Trpm2 | 0.088 |
| cathepsin B | Ctsb | 0.115 |
| inositol 1,4,5-trisphosphate receptor 1 | Itpr1 | 0.117 |
| interferon-activated gene 204 | Ifi204 | 0.138 |
| C-X-C motif chemokine ligand 1 | Cxcl1 | 0.14 |
| GABA type A receptor-associated protein-like 1 | Gabarapl1 | 0.142 |
| NLR family, apoptosis inhibitory protein 6 | Naip6 | 0.143 |
| inhibitor of kappaB kinase epsilon | Ikbke | 0.153 |
| C-X-C motif chemokine ligand 2 | Cxcl2 | 0.167 |
| interferon regulatory factor 3 | Irf3 | 0.173 |
| tumor necrosis factor, alpha-induced protein 3 | Tnfaip3 | 0.179 |
| voltage-dependent anion channel 3 | Vdac3 | 0.194 |
| DEAH-box helicase 33 | Dhx33 | 0.224 |
| nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha | Nfkbia | 0.308 |
| GABA type A receptor-associated protein-like 2 | Gabarapl2 | 0.423 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Oh, J.Y. Mesenchymal Stem/Stromal Cells Induce Myeloid-Derived Suppressor Cells in the Bone Marrow via the Activation of the c-Jun N-Terminal Kinase Signaling Pathway. Int. J. Mol. Sci. 2024, 25, 1119. https://doi.org/10.3390/ijms25021119
Lee HJ, Oh JY. Mesenchymal Stem/Stromal Cells Induce Myeloid-Derived Suppressor Cells in the Bone Marrow via the Activation of the c-Jun N-Terminal Kinase Signaling Pathway. International Journal of Molecular Sciences. 2024; 25(2):1119. https://doi.org/10.3390/ijms25021119
Chicago/Turabian StyleLee, Hyun Ju, and Joo Youn Oh. 2024. "Mesenchymal Stem/Stromal Cells Induce Myeloid-Derived Suppressor Cells in the Bone Marrow via the Activation of the c-Jun N-Terminal Kinase Signaling Pathway" International Journal of Molecular Sciences 25, no. 2: 1119. https://doi.org/10.3390/ijms25021119