
X-Linked Epilepsies: A Narrative Review

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
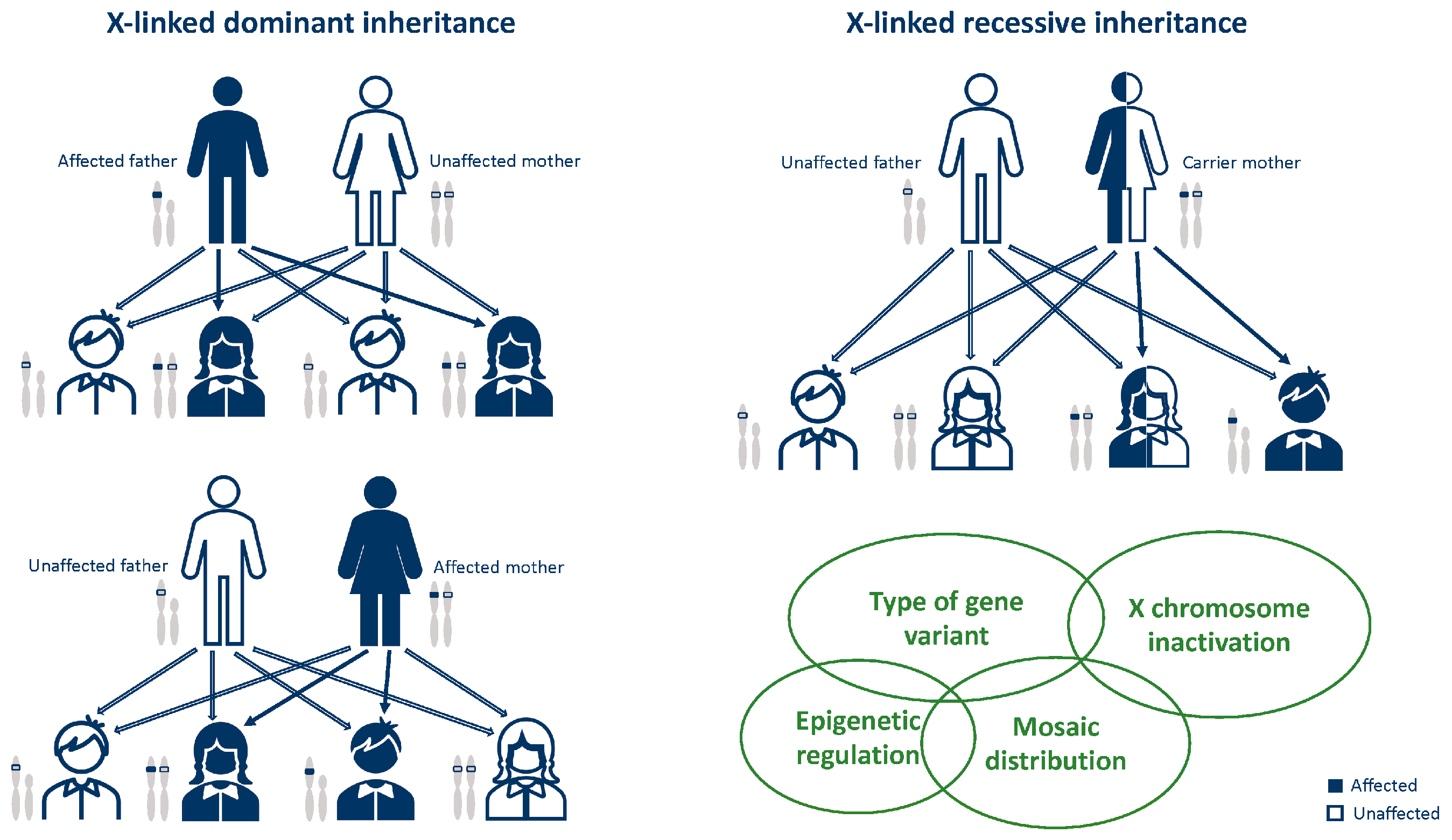
3. General Notes on X-Linked Disease Inheritance
4. Known X-Linked Developmental and Epileptic Encephalopathies
4.1. PCDH19
4.1.1. Genetic Features and Inheritance
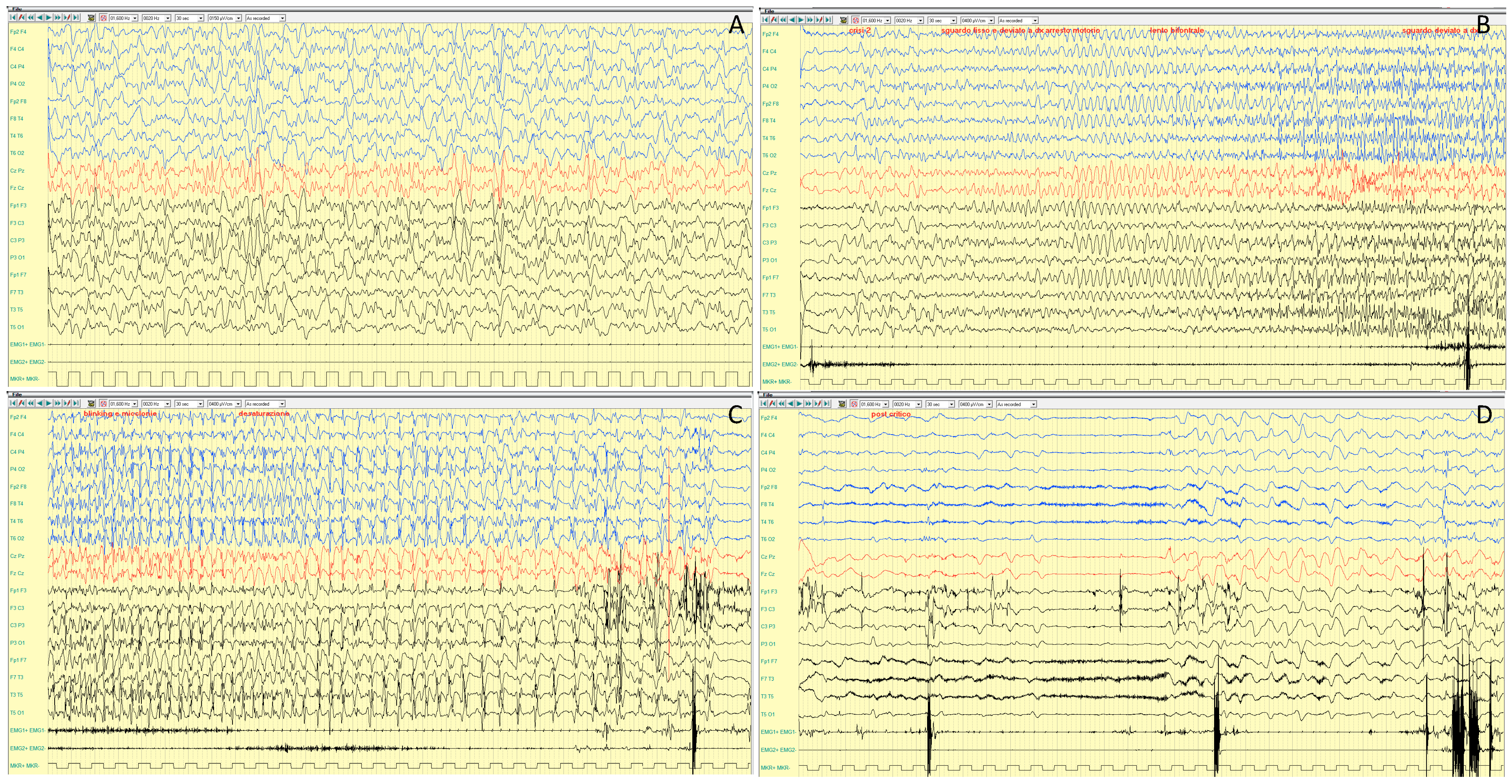
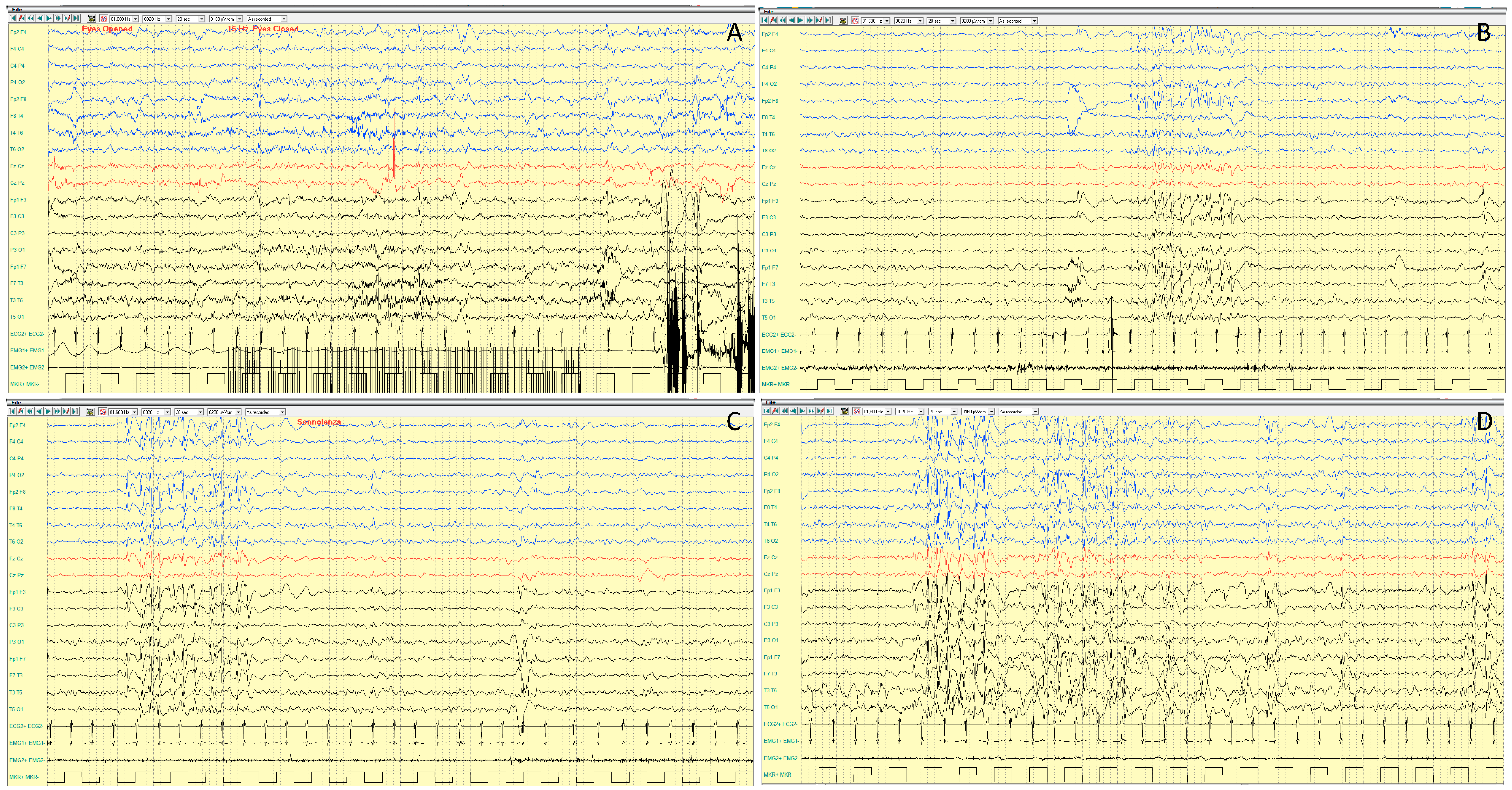
4.1.2. Epileptic Phenotypes
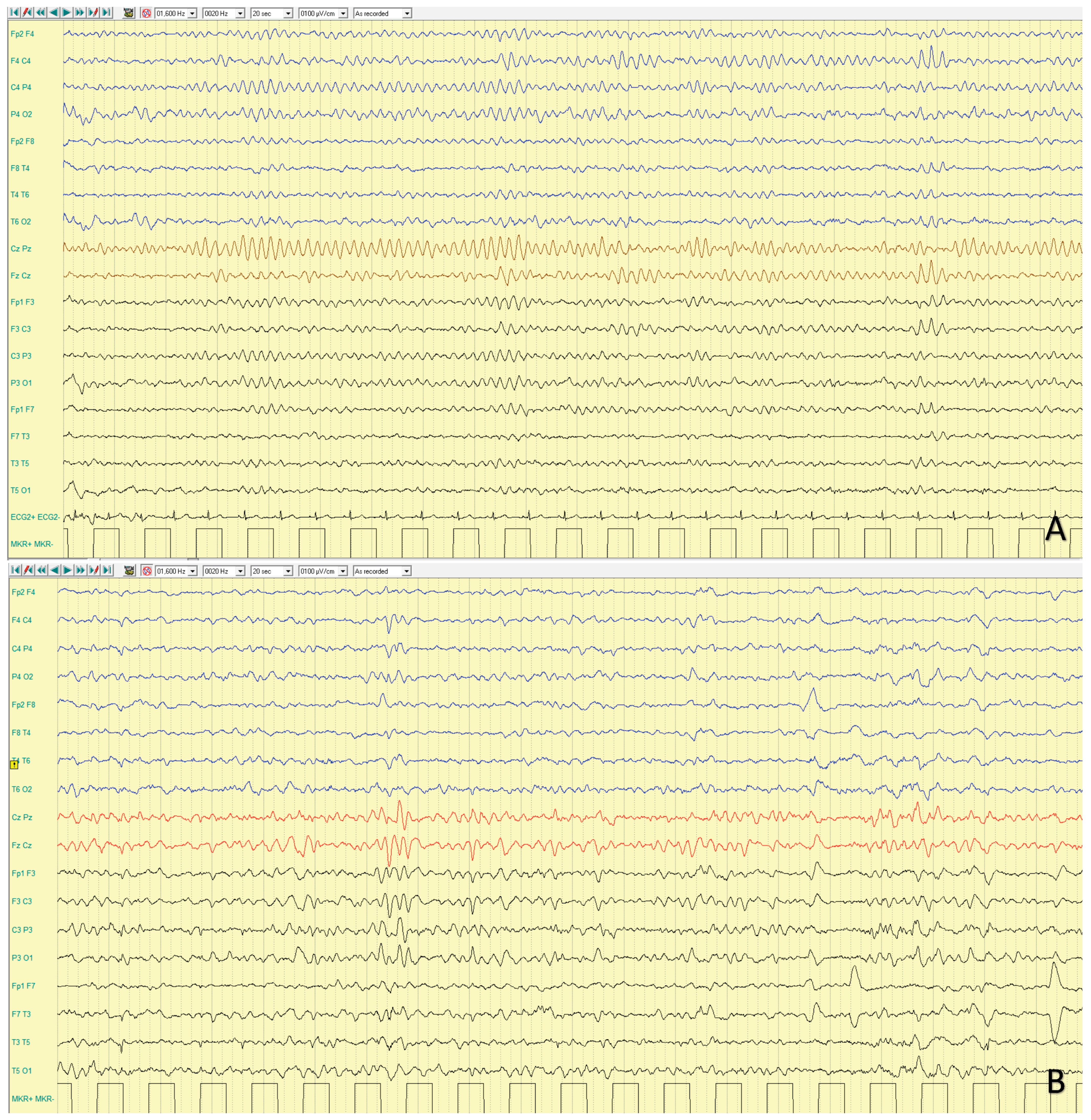
4.1.3. EEG Features
4.1.4. Neurocognitive Profile and Other Disorders
4.1.5. MRI Features
4.1.6. Treatment
4.1.7. Clinical Outcome
4.2. CDKL5
