
Genetic Epilepsies and Developmental Epileptic Encephalopathies with Early Onset: A Multicenter Study

 , , , , ,
, , , , ,  , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
2.1. Monogenic Conditions
2.1.1. Clinical Findings
Family History
Epilepsy
Electroencephalogram (EEG) Pattern at Onset
Neurological Examination
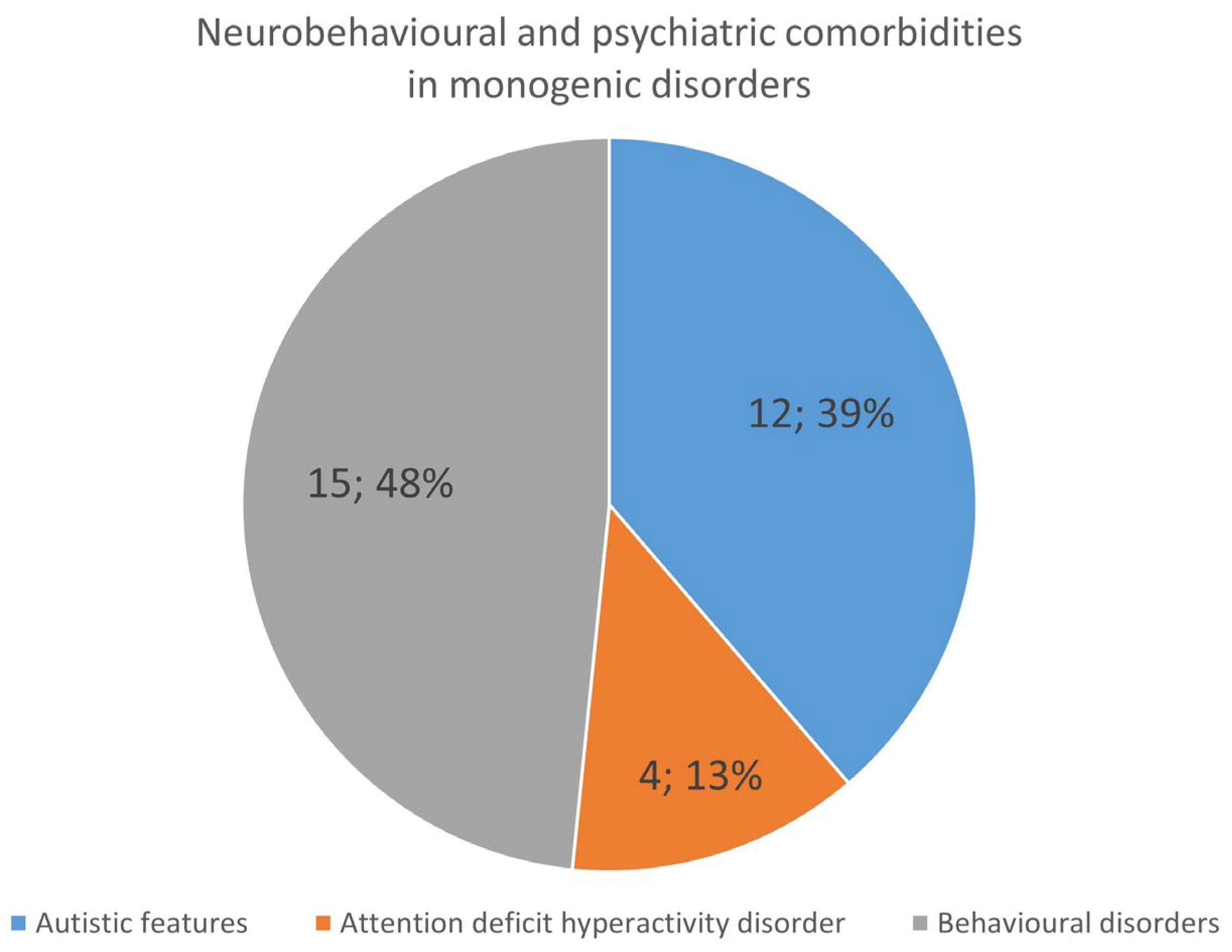
Neurodevelopmental Features and Psychiatric Comorbidities
Neuroimaging Findings
Genetic Testing
Segregation Analysis
Seizure Outcome at the Latest Follow-Up Visit
EEG Pattern at the Latest Follow-Up Visit
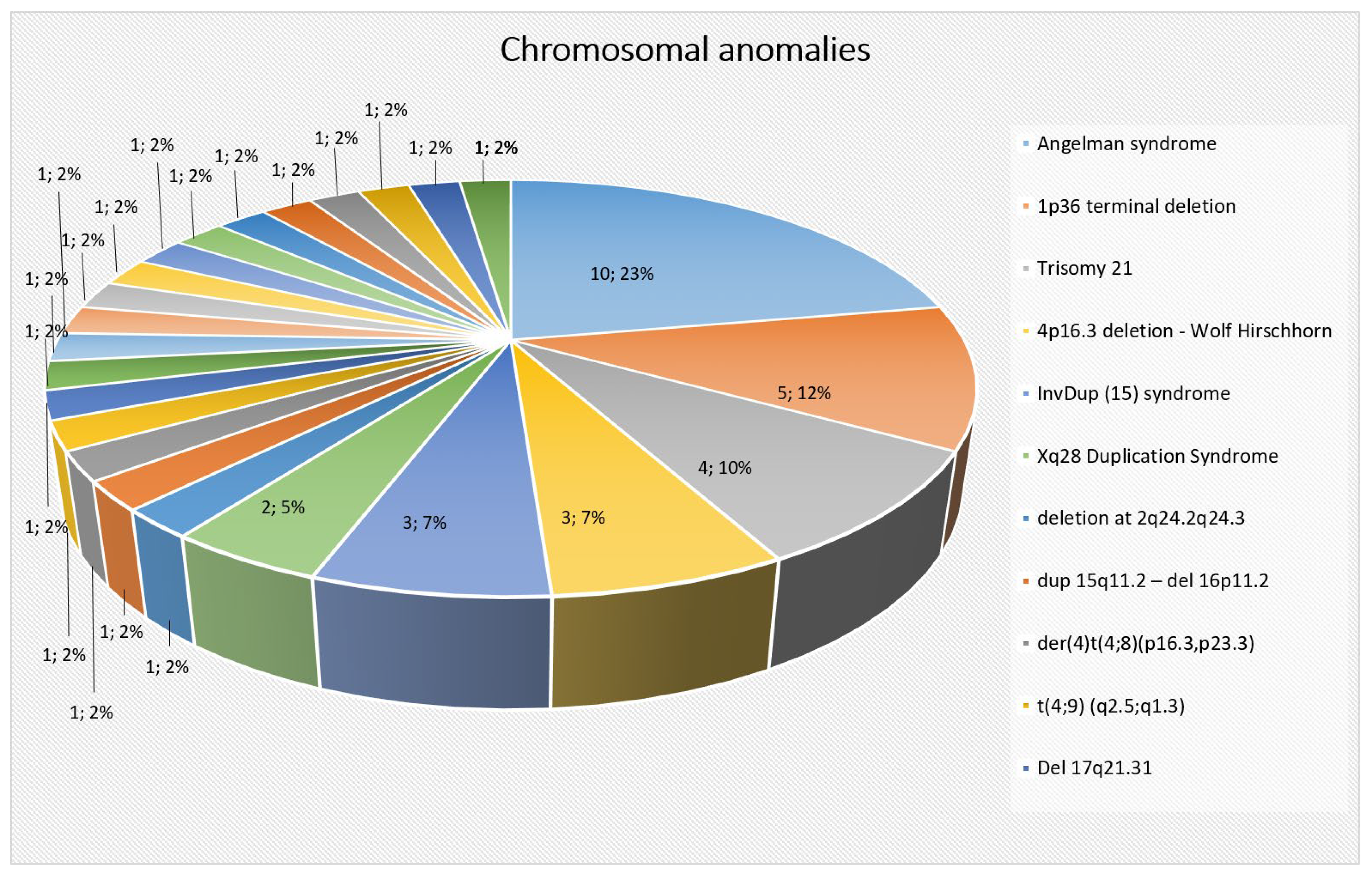
2.2. Chromosomal Abnormalities
2.2.1. Clinical Findings
Family History
Epilepsy
Electroencephalogram Pattern (EEG) at Onset
Neurological Examination
Neurodevelopmental Features and Psychiatric Comorbidities
Neuroimaging Findings
Genetic Testing
Segregation Analysis
Seizure Outcome
EEG Pattern at the Latest Follow-Up Visit
2.3. Genetic Variations of Unknown Clinical Significance
2.3.1. Clinical Findings
Family History
Epilepsy
Electroencephalogram Pattern (EEG) at Onset
Neurological Examination
Neurodevelopmental Features and Psychiatric Comorbidities
Neuroimaging Findings
Genetic Testing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | VUS | Inheritance | Family History | Age at First Seizure | Seizure Type at Onset | DD/ ID | Neurological Examination | Behavioural Problems | EEG Pattern | Brain Nuroimaging | Drug Resistance | Seizures at Last Follow-Up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (1) | F | 46,XX,del(16)(p13.3) (301 Kb deletion) | Inherited from asymptomatic mother | Negative | 36 months | Atypical absences | Present | Normal | No | At onset: NA | Normal | No | Seizure-free on oxcarbazepine and levetiracetam |
| At last follow-up: slow activity | |||||||||||||
| (2) | F | CLCN2 [NM_004366.6]: c.1783T>C: p.Cys595Arg | Inherited from mother, VUS | Negative | 15 months | Complex febrile seizure | Present | Ataxic gait and tremor | Yes (aggressive behaviour) | At onset: NA | Aspecific abnormalities: hyperintensity of right occipital cortex | Yes | Focal motor seizures |
| COL4A3BP [NM_001379029.1]: c.979+7T>C | Inherited from mother, likely benign | At last follow-up: diffuse abnormalities (mainly in the left temporal region) | |||||||||||
| SLC9A6 [NM_001379110.1] c.37C>T, p.Arg13Cys | Inherited from father, benign | ||||||||||||
| (3) | F | SCN8A [NM_001330260.2]: c.4697C>T, p.Thr1566Ile | de novo | Negative | 36 months | Status epilepticus | No | Normal | No | Focal discharges (frontal) | Normal | No | Seizure-free on carbamazepine |
| (4) | M | 248 Kb 46,XY,del(16)(p13.2) involving A2BP1 gene | NA | Negative | 13 months | Generalized tonic clonic seizure | Yes (moderate ID) | Macrocephaly, ataxic gait, dysmetria | Present | At onset: NA | Hypoplasia of cerebellum and corpus callosus | No | Seizure-free on carbamazepine, valproate and levetiracetam |
| At last follow-up: slow background activity with sharp waves over posterior regions | |||||||||||||
| (5) | M | MTOR [NM_004958.3] c.4472G>T, p.Gly1491Val | NA | Positive | 24 months | Childhood absence epilepsy (GGE) | Speech delay and specific learning difficulties | Attention deficit and obsessive trait | Normal | At onset: Diffuse discharges induced by hyperpnea | Normal | No | Seizure-free on valproate |
| At last follow-up: no abnormalities | |||||||||||||
| (6) | F | SIK1 [NM_173354.5]: c.718C>T, p.Arg240Cys | Inherited from her mother | Positive | 23 months | Generalized tonic–clonic | Normal until 23 months old, then developmental regression | Abnormal | Autistic traits, stereotypies | At onset: Several diffuse abnormalities upon falling asleep | Brain MRI: aspecific white matter changes | Yes | Absence seizures |
| At last follow-up: disorganization of background activity with diffuse discharges | |||||||||||||
| (7) | F | WDR45 [NM_007075.3]: | Mother: negative | Negative | 36 months | Myoclonic atonic | Speech delay | Normal | Inattention traits | At onset: normal | Brain MRI: normal | Yes | Generalized tonic–clonic seizures |
| c.1078G>T, p.(Asp360Tyr) | Father: not performed | At last follow-up: slow activity and multifocal discharges | |||||||||||
| (8) | M | 46,XY,del(22)(q11.21) | Inherited from his father | Negative | 13 months | Complex febrile seizures and focal motor seizures | Yes | Abnormal | No | At onset: multifocal left discharges | Brain MRI: aspecific white matter changes, microcalcifications and suspected polymicrogyria | No | Seizure-free on carbamazepine |