4.2.1. Genetic Features and Inheritance
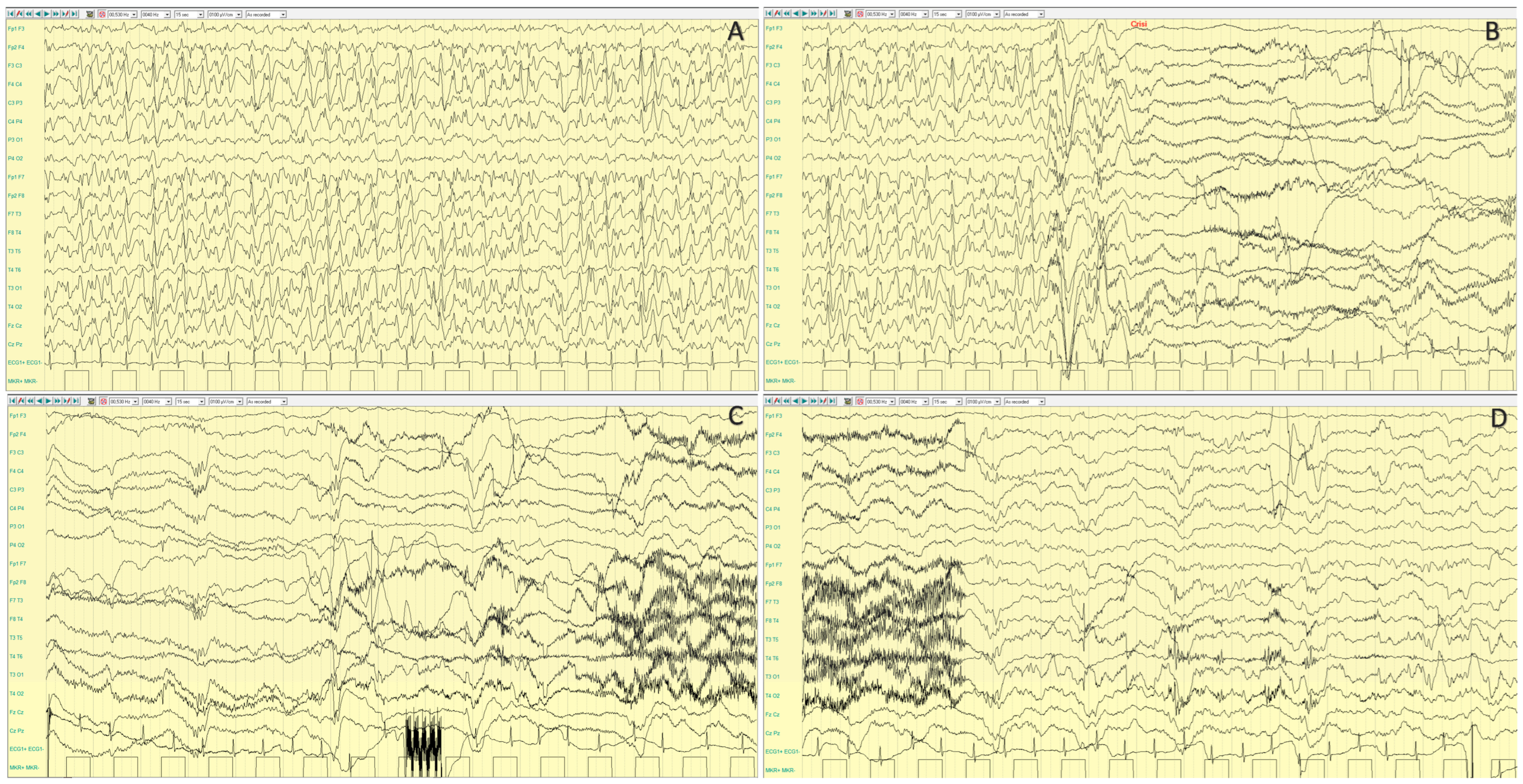
4.2.2. Epileptic Phenotypes
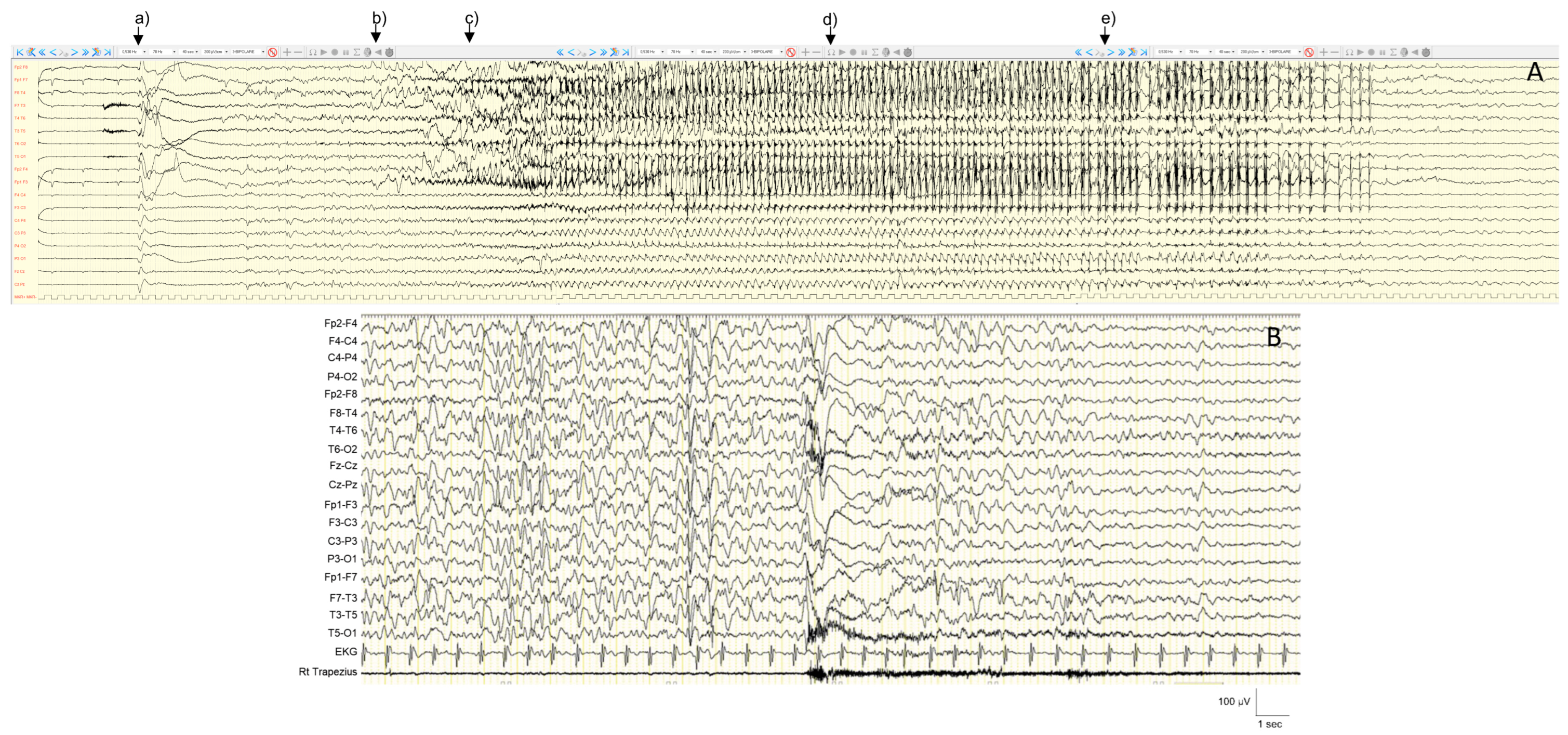
4.2.3. Neurocognitive Profile and Other Disorders
4.2.4. Treatment
4.2.5. Clinical Outcome
4.3. MECP2
4.3.1. Rett Syndrome (RTT)
4.3.2. Other MECP2-Related Disorders
- Severe neonatal encephalopathy and infantile death. MECP2 variant is usually passed on by a mildly symptomatic or asymptomatic mother. Clinically the scenarios can be spontaneous miscarriages or, if born, neonatal encephalopathy, respiratory arrest and seizures; death occurs within 2 years.
- Less severe neuro-psychiatric symptoms. In these cases, the MECP2 variants are less severe than those in female RTT patients, and symptoms are heterogeneous and overlap with features of Angelman syndrome (intellectual disability and motor abnormalities).
- MECP2 duplication syndrome, with gain in MECP2 dosage. The clinical phenotype is characterized by hypotonia, severe ID, recurrent lung infections, absent or limited speech and walking, seizures, motor spasticity and muscle stiffness, and 50% of affected individuals usually die before the age of 25 years [110,111].
4.3.3. The MECP2 Duplication Syndrome (MDS)
5. X-Linked Neuronal Migration Disorders
5.1. ARX
5.2. DCX
5.3. FLNA
5.4. X-Linked Polymicrogyria (PMG) Genes
6. Other Emerging X-Linked DEE Genes
6.1. SLC9A6
6.2. SLC35A2
6.3. SYN1
6.4. ARHGEF9
6.5. ATP6AP2
6.6. IQSEC2
6.7. NEXMIF
6.8. PIGA
6.9. ALG13
6.10. FHF2/FGF13
6.11. GRIA3
6.12. SMC1A
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deng, H.; Zheng, W.; Song, Z. Genetics, molecular biology, and phenotypes of X-linked epilepsy. Mol. Neurobiol. 2014, 49, 1166–1180. [Google Scholar] [CrossRef]
- Stevenson, R.E.; Holden, K.R.; Rogers, R.C.; Schwartz, C.E. Seizures and X-linked intellectual disability. Eur. J. Med. Genet. 2012, 55, 307–312. [Google Scholar] [CrossRef] [PubMed]
- den Veyver, I.B.V. Skewed X inactivation in X-linked disorders. Semin. Reprod. Med. 2001, 19, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. General aspects of X-linked diseases. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 7. [Google Scholar]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Darra, F.; Specchio, N.; Mei, D.; Terracciano, A.; Parmeggiani, L.; Ferrari, A.; Sicca, F.; Mastrangelo, M.; Spaccini, L.; et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012, 53, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Trivisano, M.; Pietrafusa, N.; Terracciano, A.; Marini, C.; Mei, D.; Darra, F.; Accorsi, P.; Battaglia, D.; Caffi, L.; Canevini, M.P.; et al. Defining the electroclinical phenotype and outcome of PCDH19-related epilepsy: A multicenter study. Epilepsia 2018, 59, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D. PCDH19-Related Epilepsy Syndrome: A Comprehensive Clinical Review. Pediatr. Neurol. 2020, 105, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, S.M.; Wirrell, E.; Yozawitz, E.; Wilmshurst, J.M.; Specchio, N.; Riney, K.; Pressler, R.; Auvin, S.; Samia, P.; Hirsch, E.; et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1349–1397. [Google Scholar] [CrossRef] [PubMed]
- Melani, F.; Mei, D.; Pisano, T.; Savasta, S.; Franzoni, E.; Ferrari, A.R.; Marini, C.; Guerrini, R. CDKL5 gene-related epileptic encephalopathy: Electroclinical findings in the first year of life. Dev. Med. Child. Neurol. 2011, 53, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.M.P.; Amin, S.; Bahi-Buisson, N.; Benke, T.A.; Cross, J.H.; Demarest, S.T.; Olson, H.E.; Specchio, N.; Fleming, T.R.; Aimetti, A.A.; et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: Results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2022, 21, 417–427. [Google Scholar] [CrossRef]
- Guerrini, R.; Parrini, E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia 2012, 53, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Glaze, D.G.; Percy, A.K.; Skinner, S.; Motil, K.J.; Neul, J.L.; Barrish, J.O.; Lane, J.B.; Geerts, S.P.; Annese, F.; Graham, J.; et al. Epilepsy and the natural history of Rett syndrome. Neurology 2010, 74, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 duplication syndrome. Am. J. Med. Genet. Part A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, A.; Borgatti, R.; Peron, A.; Zucca, C.; Ballarati, L.; Bonaglia, C.; Bellini, M.; Giordano, L.; Romaniello, R.; Bedeschi, M.F.; et al. Electroclinical pattern in MECP2 duplication syndrome: Eight new reported cases and review of literature. Epilepsia 2012, 53, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Yanazawa, M.; Sugiyama, N.; Miura, H.; Iizuka-Kogo, A.; Kusaka, M.; Omichi, K.; Suzuki, R.; Kato-Fukui, Y.; Kamiirisa, K.; et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 2002, 32, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Das, S.; Petras, K.; Kitamura, K.; Morohashi, K.I.; Abuelo, D.N.; Barr, M.; Bonneau, D.; Brady, A.F.; Carpenter, N.J.; et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 2004, 23, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.D.; Golden, J.A. Developing Models of Aristaless-related homeobox mutations. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Edey, J.; Soleimani-Nouri, P.; Dawson-Kavanagh, A.; Imran Azeem, M.S.; Episkopou, V. X-linked neuronal migration disorders: Gender differences and insights for genetic screening. Int. J. Dev. Neurosci. 2023, 83, 581–599. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Souville, I.; Fourniol, F.J.; Toussaint, A.; Moores, C.A.; Houdusse, A.; Lemaitre, J.Y.; Poirier, K.; Khalaf-Nazzal, R.; Hully, M.; et al. SBH-LIS European Consortium. New insights into genotype-phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 2013, 136 Pt 1, 223–244. [Google Scholar] [CrossRef]
- Chou, A.; Boerkoel, C.; du Souich, C.; Rupps, R. Phenotypic and molecular characterization of a novel DCX deletion and a review of the literature. Clin. Genet. 2009, 76, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Parrini, E.; Ramazzotti, A.; Dobyns, W.B.; Mei, D.; Moro, F.; Veggiotti, P.; Marini, C.; Brilstra, E.H.; Dalla Bernardina, B.; Goodwin, L.; et al. Periventricular heterotopia: Phenotypic heterogeneity and correlation with Filamin A mutations. Brain 2006, 129 Pt 7, 1892–1906. [Google Scholar] [CrossRef]
- Cannaerts, E.; Shukla, A.; Hasanhodzic, M.; Alaerts, M.; Schepers, D.; Van Laer, L.; Girisha, K.M.; Hojsak, I.; Loeys, B.; Verstraeten, A. FLNA mutations in surviving males presenting with connective tissue findings: Two new case reports and review of the literature. BMC Med. Genet. 2018, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Schroer, R.J.; Holden, K.R.; Tarpey, P.S.; Matheus, M.G.; Griesemer, D.A.; Friez, M.J.; Fan, J.Z.; Simensen, R.J.; Strømme, P.; Stevenson, R.E.; et al. Natural history of Christianson syndrome. Am. J. Med. Genet. Part A 2010, 152A, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Sinajon, P.; Verbaan, D.; So, J. The expanding phenotypic spectrum of female SLC9A6 mutation carriers: A case series and review of the literature. Hum. Genet. 2016, 135, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Nakamura, K.; Osaka, H.; Maegaki, Y.; Haginoya, K.; Mizumoto, S.; Kato, M.; Okamoto, N.; Iai, M.; Kondo, Y.; et al. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum. Mutat. 2013, 34, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Winawer, M.R.; Griffin, N.G.; Samanamud, J.; Baugh, E.H.; Rathakrishnan, D.; Ramalingam, S.; Zagzag, D.; Schevon, C.A.; Dugan, P.; Hegde, M.; et al. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann. Neurol. 2018, 83, 1133–1146. [Google Scholar] [CrossRef]
- Ng, B.G.; Sosicka, P.; Agadi, S.; Almannai, M.; Bacino, C.A.; Barone, R.; Botto, L.D.; Burton, J.E.; Carlston, C.; Chung, B.H.; et al. SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Hum. Mutat. 2019, 40, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.C.; Blair, H.J.; Seager, M.; Coulthard, A.; Tennant, S.; Buddles, M.; Curtis, A.; Goodship, J.A. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J. Med. Genet. 2004, 41, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Giovedì, S.; Darchen, F.; Valtorta, F.; Greengard, P.; Benfenati, F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles. II. Functional effects of the Rab3A-synapsin I interaction. J. Biol. Chem. 2004, 279, 43769–43779. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.K.; Rouleau, I.; Sénéchal, G.; Ansaldo, A.I.; Gravel, M.; Benfenati, F.; Cossette, P. X-linked focal epilepsy with reflex bathing seizures: Characterization of a distinct epileptic syndrome. Epilepsia 2015, 56, 1098–1108. [Google Scholar] [CrossRef]
- Shimojima, K.; Sugawara, M.; Shichiji, M.; Mukaida, S.; Takayama, R.; Imai, K.; Yamamoto, T. Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J. Hum. Genet. 2011, 56, 561–565. [Google Scholar] [CrossRef]
- Alber, M.; Kalscheuer, V.M.; Marco, E.; Sherr, E.; Lesca, G.; Till, M.; Gradek, G.; Wiesener, A.; Korenke, C.; Mercier, S.; et al. ARHGEF9 disease: Phenotype clarification and genotype-phenotype correlation. Neurol. Genet. 2017, 3, e148. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liao, H.; Gan, S.; Xiao, T.; Wu, L. ARHGEF9 gene variant leads to developmental and epileptic encephalopathy: Genotypic phenotype analysis and treatment exploration. Mol. Genet. Genom. Med. 2022, 10, e1967. [Google Scholar] [CrossRef]