| At last follow-up: multifocal left discharges | |||||||||||||
| (9) | M | GRIN2A: [NM_001134407.3]: c.459G>C, p.Gln153His | NA | Negative | Neonatal period | Status epilepticus with recurrent focal motor seizures | Speech delay | Normal | No | At onset: frequent theta-delta activity over left fronto-central regions. | Brain MRI: normal | Yes | Focal motor seizures |
| At last follow-up: multifocal discharges upon falling asleep | |||||||||||||
| (10) | F | KCNMA1 [NM_001161352.1]: c.413C>T, p.Ala138 Val | Both parents are heterozygous for variant | Negative | 24 months | Febrile seizures | ID with absent speech | Normal | Ideomotor slowdown, aggressiveness, and irritability | At onset: normal | NA | Yes | Atypical absence, tonic and focal motor seizures. |
| At last follow-up: pattern Lennox–Gastaut | |||||||||||||
| (11) | M | HUWE1: [NM_031407.7]: c.413C > T, p.Ala138Val | Inherited from his mother | Negative | 30 months | Febrile seizures | Speech delay | Normal | Yes (hyperactivity) | At onset: normal | Brain MRI: normal | Yes | Focal motor seizures |
| At last follow-up: theta-delta activity with multifocal discharges | |||||||||||||
| (12) | F | 1. SCN1A: [NM_001165963.4]: c.419C>T, p.Thr140Ile | NA | Negative | 1 month | Myoclonic | Yes | Spastic tetraparesis | No | At onset: NA | Brain MRI: progressive cerebral and cerebellar atrophy | Yes | Focal motor seizures |
| 2. HDAC4 [NM_001378414.1] c.928C>A, p.Val310Ile | At last follow-up: slow and disorganized background activities | ||||||||||||
| (13) | M | HCN1: [NM_021072.4]: c.1232A>G, p.Tyr411Cys | Inherited from his father | Positive | 14 months | Generalized tonic–clonic seizures with and without fever | No | Normal | No | At onset: normal | Normal | No | Seizure-free without therapy |
| At last follow-up: Multifocal abnormalities with secondary generalization | |||||||||||||
| (14) | M | 1. DOCK3 [NM_004947.5]: c.3884G>A, p.Arg1295Gln | Inherited from his mother | Positive | 16 months | Status epilepticus | No | Strabismus | No | At onset: non-convulsive status epilepticus | Normal | No | Seizure-free on valproate |
| 2. DOCK3 [NM_004947.5]: c.5500+6G>A | At last follow-up: normal | ||||||||||||
| (15) | M | SCN2A [NM_001040142.2] c.3385G> | Inherited from his father | Negative | 5 months | Focal motor seizures | No | Normal | No | At onset: slow activity | Normal | No | Seizure-free |
| At last follow-up: normal | |||||||||||||
| (16) | M | SCN1A [NM_0011659634]: c.99G>C (K33N) | Inherited from his mother | Positive | 28 months | Febrile seizures | No | Normal | No | At onset: Normal | Normal | No | Seizure-free |
| At last follow-up: normal | |||||||||||||
| (17) (HSP) | M | SCN1A [NM_0011659634]: (c.5717T>A) p.I1906N | Not reported | Not reported | 5 months | Febrile seizures | No | Normal | No | At onset: Normal | Normal | No | Seizure-free |
| At last follow-up: normal | |||||||||||||
| (18) (HSP) | F | 46,XXdel(16)(p13.11) | Not reported | Positive | First days of life | Generalized tonic–clonic seizures | Present | Spastic tetraparesis | No | At onset: Not reported | Periventricular leucomalacia | Yes | Focal motor seizures |
| At last follow-up: disorganization of background activity with focal discharges | |||||||||||||
| (19) | M | 46,XY,dup(2p21) and 46,XY,dup(16p13.3) | Not reported | Positive | 6 months | Clonic seizures | Present | Ataxia gait | Yes (autistic spectrum disorder) | At onset: Multifocal anomalies | Cerebellar atrophy | yes | Not reported |
| At last follow-up: slow activity with multifocal discharges upon falling asleep |
Seizure Outcome
EEG Pattern at the Latest Follow-Up Visit
3. Discussion
4. Materials and Methods
4.1. Inclusion Criteria
4.2. Exclusion Criteria
4.3. Data Collection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, M.; Mefford, H.C. Recent Advances in Epilepsy Genomics and Genetic Testing. F1000Research 2020, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, S.; Arzimanoglou, A.; Blümcke, I.; Scheffer, I.E.; Wiebe, S.; Zelano, J.; Walker, M.C. The aetiologies of epilepsy. Epileptic Disord. 2021, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Syvertsen, M.; Nakken, K.O.; Edland, A.; Hansen, G.; Hellum, M.K.; Koht, J. Prevalence and etiology of epilepsy in a Norwegian county-A population based study. Epilepsia 2015, 56, 699–706. [Google Scholar] [CrossRef]