- Hedera, P.; Alvarado, D.; Beydoun, A.; Fink, J.K. Novel mental retardation-epilepsy syndrome linked to Xp21.1-p11.4. Ann. Neurol. 2002, 51, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ramser, J.; Abidi, F.E.; Burckle, C.A.; Lenski, C.; Toriello, H.; Wen, G.; Lubs, H.A.; Engert, S.; Stevenson, R.E.; Meindl, A.; et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Mol Genet. 2005, 14, 1019–1027. [Google Scholar] [CrossRef]
- Gupta, H.V.; Vengoechea, J.; Sahaya, K.; Virmani, T. A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and parkinsonism. Park. Relat. Disord. 2015, 21, 1473–1475. [Google Scholar] [CrossRef]
- Zerem, A.; Haginoya, K.; Lev, D.; Blumkin, L.; Kivity, S.; Linder, I.; Shoubridge, C.; Palmer, E.E.; Field, M.; Boyle, J.; et al. The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia 2016, 57, 1858–1869. [Google Scholar] [CrossRef]
- Choi, M.H.; Yang, J.O.; Min, J.S.; Lee, J.J.; Jun, S.Y.; Lee, Y.J.; Yoon, J.Y.; Jeon, S.J.; Byeon, I.; Kang, J.W.; et al. A Novel X-Linked Variant of IQSEC2 is Associated with Lennox-Gastaut Syndrome and Mild Intellectual Disability in Three Generations of a Korean Family. Genet. Test Mol. Biomark. 2020, 24, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Hammer, T.B.; Gardella, E.; Vlaskamp, D.R.M.; Bertelsen, B.; Mandelstam, S.; de Lange, I.; Zhang, J.; Myers, C.T.; Fenger, C.; et al. NEXMIF encephalopathy: An X-linked disorder with male and female phenotypic patterns. Genet. Med. 2021, 23, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Coppola, A.; Krithika, S.; Iacomino, M.; Bobbili, D.; Balestrini, S.; Bagnasco, I.; Bilo, L.; Buti, D.; Casellato, S.; Cuccurullo, C.; et al. Dissecting genetics of spectrum of epilepsies with eyelid myoclonia by exome sequencing. Epilepsia 2023. ahead of print. [Google Scholar] [CrossRef]
- Kim, Y.O.; Yang, J.H.; Park, C.; Kim, S.K.; Kim, M.K.; Shin, M.G.; Woo, Y.J. A novel PIGA mutation in a family with X-linked, early-onset epileptic encephalopathy. Brain Dev. 2016, 38, 750–754. [Google Scholar] [CrossRef]
- Bayat, A.; Knaus, A.; Pendziwiat, M.; Afenjar, A.; Barakat, T.S.; Bosch, F.; Callewaert, B.; Calvas, P.; Ceulemans, B.; Chassaing, N.; et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 2020, 61, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Nagafuchi, H.; Enomoto, Y.; Kurosawa, K.; Tsuyusaki, Y.; Tsuji, M.; Goto, T. The efficacy of a medium-chain triglyceride ketogenic diet for drug-resistant epilepsy with PIGA germline variant. Seizure 2023, 111, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.N.; Bahi-Buisson, N.; Bienvenu, T.; Buerki, S.E.; Gardiner, F.; Cross, J.H.; Heron, B.; Kaminska, A.; Korff, C.M.; Lepine, A.; et al. The phenotypic spectrum of X-linked, infantile onset ALG13-related developmental and epileptic encephalopathy. Epilepsia 2021, 62, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Freeze, H.H.; Morava, E. Is X-linked, infantile onset ALG13-related developmental and epileptic encephalopathy a congenital disorder of glycosylation? Epilepsia 2021, 62, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.E.; Marra, C.; Derrick, A.V.; Pickrell, W.O.; Higgins, A.T.; Naude, J.T.W.; McClatchey, M.A.; Davies, S.J.; Metcalfe, K.A.; Tan, H.J.; et al. Missense variants in the N-terminal domain of the A isoform of FHF2/FGF13 cause an X-linked developmental and epileptic encephalopathy. Am. J. Hum. Genet. 2021, 108, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.L.; Majethia, P.; Shrikiran, A.; Siddiqui, S.; Dalal, A.; Shukla, A. Further evidence of affected females with a heterozygous variant in FGF13 causing X-linked developmental and epileptic encephalopathy 90. Eur. J. Med. Genet. 2022, 65, 104403. [Google Scholar] [CrossRef] [PubMed]
- Trivisano, M.; Santarone, M.E.; Micalizzi, A.; Ferretti, A.; Dentici, M.L.; Novelli, A.; Vigevano, F.; Specchio, N. GRIA3 missense mutation is cause of an X-linked developmental and epileptic encephalopathy. Seizure 2020, 82, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Chen, J.; Valenzuela, F.E.A.; Brown, C.; Masser-Frye, D.; Jones, M.; Romero, L.P.; Rinaldi, B.; Li, W.L.; Li, Q.Q.; et al. X-linked neonatal-onset epileptic encephalopathy associated with a gain-of-function variant p.R660T in GRIA3. PLoS Genet. 2021, 17, e1009608. [Google Scholar] [CrossRef] [PubMed]
- Selicorni, A.; Mariani, M.; Lettieri, A.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes 2021, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Joss, S.; Metcalfe, K.A.; Somarathi, S.; Cruden, J.; Devlin, A.M.; Donaldson, A.; DiDonato, N.; Fitzpatrick, D.; Kaiser, F.J.; et al. Heterozygous truncation mutations of the SMC1A gene cause a severe early onset epilepsy with cluster seizures in females: Detailed phenotyping of 10 new cases. Epilepsia 2017, 58, 565–575. [Google Scholar] [CrossRef]
- Bozarth, X.L.; Lopez, J.; Fang, H.; Lee-Eng, J.; Duan, Z.; Deng, X. Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes 2023, 14, 852. [Google Scholar] [CrossRef] [PubMed]
- Dibbens, L.M.; Tarpey, P.S.; Hynes, K.; Bayly, M.A.; Scheffer, I.E.; Smith, R.; Bomar, J.; Sutton, E.; Vandeleur, L.; Shoubridge, C.; et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 2008, 40, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Thiffault, I.; Farrow, E.; Smith, L.; Lowry, J.; Zellmer, L.; Black, B.; Abdelmoity, A.; Miller, N.; Soden, S.; Saunders, C. PCDH19-related epileptic encephalopathy in a male mosaic for a truncating variant. Am. J. Med. Genet. Part A 2016, 170, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, A.; Trivisano, M.; Cusmai, R.; De Palma, L.; Fusco, L.; Compagnucci, C.; Bertini, E.; Vigevano, F.; Specchio, N. PCDH19-related epilepsy in two mosaic male patients. Epilepsia 2016, 57, e51–e55. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, N.; Losito, E.; Pinard, J.M.; Gautier, A.; Villeneuve, N.; Arbues, A.S.; An, I.; Desguerre, I.; Dulac, O.; Chiron, C.; et al. Early and long-term electroclinical features of patients with epilepsy and PCDH19 mutation. Epileptic Disord. 2018, 20, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Kolc, K.L.; Sadleir, L.G.; Scheffer, I.E.; Ivancevic, A.; Roberts, R.; Pham, D.H.; Gecz, J. A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Mol. Psychiatry 2019, 24, 241–251. [Google Scholar] [CrossRef] [PubMed]
- van Harssel, J.J.; Weckhuysen, S.; van Kempen, M.J.; Hardies, K.; Verbeek, N.E.; de Kovel, C.G.; Gunning, W.B.; van Daalen, E.; de Jonge, M.V.; Jansen, A.C.; et al. Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders. Neurogenetics 2013, 14, 23–34. [Google Scholar] [CrossRef]
- Kurian, M.; Korff, C.M.; Ranza, E.; Bernasconi, A.; Lübbig, A.; Nangia, S.; Ramelli, G.P.; Wohlrab, G.; Nordli, D.R., Jr.; Bast, T. Focal cortical malformations in children with early infantile epilepsy and PCDH19 mutations: Case report. Dev. Med. Child Neurol. 2018, 60, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Lotte, J.; Bast, T.; Borusiak, P.; Coppola, A.; Cross, J.H.; Dimova, P.; Fogarasi, A.; Graneß, I.; Guerrini, R.; Hjalgrim, H.; et al. Effectiveness of antiepileptic therapy in patients with PCDH19 mutations. Seizure 2016, 35, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Higurashi, N.; Takahashi, Y.; Kashimada, A.; Sugawara, Y.; Sakuma, H.; Tomonoh, Y.; Inoue, T.; Hoshina, M.; Satomi, R.; Ohfu, M.; et al. Immediate suppression of seizure clusters by corticosteroids in PCDH19 female epilepsy. Seizure 2015, 27, 1–5. [Google Scholar] [CrossRef]
- Sadleir, L.G.; Kolc, K.L.; King, C.; Mefford, H.C.; Dale, R.C.; Gecz, J.; Scheffer, I.E. Levetiracetam efficacy in PCDH19 Girls Clustering Epilepsy. Eur. J. Paediatr. Neurol. 2020, 24, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Nissenkorn, A.; Kluger, G.; Schubert-Bast, S.; Bayat, A.; Bobylova, M.; Bonanni, P.; Ceulemans, B.; Coppola, A.; Di Bonaventura, C.; Feucht, M.; et al. Perampanel as precision therapy in rare genetic epilepsies. Epilepsia 2023, 64, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.; Vaccarezza, M.; Cersósimo, R.; Rios, V.; Soraru, A.; Arroyo, H.; Agosta, G.; Escobal, N.; Demartini, M.; Maxit, C.; et al. Long-term follow-up of the ketogenic diet for refractory epilepsy: Multicenter Argentinean experience in 216 pediatric patients. Seizure 2011, 20, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Appavu, B.; Vanatta, L.; Condie, J.; Kerrigan, J.F.; Jarrar, R. Ketogenic diet treatment for pediatric super-refractory status epilepticus. Seizure 2016, 41, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Moncayo, J.A.; Vargas, M.N.; Castillo, I.; Granda, P.V.; Duque, A.M.; Argudo, J.M.; Matcheswalla, S.; Lopez Dominguez, G.E.; Monteros, G.; Andrade, A.F.; et al. Adjuvant Treatment for Protocadherin 19 (PCDH19) Syndrome. Cureus 2022, 14, e27154. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Tao, J.; Donnelly, A.; Hollway, G.; Schwinger, E.; Kübart, S.; Menzel, C.; Hoeltzenbein, M.; Tommerup, N.; Eyre, H.; et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003, 72, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.P.; Schwinger, E.; Gécz, J.; et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 42, 103–107. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131 Pt 10, 2647–2661. [Google Scholar] [CrossRef]
- White, R.; Ho, G.; Schmidt, S.; Scheffer, I.E.; Fischer, A.; Yendle, S.C.; Bienvenu, T.; Nectoux, J.; Ellaway, C.J.; Darmanian, A.; et al. Cyclin-dependent kinase-like 5 (CDKL5) mutation screening in Rett syndrome and related disorders. Twin Res. Hum. Genet. 2010, 13, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Jansen, A.; Bauters, M.; Froyen, G.; Fryns, J.P. Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. Am. J. Med. Genet. Part A 2007, 143, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.; Di Rosa, G.; Polli, R.; Bettella, E.; Tricomi, G.; Tortorella, G.; Murgia, A. A novel CDKL5 mutation in a 47,XXY boy with the early-onset seizure variant of Rett syndrome. Am. J. Med. Genet. Part A. 2009, 149A, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Archer, H.L.; Evans, J.; Edwards, S.; Colley, J.; Newbury-Ecob, R.; O’Callaghan, F.; Huyton, M.; O’Regan, M.; Tolmie, J.; Sampson, J.; et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J. Med. Genet. 2006, 43, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Buoni, S.; Zannolli, R.; Colamaria, V.; Macucci, F.; di Bartolo, R.M.; Corbini, L.; Orsi, A.; Zappella, M.; Hayek, J. Myoclonic encephalopathy in the CDKL5 gene mutation. Clin. Neurophysiol. 2006, 117, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Verducci, C.; Thiele, E.A.; Laux, L.C.; Patel, A.D.; Filloux, F.; Szaflarski, J.P.; Wilfong, A.; Clark, G.D.; Park, Y.D.; et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. 2018, 86, 131–137. [Google Scholar] [CrossRef]
- Dale, T.; Downs, J.; Wong, K.; Leonard, H. The perceived effects of cannabis products in the management of seizures in CDKL5 Deficiency Disorder. Epilepsy Behav. 2021, 122, 108152. [Google Scholar] [CrossRef]
- Devinsky, O.; King, L.; Schwartz, D.; Conway, E.; Price, D. Effect of fenfluramine on convulsive seizures in CDKL5 deficiency disorder. Epilepsia 2021, 62, e98–e102. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.; Wong, K.; Olson, H.E.; Bergin, A.M.; Downs, J.; Leonard, H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: Experience of >100 patients. Epilepsia 2017, 58, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.; Wong, K.; Downs, J.; Bebbington, K.; Demarest, S.; Leonard, H. Vagus nerve stimulation for the treatment of refractory epilepsy in the CDKL5 Deficiency Disorder. Epilepsy Res. 2018, 146, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Ferretti, A.; Terrone, G.; Santoro, C.; Bravaccio, C.; Striano, S.; Coppola, A.; Striano, P. Clinical evolution and epilepsy outcome in three patients with CDKL5-related developmental encephalopathy. Epileptic Disord. 2019, 21, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Lee, S.S.; Zhang, X.; Houwink-Manville, I.; Song, H.R.; Amir, R.E.; Budden, S.; Naidu, S.; Pereira, J.L.; Lo, I.F.; et al. Rett syndrome and beyond: Recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am. J. Hum. Genet. 1999, 65, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Kankirawatana, P.; Leonard, H.; Ellaway, C.; Scurlock, J.; Mansour, A.; Makris, C.M.; Dure, L.S., IV; Friez, M.; Lane, J.; Kiraly-Borri, C.; et al. Early progressive encephalopathy in boys and MECP2 mutations. Neurology 2006, 67, 164–166. [Google Scholar] [CrossRef]
- Leuzzi, V.; Di Sabato, M.L.; Zollino, M.; Montanaro, M.L.; Seri, S. Early-onset encephalopathy and cortical myoclonus in a boy with MECP2 gene mutation. Neurology 2004, 63, 1968–1970. [Google Scholar] [CrossRef]
- Cohen, D.; Lazar, G.; Couvert, P.; Desportes, V.; Lippe, D.; Mazet, P.; Héron, D. MECP2 mutation in a boy with language disorder and schizophrenia. Am. J. Psychiatry 2002, 159, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Yntema, H.G.; Oudakker, A.R.; Romein, T.; Sistermans, E.; Nillessen, W.; van Bokhoven, H.; de Vries, B.B.; Hamel, B.C. De novo MECP2 frameshift mutation in a boy with moderate mental retardation, obesity and gynaecomastia. Clin. Genet. 2002, 61, 359–362. [Google Scholar] [CrossRef]
- Tan, W.H.; Bird, L.M.; Thibert, R.L.; Williams, C.A. If not Angelman, what is it? A review of Angelman-like syndromes. Am. J. Med. Genet. Part A 2014, 164A, 975–992. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Bernardo, P.; Cobb, S.; Coppola, A.; Tomasevic, L.; Di Lazzaro, V.; Bravaccio, C.; Manganelli, F.; Dubbioso, R. Neurophysiological Signatures of Motor Impairment in Patients with Rett Syndrome. Ann. Neurol. 2020, 87, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.J.; Tsytsarev, V.; Erzurumlu, R.S. Structural and functional differences in the barrel cortex of Mecp2 null mice. J. Comp. Neurol. 2017, 525, 3951–3961. [Google Scholar] [CrossRef] [PubMed]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Pintaudi, M.; Calevo, M.G.; Vignoli, A.; Parodi, E.; Aiello, F.; Baglietto, M.G.; Hayek, Y.; Buoni, S.; Renieri, A.; Russo, S.; et al. Epilepsy in Rett syndrome: Clinical and genetic features. Epilepsy Behav. 2010, 19, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Nissenkorn, A.; Gak, E.; Vecsler, M.; Reznik, H.; Menascu, S.; Ben Zeev, B. Epilepsy in Rett syndrome—The experience of a National Rett Center. Epilepsia 2010, 51, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Nissenkorn, A.; Levy-Drummer, R.S.; Bondi, O.; Renieri, A.; Villard, L.; Mari, F.; Mencarelli, M.A.; Lo Rizzo, C.; Meloni, I.; Pineda, M.; et al. Epilepsy in Rett syndrome—Lessons from the Rett networked database. Epilepsia 2015, 56, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Coppola, A.; Terrone, G.; Riccio, M.P.; Santoro, C.; Del Giudice, E.; Bravaccio, C. Epilepsy in Rett Syndrome: Can seizures play an encephalopathic effect in this disorder? Minerva Pediatr. 2019, 71, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Bonanni, P.; Parmeggiani, L.; Santucci, M.; Parmeggiani, A.; Sartucci, F. Cortical reflex myoclonus in Rett syndrome. Ann. Neurol. 1998, 43, 472–479. [Google Scholar] [CrossRef]
- Smeets, E.E.; Pelc, K.; Dan, B. Rett Syndrome. Mol. Syndromol. 2012, 2, 113–127. [Google Scholar] [CrossRef]
- Gayatri, N.A.; Livingston, J.H. Aggravation of epilepsy by anti-epileptic drugs. Dev. Med. Child Neurol. 2006, 48, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, G.; Bernardo, P.; Raiano, E.; Pinelli, M.; Alagia, M.; Esposito, M.; Della Casa, R.; Strisciuglio, P.; Brunetti-Pierri, N.; Bravaccio, C. Pain and sleep disturbances in Rett syndrome and other neurodevelopmental disorders. Acta Paediatr. 2019, 108, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Raiano, E.; Cappuccio, G.; Dubbioso, R.; Bravaccio, C.; Vergara, E.; Peluso, S.; Manganelli, F.; Esposito, M. The Treatment of Hypersalivation in Rett Syndrome with Botulinum Toxin: Efficacy and Clinical Implications. Neurol. Ther. 2019, 8, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Benke, T.A.; Marsh, E.D.; Skinner, S.A.; Merritt, J.; Lieberman, D.N.; Standridge, S.; Feyma, T.; Heydemann, P.; Peters, S.; et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2019, 180, 55–67. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, J.S.; Bernardino, A.; Nishimura, A.; Gomes, R.R.; Zatz, M. Rett syndrome in a boy with a 47,XXY karyotype confirmed by a rare mutation in the MECP2 gene. Neuropediatrics 2001, 32, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G.; Demidova, I.A.; Kolotii, A.D.; Kurinnaia, O.S.; Kravets, V.S.; Soloviev, I.V.; Yurov, Y.B.; Iourov, I.Y. Klinefelter syndrome mosaicism in boys with neurodevelopmental disorders: A cohort study and an extension of the hypothesis. Mol. Cytogenet. 2022, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Bird, A.P. MeCP2 mutations: Progress towards understanding and treating Rett syndrome. Genome Med. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8, 170216. [Google Scholar] [CrossRef]