- Falco-Walter, J. Epilepsy-Definition, Classification, Pathophysiology, and Epidemiology. Semin. Neurol. 2020, 40, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Weber, Y.G.; Biskup, S.; Helbig, K.L.; Von Spiczak, S.; Lerche, H. The role of genetic testing in epilepsy diagnosis and management. Expert Rev. Mol. Diagn. 2017, 17, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Rastin, C.; Schenkel, L.C.; Sadikovic, B. Complexity in Genetic Epilepsies: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 14606. [Google Scholar] [CrossRef]
- Ji, J.; Leung, M.L.; Baker, S.; Deignan, J.L.; Santani, A. Clinical exome reanalysis: Current practice and beyond. Mol. Diagn. Ther. 2021, 25, 529–536. [Google Scholar] [CrossRef]
- Brock, D.C.; Abbott, M.; Reed, L.; Kammeyer, R.; Gibbons, M.; Angione, K.; Bernard, T.J.; Gaskell, A.; Demarest, S. Epilepsy panels in clinical practice: Yield, variants of uncertain significance, and treatment implications. Epilepsy Res. 2023, 193, 107167. [Google Scholar] [CrossRef]
- Johannesen, K.M.; Tümer, Z.; Weckhuysen, S.; Barakat, T.S.; Bayat, A. Solving the unsolved genetic epilepsies: Current and future perspectives. Epilepsia 2023, 64, 3143–3154. [Google Scholar] [CrossRef]
- Koh, H.Y.; Smith, L.; Wiltrout, K.N.; Podury, A.; Chourasia, N.; D’Gama, A.M.; Park, M.; Knight, D.; Sexton, E.L.; Koh, J.J.; et al. Utility of Exome Sequencing for Diagnosis in Unexplained Pediatric-Onset Epilepsy. JAMA Netw. Open 2023, 6, e2324380. [Google Scholar] [CrossRef] [PubMed]
- Spagnoli, C.; Frattini, D.; Rizzi, S.; Salerno, G.G.; Fusco, C. Early infantile SCN1A epileptic encephalopathy: Expanding the genotype-phenotype correlations. Seizure 2019, 65, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.; Peron, A.; Spaccini, L.; Novara, F.; Scelsa, B.; Introvini, P.; Raviglione, F.; Faiola, S.; Zuffardi, O. Neonatal suppression-burst without epileptic seizures: Expanding the electroclinical phenotype of STXBP1-related, early-onset encephalopathy. Epileptic Disord. 2013, 15, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Peron, A.; Spaccini, L.; Norris, J.; Bova, S.M.; Selicorni, A.; Weber, G.; Wood, T.; Schwartz, C.E.; Mastrangelo, M. Snyder-Robinson syndrome: A novel nonsense mutation in spermine synthase and expansion of the phenotype. Am. J. Med. Genet. A 2013, 161A, 2316–2320. [Google Scholar] [CrossRef] [PubMed]
- Peron, A.; Iascone, M.; Salvatici, E.; Cavirani, B.; Marchetti, D.; Corno, S.; Vignoli, A. PIGW-related glycosylphosphatidylinositol deficiency: Description of a new patient and review of the literature. Am. J. Med. Genet. A 2020, 182, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum. Mutat. 2020, 41, 1183. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, F.; Giorgini, V.; Masciadri, M.; Bonati, M.T.; Marchi, M.; Cracco, I.; Gentilini, D.; Peron, A.; Savini, M.N.; Spaccini, L.; et al. Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes. Int. J. Mol. Sci. 2019, 20, 3621. [Google Scholar] [CrossRef]
- Scala, M.; Zonneveld-Huijssoon, E.; Brienza, M.; Mecarelli, O.; van der Hout, A.H.; Zambrelli, E.; Turner, K.; Zara, F.; Peron, A.; Vignoli, A.; et al. De novo ARHGEF9 missense variants associated with neurodevelopmental disorder in females: Expanding the genotypic and phenotypic spectrum of ARHGEF9 disease in females. Neurogenetics 2021, 22, 87–94. [Google Scholar] [CrossRef]
- Carter, L.B.; Battaglia, A.; Cherry, A.; Manning, M.A.; Ruzhnikov, M.R.; Bird, L.M.; Dowsett, L.; Graham, J.M., Jr.; Alkuraya, F.S.; Hashem, M.; et al. Perinatal distress in 1p36 deletion syndrome can mimic hypoxic ischemic encephalopathy. Am. J. Med. Genet. A 2019, 179, 1543–1546. [Google Scholar] [CrossRef]
- Spagnoli, C.; Salerno, G.G.; Iodice, A.; Frattini, D.; Pisani, F.; Fusco, C. KCNQ2 encephalopathy: A case due to a de novo deletion. Brain Dev. 2018, 40, 65–68. [Google Scholar] [CrossRef]
- Fusco, C.; Frattini, D.; Bassi, M.T. A novel KCNQ3 gene mutation in a child with infantile convulsions and partial epilepsy with centrotemporal spikes. Eur. J. Paediatr. Neurol. 2015, 19, 102–103. [Google Scholar] [CrossRef]
- Maini, I.; Iodice, A.; Spagnoli, C.; Salerno, G.G.; Bertani, G.; Frattini, D.; Fusco, C. Expanding phenotype of PRRT2 gene mutations: A new case with epilepsy and benign myoclonus of early infancy. Eur. J. Paediatr. Neurol. 2016, 20, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Iodice, A.; Spagnoli, C.; Frattini, D.; Salerno, G.G.; Rizzi, S.; Fusco, C. Biallelic SZT2 mutation with early onset of focal status epilepticus: Useful diagnostic clues other than epilepsy, intellectual disability and macrocephaly. Seizure 2019, 69, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Coryell, J.; Gaillard, W.D.; Shellhaas, R.A.; Grinspan, Z.M.; Wirrell, E.C.; Knupp, K.G.; Wusthoff, C.J.; Keator, C.; Sullivan, J.E.; Loddenkemper, T.; et al. Neuroimaging of Early Life Epilepsy. Pediatrics 2018, 142, e20180672. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.B.; Yoong, M.; Sumpter, R.E.; Verity, K.; Shetty, J.; McLellan, A.; Chin, R.F.M. Incidence of early-onset epilepsy: A prospective population-based study. Seizure 2020, 75, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.A.; Annegers, J.F.; Kurland, L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993, 34, 453–468. [Google Scholar] [CrossRef] [PubMed]
- De Wachter, M.; Schoonjans, A.S.; Weckhuysen, S.; Van Schil, K.; Löfgren, A.; Meuwissen, M.; Jansen, A.; Ceulemans, B. From diagnosis to treatment in genetic epilepsies: Implementation of precision medicine in real-world clinical practice. Eur. J. Paediatr. Neurol. 2023, 48, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, C.; Ki, C.S.; Lee, J. Determining the best candidates for next-generation sequencing-based gene panel for evaluation of early-onset epilepsy. Mol. Genet. Genom. Med. 2020, 8, e1376. [Google Scholar] [CrossRef]
- Jang, S.S.; Kim, S.Y.; Kim, H.; Hwang, H.; Chae, J.H.; Kim, K.J.; Kim, J.I.; Lim, B.C. Diagnostic Yield of Epilepsy Panel Testing in Patients With Seizure Onset Within the First Year of Life. Front. Neurol. 2019, 10, 988. [Google Scholar] [CrossRef]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood- onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef]
- Wilmshurst, J.M.; Gaillard, W.D.; Vinayan, K.P.; Tsuchida, T.N.; Plouin, P.; Van Bogaert, P.; Carrizosa, J.; Elia, M.; Craiu, D.; Jovic, N.J.; et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia 2015, 56, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.; Galosi, S.; Cesario, S.; Renzi, A.; Campea, L.; Leuzzi, V. Presenting Patterns of Genetically Determined Developmental Encephalopathies With Epilepsy and Movement Disorders: A Single Tertiary Center Retrospective Cohort Study. Front. Neurol. 2022, 13, 855134. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D. Congenital Disorders of Autophagy: What a Pediatric Neurologist Should Know. Neuropediatrics 2018, 49, 18–25. [Google Scholar] [CrossRef]
- Girard, J.M.; Turnbull, J.; Ramachandran, N.; Minassian, B.A. Progressive myoclonus epilepsy. Handb. Clin. Neurol. 2013, 113, 1731–1736. [Google Scholar] [PubMed]
- Ikeda, A.; Kumaki, T.; Tsuyusaki, Y.; Tsuji, M.; Enomoto, Y.; Fujita, A.; Saitsu, H.; Matsumoto, N.; Kurosawa, K.; Goto, T. Genetic and clinical features of pediatric-onset hereditary spastic paraplegia: A single-center study in Japan. Front. Neurol. 2023, 14, 1085228. [Google Scholar] [CrossRef] [PubMed]
- D’Gama, A.M.; Mulhern, S.; Sheidley, B.R.; Boodhoo, F.; Buts, S.; Chandler, N.J.; Cobb, J.; Curtis, M.; Higginbotham, E.J.; Holland, J.; et al. Evaluation of the feasibility, diagnostic yield, and clinical utility of rapid genome sequencing in infantile epilepsy (Gene-STEPS): An international, multicentre, pilot cohort study. Lancet Neurol. 2023, 22, 812–825. [Google Scholar] [CrossRef]
- Ho, N.T.; Kroner, B.; Grinspan, Z.; Fureman, B.; Farrell, K.; Zhang, J.; Buelow, J.; Hesdorffer, D.C.; Rare Epilepsy Network Steering Committee. Comorbidities of Rare Epilepsies: Results from the Rare Epilepsy Network. J. Pediatr. 2018, 203, 249–258.e5. [Google Scholar] [CrossRef]
- Morrison-Levy, N.; Borlot, F.; Jain, P.; Whitney, R. Early-Onset Developmental and Epileptic Encephalopathies of Infancy: An Overview of the Genetic Basis and Clinical Features. Pediatr. Neurol. 2021, 116, 85–94. [Google Scholar] [CrossRef]
- Battaglia, A.; Hoyme, H.E.; Dallapiccola, B.; Zackai, E.; Hudgins, L.; McDonald-McGinn, D.; Bahi-Buisson, N.; Romano, C.; Williams, C.A.; Brailey, L.L.; et al. Further delineation of deletion 1p36 syndrome in 60 patients: A recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics 2008, 121, 404–410. [Google Scholar] [CrossRef]
- Kolc, K.L.; Sadleir, L.G.; Scheffer, I.E.; Ivancevic, A.; Roberts, R.; Pham, D.H.; Gecz, J. A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Mol. Psychiatry 2019, 24, 241–251. [Google Scholar] [CrossRef]