- Echenne, B.; Roubertie, A.; Lugtenberg, D.; Kleefstra, T.; Hamel, B.C.; Van Bokhoven, H.; Lacombe, D.; Philippe, C.; Jonveaux, P.; de Brouwer, A.P. Neurologic aspects of MECP2 gene duplication in male patients. Pediatr. Neurol. 2009, 41, 187–191. [Google Scholar] [CrossRef]
- Marafi, D.; Suter, B.; Schultz, R.; Glaze, D.; Pavlik, V.N.; Goldman, A.M. Spectrum and time course of epilepsy and the associated cognitive decline in MECP2 duplication syndrome. Neurology 2019, 92, e108–e114. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Chuang, T.P.; Chiang, M.F.; Ho, C.S.; Hsiao, C.D.; Huang, Y.W.; Wu, T.Y.; Wu, J.Y.; Chen, Y.T.; Chen, T.C.; et al. De novo MECP2 duplication derived from paternal germ line result in dysmorphism and developmental delay. Gene 2014, 533, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Hanchard, N.A.; Carvalho, C.M.; Bader, P.; Thome, A.; Omo-Griffith, L.; del Gaudio, D.; Pehlivan, D.; Fang, P.; Schaaf, C.P.; Ramocki, M.B.; et al. A partial MECP2 duplication in a mildly affected adult male: A putative role for the 3′ untranslated region in the MECP2 duplication phenotype. BMC Med. Genet. 2012, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Okamoto, N.; Ito, M.; Arai, Y.; Momosaki, K.; Togawa, M.; Maegaki, Y.; Sugawara, M.; Shimojima, K.; Osawa, M.; et al. MECP2 duplication syndrome in both genders. Brain Dev. 2013, 35, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Auber, B.; Burfeind, P.; Thiels, C.; Alsat, E.A.; Shoukier, M.; Liehr, T.; Nelle, H.; Bartels, I.; Salinas-Riester, G.; Laccone, F. An unbalanced translocation resulting in a duplication of Xq28 causes a Rett syndrome-like phenotype in a female patient. Clin. Genet. 2010, 77, 593–597. [Google Scholar] [CrossRef] [PubMed]
- El Chehadeh, S.; Touraine, R.; Prieur, F.; Reardon, W.; Bienvenu, T.; Chantot-Bastaraud, S.; Doco-Fenzy, M.; Landais, E.; Philippe, C.; Marle, N.; et al. Xq28 duplication including MECP2 in six unreported affected females: What can we learn for diagnosis and genetic counselling? Clin. Genet. 2017, 91, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.U.; Fu, C.; Suter, B.; Marsh, E.; Benke, T.A.; Skinner, S.A.; Lieberman, D.N.; Standridge, S.; Jones, M.; Beisang, A.; et al. Characterizing the phenotypic effect of Xq28 duplication size in MECP2 duplication syndrome. Clin. Genet. 2019, 95, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Clayton-Smith, J.; Walters, S.; Hobson, E.; Burkitt-Wright, E.; Smith, R.; Toutain, A.; Amiel, J.; Lyonnet, S.; Mansour, S.; Fitzpatrick, D.; et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur. J. Hum. Genet. 2009, 17, 434–443. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135 Pt 5, 1348–1369. [Google Scholar] [CrossRef] [PubMed]
- Drongitis, D.; Caterino, M.; Verrillo, L.; Santonicola, P.; Costanzo, M.; Poeta, L.; Attianese, B.; Barra, A.; Terrone, G.; Lioi, M.B.; et al. Deregulation of microtubule organization and RNA metabolism in Arx models for lissencephaly and developmental epileptic encephalopathy. Hum. Mol. Genet. 2022, 31, 1884–1908. [Google Scholar] [CrossRef]
- Dobyns, W.B.; Berry-Kravis, E.; Havernick, N.J.; Holden, K.R.; Viskochil, D. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am. J. Med. Genet. 1999, 86, 331–337. [Google Scholar] [CrossRef]
- Mattiske, T.; Moey, C.; Vissers, L.E.; Thorne, N.; Georgeson, P.; Bakshi, M.; Shoubridge, C. An Emerging Female Phenotype with Loss-of-Function Mutations in the Aristaless-Related Homeodomain Transcription Factor ARX. Hum. Mutat. 2017, 38, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, C.; Tan, M.H.; Seiboth, G.; Gécz, J. ARX homeodomain mutations abolish DNA binding and lead to a loss of transcriptional repression. Hum. Mol. Genet. 2012, 21, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.; Nasrallah, M.P.; Lim, Y.; Golden, J.A. Distinct DNA binding and transcriptional repression characteristics related to different ARX mutations. Neurogenetics 2012, 13, 23–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shoubridge, C.; Fullston, T.; Gécz, J. ARX spectrum disorders: Making inroads into the molecular pathology. Hum. Mutat. 2010, 31, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Represa, A. Why Malformations of Cortical Development Cause Epilepsy. Front. Neurosci. 2019, 13, 250. [Google Scholar] [CrossRef]
- Matsumoto, N.; Leventer, R.J.; Kuc, J.A.; Mewborn, S.K.; Dudlicek, L.L.; Ramocki, M.B.; Pilz, D.T.; Mills, P.L.; Das, S.; Ross, M.E.; et al. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur. J. Hum. Genet. 2001, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.D.; Bernasconi, A.; Das, S.; Bastos, A.; Valerio, R.M.; Palmini, A.; Costa da Costa, J.; Scheffer, I.E.; Berkovic, S.; Guerrini, R.; et al. Subcortical band heterotopia (SBH) in males: Clinical, imaging and genetic findings in comparison with females. Brain 2002, 125 Pt 11, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.G.; Lin, P.T.; Flanagan, L.A.; Walsh, C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 1999, 23, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Dobyns, W.B.; Guerrini, R. Malformations of cortical development and epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022392. [Google Scholar] [CrossRef]
- Moffat, J.J.; Ka, M.; Jung, E.M.; Kim, W.Y. Genes and brain malformations associated with abnormal neuron positioning. Mol. Brain 2015, 8, 72. [Google Scholar] [CrossRef]
- Carabalona, A.; Beguin, S.; Pallesi-Pocachard, E.; Buhler, E.; Pellegrino, C.; Arnaud, K.; Hubert, P.; Oualha, M.; Siffroi, J.P.; Khantane, S.; et al. A glial origin for periventricular nodular heterotopia caused by impaired expression of Filamin-A. Hum. Mol. Genet. 2012, 21, 1004–1017. [Google Scholar] [CrossRef]
- Lu, Y.T.; Hsu, C.Y.; Liu, Y.T.; Chan, C.K.; Chuang, Y.C.; Lin, C.H.; Chang, K.P.; Ho, C.J.; Ng, C.C.; Lim, K.S.; et al. The clinical and imaging features of FLNA positive and negative periventricular nodular heterotopia. Biomed. J. 2022, 45, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Roll, P.; Rudolf, G.; Pereira, S.; Royer, B.; Scheffer, I.E.; Massacrier, A.; Valenti, M.P.; Roeckel-Trevisiol, N.; Jamali, S.; Beclin, C.; et al. SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 2006, 15, 1195–1207. [Google Scholar] [CrossRef]
- Numata, M.; Petrecca, K.; Lake, N.; Orlowski, J. Identification of a mitochondrial Na+/H+ exchanger. J. Biol. Chem. 1998, 273, 6951–6959. [Google Scholar] [CrossRef]
- Kerner-Rossi, M.; Gulinello, M.; Walkley, S.; Dobrenis, K. Pathobiology of Christianson syndrome: Linking disrupted endosomal-lysosomal function with intellectual disability and sensory impairments. Neurobiol. Learn. Mem. 2019, 165, 106867. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, G.D.; Selmer, K.K.; Roxrud, I.; Smith, R.; Kyllerman, M.; Eiklid, K.; Kroken, M.; Mattingsdal, M.; Egeland, T.; Stenmark, H.; et al. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am. J. Hum. Genet. 2008, 82, 1003–1010. [Google Scholar] [CrossRef]
- Sakagami, H.; Sanda, M.; Fukaya, M.; Miyazaki, T.; Sukegawa, J.; Yanagisawa, T.; Suzuki, T.; Fukunaga, K.; Watanabe, M.; Kondo, H. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neurosci. Res. 2008, 60, 199–212. [Google Scholar] [CrossRef]
- Cioclu, M.C.; Coppola, A.; Tondelli, M.; Vaudano, A.E.; Giovannini, G.; Krithika, S.; Iacomino, M.; Zara, F.; Sisodiya, S.M.; Meletti, S. Cortical and Subcortical Network Dysfunction in a Female Patient with NEXMIF Encephalopathy. Front. Neurol. 2021, 12, 722664. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Inoue, N.; Westfall, B.; Taron, C.H.; Orlean, P.; Takeda, J.; Kinoshita, T. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 1998, 17, 877–885. [Google Scholar] [CrossRef]