- Trivisano, M.; Striano, P.; Sartorelli, J.; Giordano, L.; Traverso, M.; Accorsi, P.; Cappelletti, S.; Claps, D.J.; Vigevano, F.; Zara, F.; et al. CHD2 mutations are a rare cause of generalized epilepsy with myoclonic-atonic seizures. Epilepsy Behav. 2015, 51, 53–56. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Mendoza-Londono, R.; Elsea, S.H. MBD5 Haploinsufficiency. 27 October 2016 [updated 28 April 2022]. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar]
- Emerson, E. Prevalence of psychiatric disorders in children and adolescents with and without intellectual disability. J. Intellect. Disabil. Res. 2003, 47 Pt 1, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Helmstaedter, C.; Aldenkamp, A.P.; Baker, G.A.; Mazarati, A.; Ryvlin, P.; Sankar, R. Disentangling the relationship between epilepsy and its behavioral comorbidities- the need for prospective studies in new-onset epilepsies. Epilepsy Behav. 2014, 31, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J. Mechanisms of Psychiatric Comorbidities in Epilepsy. In Psychiatric and Behavioral Aspects of Epilepsy. Current Perspectives and Mechanisms; Jones, N.C., Kanner, A.M., Eds.; Current Topics in Behavioral Neurosciences 55; Springer Nature: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Revdal, E.; Kolstad, B.P.; Winsvold, B.S.; Selmer, K.K.; Morken, G.; Brodtkorb, E. Psychiatric comorbidity in relation to clinical characteristics of epilepsy: A retrospective observational study. Seizure 2023, 110, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; McTague, A. Epilepsy and developmental disorders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 2020, 24, 15–23. [Google Scholar] [CrossRef]
- Guerrini, R.; Conti, V.; Mantegazza, M.; Balestrini, S.; Galanopoulou, A.S.; Benfenati, F. Developmental and epileptic encephalopathies: From genetic heterogeneity to phenotypic continuum. Physiol. Rev. 2023, 103, 433–513. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.K.; Helbig, I.; Metcalf, C.S.; Lubbers, L.S.; Isom, L.L.; Demarest, S.; Goldberg, E.M.; George, A.L., Jr.; Lerche, H.; Weckhuysen, S.; et al. Precision medicine for genetic epilepsy on the horizon: Recent advances, present challenges, and suggestions for continued progress. Epilepsia 2022, 63, 2461–2475. [Google Scholar] [CrossRef] [PubMed]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 2018, 70, 142–173. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef]
- Coppola, A.; Cellini, E.; Stamberger, H.; Saarentaus, E.; Cetica, V.; Lal, D.; Djémié, T.; Bartnik-Glaska, M.; Ceulemans, B.; Helen Cross, J.; et al. Diagnostic implications of genetic copy number variation in epilepsy plus. Epilepsia 2019, 60, 689–706. [Google Scholar] [CrossRef]
- Heilstedt, H.A.; Ballif, B.C.; Howard, L.A.; Kashork, C.D.; Shaffer, L.G. Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Clin. Genet. 2003, 64, 310–316. [Google Scholar] [CrossRef]
- Torres, F.; Barbosa, M.; Maciel, P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: Critical overview and analysis of clinical implications. J. Med. Genet. 2015, 53, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Burk, K.C.; Kaneko, M.; Quindipan, C.; Vu, M.H.; Cepin, M.F.; Santoro, J.D.; Van Hirtum-Das, M.; Holder, D.; Raca, G. Diagnostic Yield of Epilepsy-Genes Sequencing and Chromosomal Microarray in Pediatric Epilepsy. Pediatr. Neurol. 2023, 150, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Steenhof, M.; Kibæk, M.; Larsen, M.J.; Christensen, M.; Lund, A.M.; Brusgaard, K.; Hertz, J.M. Compound heterozygous mutations in two different domains of ALDH18A1 do not affect the amino acid levels in a patient with hereditary spastic paraplegia. Neurogenetics 2018, 19, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, X.; Tian, H.; Wang, L.; Guo, B.; Wang, Y.; Li, W.; Wang, F.; Sun, T. SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front. Neurol. 2021, 12, 743726. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60 (Suppl. S3), S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Burgess, R.; Afawi, Z.; Damiano, J.A.; Berkovic, S.F.; Hildebrand, M.S.; Scheffer, I.E. De novo SCN1A pathogenic variants in the GEFS+ spectrum: Not always a familial syndrome. Epilepsia 2017, 58, e26–e30. [Google Scholar] [CrossRef]
- Brunklaus, A.; Brünger, T.; Feng, T.; Fons, C.; Lehikoinen, A.; Panagiotakaki, E.; Vintan, M.A.; Symonds, J.; Andrew, J.; Arzimanoglou, A.; et al. The gain of function SCN1A disorder spectrum: Novel epilepsy phenotypes and therapeutic implications. Brain 2022, 145, 3816–3831. [Google Scholar] [CrossRef]
- Fang, Z.; Hu, C.; Zhou, S.; Yu, L. PIGW-related glycosylphosphatidylinositol deficiency: A case report and literature review. Neurol. Sci. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Dontaine, P.; Kottos, E.; Dassonville, M.; Balasel, O.; Catros, V.; Soblet, J.; Perlot, P.; Vilain, C. Digestive involvement in a severe form of Snyder-Robinson syndrome: Possible expansion of the phenotype. Eur. J. Med. Genet. 2021, 64, 104097. [Google Scholar] [CrossRef]
- Balestrini, S.; Mei, D.; Sisodiya, S.M.; Guerrini, R. Steps to Improve Precision Medicine in Epilepsy. Mol. Diagn. Ther. 2023, 27, 661–672. [Google Scholar] [CrossRef]
- Byrne, S.; Enright, N.; Delanty, N. Precision therapy in the genetic epilepsies of childhood. Dev. Med. Child. Neurol. 2021, 63, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.T.; Brooks-Kayal, A. From molecules to medicines: The dawn of targeted therapies for genetic epilepsies. Nat. Rev. Neurol. 2018, 14, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Helbig, I.; Ellis, C.A. Personalized medicine in genetic epilepsies—Possibilities, challenges, and new frontiers. Neuropharmacology 2020, 172, 107970. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.T.; Langfitt, J.T.; Testa, F.M.; Levy, S.R.; DiMario, F.; Westerveld, M.; Kulas, J. Global cognitive function in children with epilepsy: A community-based study. Epilepsia 2008, 49, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Lu, H.M.; Chen, J.; Li, S.; Elliott, A.M. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. J. Mol. Diagn. 2016, 18, 923–932. [Google Scholar] [CrossRef]
- Sands, T.T.; Choi, H. Genetic Testing in Pediatric Epilepsy. Curr. Neurol. Neurosci. Rep. 2017, 17, 45. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Z.J.; Liu, L.; Xu, H.Q.; Shi, Y.W.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef]
- Guerrini, R.; Balestrini, S.; Wirrell, E.C.; Walker, M.C. Monogenic Epilepsies: Disease Mechanisms, Clinical Phenotypes, and Targeted Therapies. Neurology 2021, 97, 817–831. [Google Scholar] [CrossRef]
- Griffiths, R.; Huntley, M. Griffiths Mental Development Scales-Revised: Birth to 2 Years (GMDS 0–2); APA PsycTests: Washington, DC, USA, 1996. [Google Scholar] [CrossRef]
- Wechsler, D. Wechsler Preschool and Primary Scale of Intelligence, 3rd ed.; APA PsycTests: Washington, DC, USA, 2002. [Google Scholar] [CrossRef]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; The Psychological Corporation: San Antonio, TX, USA, 2003. [Google Scholar]
- Wechsler, D. Weschler Intelligence Scale for Children, 3rd ed.; The Psychological Corporation: San Antonio, TX, USA, 1991. [Google Scholar]
- Rydz, D.; Shevell, M.I.; Majnemer, A.; Oskoui, M. Developmental screening. J. Child. Neurol. 2005, 20, 4–21. [Google Scholar] [CrossRef]
- American Psychiatric Association. Intellectual disabilities. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Yang, L.; You, C.; Qiu, S.; Yang, X.; Li, Y.; Liu, F.; Zhang, D.; Niu, Y.; Xu, L.; Xu, N.; et al. Novel and de novo point and large microdeletion mutation in PRRT2-related epilepsy. Brain Behav. 2020, 10, e01597. [Google Scholar] [CrossRef]
- Vlaskamp, D.R.M.; Callenbach, P.M.C.; Rump, P.; Giannini, L.A.A.; Brilstra, E.H.; Dijkhuizen, T.; Vos, Y.J.; van der Kevie-Kersemaekers, A.F.; Knijnenburg, J.; de Leeuw, N.; et al. PRRT2-related phenotypes in patients with a 16p11.2 deletion. Eur. J. Med. Genet. 2019, 62, 265–269. [Google Scholar] [CrossRef]
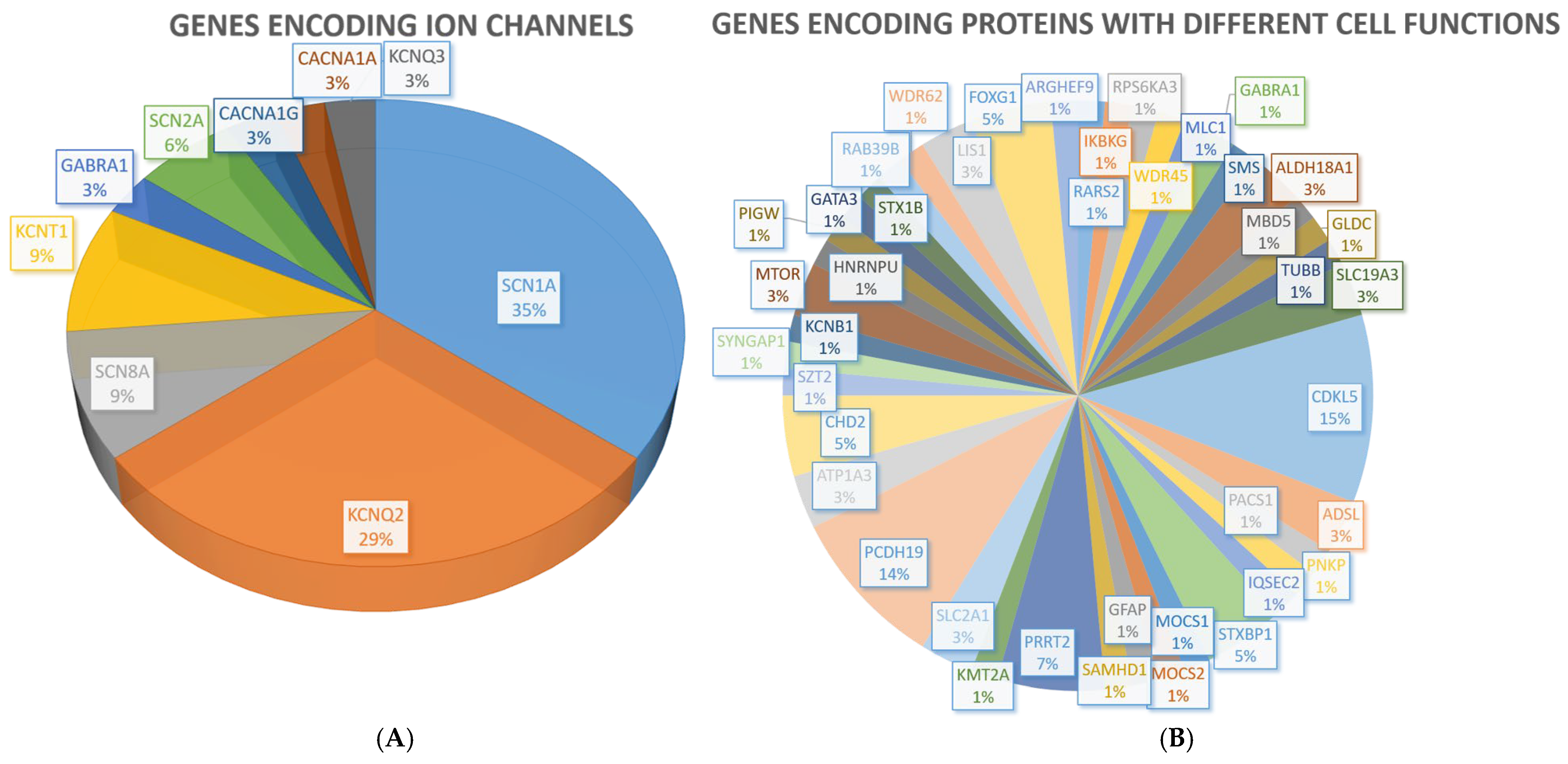

| Function | Gene Name |
|---|---|
| Ion channels | Sodium voltage-gated channel alpha subunit 1 (SCN1A) Sodium voltage-gated channel alpha subunit 8 (SCN8A) Sodium channel, voltage-gated, type II, alpha subunit (SCN2A) Potassium voltage-gated channel subfamily Q member 2 (KCNQ2) Potassium sodium-activated channel subfamily T member 1 (KCNT1) Calcium voltage-gated channel subunit alpha1 G (CACNA1G) |
| Enzymes | Cyclin-dependent kinase-like 5 (CDKL5) Chromodomain helicase DNA-binding protein 2 (CHD2) Lissencephaly 1 (LIS1) or platelet-activating factor acetylhydrolase 1b regulatory subunit 1 (PAFAH1B1) Aldehyde dehydrogenase 18 family member A1 (ALDH18A1) Adenylosuccinate lyase (ADSL) Mechanistic target of rapamycin (MTOR) Arginyl-tRNA synthetase 2, mitochondrial (RARS2) Inhibitor of nuclear factor kappa B kinase regulatory subunit gamma (IKBKG) Ribosomal protein S6 kinase A3 (RPS6KA3) Spermine synthase (SMS) Glycine decarboxylase (GLDC) Polynucleotide kinase 3′-phosphatase (PNKP) Molybdenum cofactor synthesis 1 (MOCS1) Molybdenum cofactor synthesis 2 (MOCS2) Phosphatidylinositol glycan anchor biosynthesis class W (PIGW) Cdc42 guanine nucleotide exchange factor 9 (ARHGEF9) |
| Receptors | Gamma-aminobutyric acid type A receptor subunit alpha 1 (GABRA1) Gamma-aminobutyric acid type A receptor subunit gamma2 (GABRG2) |
| Cell adhesion molecules | Protocadherin-19 (PCDH19) |
| Synaptic function | Proline-rich transmembrane protein 2 (PRRT2) IQ motif and Sec7 domain 2 (IQSEC2) Synaptic Ras GTPase-activating protein 1 (SYNGAP1) Syntaxin-1B (STX1B) |
| Trafficking | Syntaxin-binding protein 1 (STXBP1) Phosphofurin acidic cluster sorting protein 1 (PACS1) RAB39B member RAS oncogene family (RAB39B) |
| Transcription factors | Forkhead box G1 (FOXG1) GATA binding protein 3 (GATA3) |
| Transcriptional regulators | Methyl-CpG binding domain protein 5 (MBD5) |
| DNA binding | Lysine (K)-specific methyltransferase 2A (KMT2A) Heterogeneous nuclear ribonucleoprotein U (HNRNPU) |
| Transporters | Solute carrier family 2 (facilitated glucose transporter), member 1 (SLC2A1) Solute carrier family 19 member 3 (SLC19A3) |
| ATPase | ATPase Na+/K+-transporting subunit alpha 3 (ATP1A3) |
| Autophagy | WD repeat domain 45 (WDR45) |
| Cell proliferation/apoptosis | SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1 (SAMHD1) |
| Cell junction | Megalencephalic leukoencephalopathy with subcortical cysts 1 (MLC1) |
| Structural component of microtubules | Tubulin beta class I (TUBB) |
| Microtubule-associated proteins | WD repeat domain 62 (WDR62) |
| Cytoskeleton | Glial fibrillary acidic protein (GFAP) |
| Intracellular signaling | Seizure threshold 2 (SZT2) |
| Neurological Examination | Genes |
|---|---|
| Normal | PRRT2, KCNQ2, SCN1A, SCN1B, SCN8A, PCDH19, RAB39B, STX1B, SCN2A, CHD2, KCNB1, KCNQ3, IQSEC2, PACS1 |
| Macrocephaly | HNRNPU |
| Microcephaly | CDKL5, ATP1A3, PNKP, KCNT1 |
| Hypotonia | RPS6KA3, CDKL5, MTOR, PIGW, SYNGAP1, KCNQ2, SZT2, ATP1A3, SCN1A, SAMHD1, GLDC, MBD5 |
| Spastic tetraparesis | CDKL5, LIS1, WDR62, HNRNPU, MOCS1, RARS2, KMT2A, GFAP, STXBP1, SCN8A, PNKP, KCNT1, ADSL, KCNQ2 |
| Hemiparesis | MOCS2 |
| Spastic paraplegia | ALDH18A1 |
| Pyramidal signs | SLC2A1, STXBP1, KCNQ2, SCN2A, CACNA1G,MLC1 |
| Gait abnormalities (including ataxia) | RPS6KA3, ARGHEF9, GABRG2, KCNT1, SCN1A, MTOR, STXBP1, PIGW, CACNA1A, TUBB, SMS, GABRA1, MLC1 |
| Extrapyramidal signs | SCN1A |
| Movement disorders | WDR45 (hand stereotypies), FOXG1 (dystonia), GABRG2 (hand stereotypies), KCNT1 (hand stereotypies), SCN1A (tics), PIGW (hand stereotypies), KCNQ2 (hand stereotypies), SZT2 (hand stereotypies), SLC2A1 (dyskinesia), STXBP1 (dystonia), GLDC (hand stereotypies) |
| Disorders of the visual system | PCDH19 (ptosis), TUBB |
| Disorders of ocular motility | WDR45 (strabismus), GATA3 (strabismus), HNRNPU, KCNQ2 (strabismus and nystagmus) |
| Skin hyperpigmentation | IKBKG |
| Hearing loss | GATA3 |
| Congenital clubfoot | SCN1A |
| ASD | ADHD | Irritability and Psychomotor Agitation | Attachment Disorder | OCD | Psychotic Disorders | |
|---|---|---|---|---|---|---|
| WDR45 | + | |||||
| MBD5 | + | + | ||||
| KCNQ2 | + | |||||
| CACNA1G | + | |||||
| GLDC | + | |||||
| ADSL | + | |||||
| PACS1 | + | |||||
| KCNQ2 | + | |||||
| PNKP | + | |||||
| STXBP1 | + | |||||
| MOCS1 | + | |||||
| PCDH19 | + | + | + | + | ||
| CHD2 | + | |||||
| SYNGAP1 | + | |||||
| HNRNPU | + | |||||
| GATA3 | + | |||||
| STX1B | + | |||||
| RAB39B | + | |||||
| SCN1A | + | + | + | |||
| MTOR | + | |||||
| PRRT2 | + | |||||
| GABRG2 | + | |||||
| FOXG1 | + |
| Brain MRI Findings | Genes |
|---|---|
| Normal | GABRA1, CACNA1G, KCNT1, SCN2A, STXBP1, SCN1B, SLC2A1, PCDH19, PRRT2, KCNQ3, CHD2, SYNGAP1, KCNB1, ATP1A3, HNRNPU |
| Cerebral atrophy | WDR45, SLC19A3, ADSL, MOCS1, SCN1A, GABRA1, PIGW, KCNT1, CDKL5, FOXG1 |
| Cerebellar atrophy | WDR45, SLC19A3, ADSL, CACNA1A, SCN1A |
| Cerebellar hypoplasia | RARS2 (pons) |
| Corpus callosum dysgenesis | WDR45, SMS, KCNQ2, KMT2A |
| Corpus callosum hypoplasia | SZT2 |
| Malacic lesions | IKBKG |
| Periventricular white matter changes | RPS6KA3 (Coffin–Lowry syndrome) |
| Hypointense signal in the substantia nigra and globus pallidus | WDR45 |
| Large subcortical cysts | MLC1 |
| Ventricular dilatation (ventriculomegaly) | SMS |
| Cerebrospinal fluid space enlargements | ALDH18A1, CDKL5, STXBP1, PRRT2, CDKL5 |
| Optic nerves thinning | TUBB |
| Malformations of cortical development | TUBB, MTOR, WDR62 (schizencephaly), LIS1, PNKP |
| White matter abnormalities (including hypomyelination) | SLC19A3, KCNT1, SCN8A, GFAP, SAMHD1, KMT2A, PCDH19, SZT2, KCNQ2, CDKL5 |
| Large cisterna magna | KCNQ2 |
| Diffusion restriction in the posterior limb of the internal capsule | GLDC |
| Basal Ganglia involvement | MOCS, GFAP, FOXG1 (lacunar infarct) |
| Brain Calcification | SAMHD1 |
| Mesial temporal sclerosis | ATP1A3 |
| Hydromyelia | ARGHEF9 |
| Neurological Examination | Chromosomal Abnormalities |
|---|---|
| Normal | 46,XX,dup(14)(q11.2q12), 46,XX,dup(16)(p13.11p12.3), 46,XY,del(16)(p11.2) |
| Microcephaly | Angelman syndrome, 1p36 terminal deletion syndrome, 46,XX,del(9qter), 46,XX,del(8)(p23.3p23.2) and 46,Xxdup(13)(q32.1q34), 46,XY,del(6)(q26-qter) |
| Hypotonia | Trisomy 21, Angelman syndrome, InvDup (15) syndrome, 46,XX,del(9qter), 1p36 terminal deletion syndrome, 46,XX,del(1q44), Xq28 duplication syndrome, 46,XY,del(6)(q26-qter) |
| Spastic tetraparesis | 46,XX,del(4p16.3), Angelman syndrome, Xq28 duplication syndrome, 1p36 terminal deletion syndrome, 46,XY,del(17p13.3), 46,XX,del(9)(q33.3q34.11), trisomy 13 |
| Pyramidal signs | InvDup(15) syndrome, 46,XX,del(1q44) |
| Movement disorders | Angelman syndrome (tremulousness of the limbs), InvDup (15) syndrome (hand stereotypies), 46,XX,del(5)(q11.2q13.2) and 46,XX,dup(5q13.2) (hand stereotypies), 1p36 terminal deletion syndrome (hand stereotypies) |
| Gait abnormalities (including ataxia) | Angelman syndrome, 1p36 terminal deletion syndrome, 46,XX,del(9qter), 46,XX,del(6)(q21q22.31), 46,XY,del(6)(q26-qter) |
| Dyspraxia | del 17q21.31 |
| Disorders of ocular motility | 46,XX,del(5)(q11.2q13.2) and 46,XX,dup(5q13.2) (strabismus and nystagmus), 1p36 terminal deletion syndrome, 46,XX,del(1q44), 46,XY,del(6)(q26-qter) (nystagmus) |
| Hearing loss | 46,XX,del(8)(p23.3p23.2) and 46,Xxdup(13)(q32.1q34) |
| Scoliosis | Angelman syndrome |
| Brain MRI Findings | Chromosomal Abnormalities |
|---|---|
| Normal | 46,XY,del(2)(q24.2q24.3), 46,XY,del(16p11.2), 46,XX,dup(15q11.2) 46,XX,del(16p11.2), 46,XX,dup(14)(q11.2q12, 46,XX,del(4p16.3), 46,XX,del(8)(p23.3p23.2) and 46,Xxdup(13)(q32.1q34), 46,XX,del(6)(q21q22.31), Angelman syndrome |
| Cerebellar atrophy | Wolf–Hirschhorn syndrome |
| Cerebellar hypoplasia | Trisomy 21 (vermis), Wolf–Hirschhorn syndrome, Xq28 duplication syndrome |
| Corpus callosum dysgenesis | Angelman syndrome (dysmorphic), trisomy 13, 46,XX,del(1q44) |
| Cerebrospinal fluid space enlargements | Trisomy 21 |
| Enlarged subarachnoid spaces | Xq28 duplication syndrome, InvDup(15) syndrome |
| Malformations of cortical development | 46,XY,del(6)(q26-qter), 46,XY,del(17p13.3) |
| White matter abnormalities (including hypomyelination) | Trisomy 21, 1p36 terminal deletion syndrome, Angelman syndrome, 46,XX,del(9qter) |
| Basal ganglia involvement | 46,XX,del(9qter) |
| Vascular lesion due to Takayasu arteritis | Wolf–Hirschhorn syndrome |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavirani, B.; Spagnoli, C.; Caraffi, S.G.; Cavalli, A.; Cesaroni, C.A.; Cutillo, G.; De Giorgis, V.; Frattini, D.; Marchetti, G.B.; Masnada, S.; et al. Genetic Epilepsies and Developmental Epileptic Encephalopathies with Early Onset: A Multicenter Study. Int. J. Mol. Sci. 2024, 25, 1248. https://doi.org/10.3390/ijms25021248
Cavirani B, Spagnoli C, Caraffi SG, Cavalli A, Cesaroni CA, Cutillo G, De Giorgis V, Frattini D, Marchetti GB, Masnada S, et al. Genetic Epilepsies and Developmental Epileptic Encephalopathies with Early Onset: A Multicenter Study. International Journal of Molecular Sciences. 2024; 25(2):1248. https://doi.org/10.3390/ijms25021248
Chicago/Turabian StyleCavirani, Benedetta, Carlotta Spagnoli, Stefano Giuseppe Caraffi, Anna Cavalli, Carlo Alberto Cesaroni, Gianni Cutillo, Valentina De Giorgis, Daniele Frattini, Giulia Bruna Marchetti, Silvia Masnada, and et al. 2024. "Genetic Epilepsies and Developmental Epileptic Encephalopathies with Early Onset: A Multicenter Study" International Journal of Molecular Sciences 25, no. 2: 1248. https://doi.org/10.3390/ijms25021248
APA StyleCavirani, B., Spagnoli, C., Caraffi, S. G., Cavalli, A., Cesaroni, C. A., Cutillo, G., De Giorgis, V., Frattini, D., Marchetti, G. B., Masnada, S., Peron, A., Rizzi, S., Varesio, C., Spaccini, L., Vignoli, A., Canevini, M. P., Veggiotti, P., Garavelli, L., & Fusco, C. (2024). Genetic Epilepsies and Developmental Epileptic Encephalopathies with Early Onset: A Multicenter Study. International Journal of Molecular Sciences, 25(2), 1248. https://doi.org/10.3390/ijms25021248