- Puranam, R.S.; He, X.P.; Yao, L.; Le, T.; Jang, W.; Rehder, C.W.; Lewis, D.V.; McNamara, J.O. Disruption of Fgf13 causes synaptic excitatory-inhibitory imbalance and genetic epilepsy and febrile seizures plus. J. Neurosci. 2015, 35, 8866–8881. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Locus | Inheritance | Gene–Phenotype Relationships (Incidence If Available) | Main Epileptic Phenotype | Epilepsy Onset (Age) | SE | Main EEG Features | Neuro-Developmental Outcome | MRI Features | Other Features | Specific Treatment | Drug Resistance | References | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Interictal | Ictal | ||||||||||||||
| PCDH19 | Xq22.1 | XL | DEE 9 (1/42,000 live births) | Clusters of FS, focal S: hypomotor and affective S | 3–36 mo | 10% to 53% | Bilateral SW, rare focal epileptiform abnormalities | Diffuse or frontal and temporal focal discharges (both hemispheres) | ID, ASD, psychiatric disorders. Normal cognitive outcome is also reported | Normal | None | BDZ; Corticosteroids during the clusters; stiripentol | Not constant | Marini, 2012 [6]; Trivisano, 2018 [7]; Samanta, 2019 [8]; Zuberi, 2022 [9] | |
| CDKL5 | Xp22.13 | XLD | DEE 2 (1/40,000-1/60,000 live births) | IS, tonic and myoclonic S, ‘prolonged’ GTCS | 3–4 mo | rare | Slow background; SB; multifocal epileptiform activity | Bilateral synchronous initial flattening, followed by repetitive SW and spikes | Severe developmental delay, ID | Normal; progressive brain atrophy | Microcephaly, hypotonia | Ganaxolone | Yes | Zuberi, 2022 [9]; Melani, 2011 [10]; Knight, 2022 [11] | |
| MECP2 | Xq28 | RTT | XL, XLR, XLD | RTT (1 in 10,000 live female births) ASD susceptibility, X-linked 3; Encephalopathy, neonatal severe; IDD, X-linked syndromic 13; IDD, X-linked syndromic, Lubs type | Focal or GTCS | 5–10 Y | rare | Unusual fast/theta rhythmic activity | No peculiar pattern | Psycho-motor regression, ID, ASD | Normal | Microcephaly, breathing abnormalities, hand stereotypies, myoclonus, mouthing, hypotonia | NR | 30% | Guerrini, 2012 [12]; Glaze, 2010 [13] |
| MECP2-dup | Focal or GTCS; atonic seizures or drop attacks | 9–13 Y | rare | Unusual fast/theta rhythmic activity, multifocal spike discharges, generalized SW activity, “extreme spindles” during the sleep | Unusual fast rhythms and erratic myoclonic jerks evolving in spike and wave discharges with anterior predominance | ID, ASD | Mild facial dysmorphisms, recurrent infections, dyskinetic movements, hypotonia, evolving to spasticity | NR | Yes | Guerrini, 2012 [12]; Ramocki, 2010 [14]; Vignoli, 2012 [15] | |||||
| ARX | Xp21.3 | XLR | DEE 1; Hydranencephaly with abnormal genitalia; IDD, X-linked; Lissencephaly, X-linked 2; Partington syndrome; Proud syndrome | Polymorphic: IS, myoclonic S | First mo | NR | Hypsarrhythmia, multifocal, asynchronous, epileptiform activity | NR | PMD, ID | Corpus callosum agenesis, lissencephaly | Microcephaly, hypotonia, abnormal genitalia and Partington syndrome | NR | Yes | Kitamura, 2002 [16]; Kato, 2004 [17]; Marsh, 2012 [18]; Edey, 2023 [19] | |
| DCX | Xq23 | XL | Lissencephaly, X-linked; Subcortical laminal heterotopia, X-linked | IS | first Y | NR | NR | PMD, ID | Lis-sencephaly, SBH | NR | NR | Yes | Bahi-Buisson, 2013 [20]; Chou A, 2009 [21] | ||
| FLNA | Xq28 | XL, XLR, XLD | Cardiac valvular dysplasia, X-linked; Congenital short bowel syndrome; Fronto-metaphyseal dysplasia 1; Heterotopia, periventricular, 1; Intestinal pseudo-obstruction, neuronal; Melnick-Needles syndrome | Focal S | First mo-adult | NR | Focal slowing-epileptic discharges or normal | NR | Normal to ID | BPNH | Dysmorphic features, cardiac disease, skin and joints abnormalities | Mild to DRE | Parrini, 2006 [22]; Cannaerts, 2018 [23] | ||
| SLC9A6 | Xq26.3 | XL | IDD, X-linked syndromic, Christianson type | Focal or GTCS | NR | slow rhythmic activity of high amplitude | NR | ID or regression, behavior disorder | Asymmetric atrophy most prominent in left frontal and parietal cortex | Hypotonia, microcephaly, impaired ocular movements, ataxia and facial dysmorphisms | NR | Yes | Schroer, 2010 [24], Sinajon, 2016 [25] | ||
| SLC35A2 | Xp11.23 | SMo, XLD | Congenital disorder of glycosylation, type IIm | IS | First Y | NR | hypsarrythmic | NR | ID | white matter abnormalities and cerebellar atrophy; FCD | hypotonia, facial dysmorphisms, skeletal abnormalities, microcephaly | NR | Kodera, 2013 [26]; Winawer, 2018 [27]; Ng, 2019 [28] | ||
| SYN1 | Xp11.23 | XL | Epilepsy, X-linked 1, with variable learning disabilities and behavior disorders; IDD, X-linked 50 | Reflex S, FS | NR | Focus over temporo-insular regions. | Learning difficulties, specific language impairment and mild ASD | NR | Garcia, 2004 [29], Giovedi, 2004 [30] Nguyen, 2015 [31] | ||||||
| ARHGEF9 | Xq11.1 | XL | DEE 8 | GTCS | NR | Poly-microgyria, hippocampal sclerosis and delayed myelination | NR | Yes | Shimojima, 2011 [32], Alber, 2017 [33]; Yang, 2022 [34] | ||||||
| ATP6AP2 | Xp11.4 | XLR | Parkinsonism with spasticity, X-linked; Congenital disorder of glycosylation, type II; IDD, X-linked syndromic, Hedera type | GTCS, atonic, myoclonic | 4–14 mo | NR | Generalized polyspike and waves discharges before seizures | NR | Cerebellar atrophy and/or thinning of the CC | Scoliosis, ataxia and hyporeflexia, Parkinsonism | NR | Hedera, 2002 [35]; Ramser, 2005 [36]; Gupta, 2015 [37] | |||
| IQSEC2 | Xp11.22 | XLD | IDD, X-linked 1 | Atonic, myoclonic, IS atypical absences, GTCS | 8 mo–4 Y | NR | Not specific | NR | Yes | Zerem, 2016 [38]; Choi, 2020 [39] | |||||
| NEXMIF | Xq13.2 | XLD | IDD X-linked 98 | EEM, EMAtS | NR | Generalized SW or polyspikes Photo-sensitivity, ECS | ID, ASD | Normal | Hypotonia, ataxia, microcephaly, gastro-esophageal reflux disease, strabismus, dysmorphic features | NR | NR | Stamberger, 2021 [40]; Coppola, 2023 [41] | |||
| PIGA | Xp22.2 | XLR | Multiple congenital anomalies-hypotonia-seizures syndrome; Neurodevelopmental disorder with epilepsy and hemochromatosis; Paroxysmal nocturnal hemoglobinuria | Myoclonic, tonic S with apnea, GTCD, Focal S, migrating focal S, atonic, gelastic, IS, FS | Firs Y | Diffuse background slowing, multifocal or diffuse spike/sharp and SW with high amplitude | NR | PMD, ID | Cortical and subcortical volume loss, cerebellar and/or brainstem hypoplasia, white matter immaturity, corpus callosum dysgenesis; FCD, HS | Congenital heart anomalies | KD, pyridoxine | Yes | Kim, 2016 [42]; Bayat, 2020 [43]; Ikeda, 2023 [44] | ||
| ALG13 | Xq23 | XL | DEE 36 | IS, tonic S, LGS | 6 mo | NR | hypsarrythmic pattern | paroxysms of fast activity with electrodecrement without clinical correlate | PMD with regression | matter volume loss, corpus callosum dysgenesis and cortical atrophy | hypotonia, movement disorders, dysmorphic features. | NR | yes | Datta, 2020 [45]; Berry, 2020 [46] | |
| FHF2/ FGF13 | Xq26.3-q27.1 | XLD, XLR | DEE 90 | motor features, apneas, oro-alimentary automatism, IS, GTCS, GEFS+ | NR | NR | temporal focus | NR | NR | NR | NR | Surgery | yes | Fry, 2021 [47]; Narayanan, 2022 [48] | |
| GRIA3 | Xq25 | XLR | ID, X-linked syndromic, Wu type | Myoclonic S, is, atypical absences | First mo | NCSE | Multifocal and generalized abnormalities; subcontinuous diffuse epileptiform abnormalities with higher amplitude over bilateral frontal regions | NR | ID, ASD | NR | hypotonia, asthenic body habitus with poor muscle bulk, hyporeflexia. | PER | NR | Trivisano, 2020 [49]; Sun, 2021 [50] | |
| SMC1A | Xp11.22 | XLD | Cornelia de Lange syndrome 2; DEE 85, with or without midline brain defects | clustering seizures | NR | NR | NR | NR | ID | NR | NR | NR | Yes | Selicorni, 2021 [51]; Symonds, 2017 [52]; Bozarth, 2023 [53] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardo, P.; Cuccurullo, C.; Rubino, M.; De Vita, G.; Terrone, G.; Bilo, L.; Coppola, A. X-Linked Epilepsies: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 4110. https://doi.org/10.3390/ijms25074110
Bernardo P, Cuccurullo C, Rubino M, De Vita G, Terrone G, Bilo L, Coppola A. X-Linked Epilepsies: A Narrative Review. International Journal of Molecular Sciences. 2024; 25(7):4110. https://doi.org/10.3390/ijms25074110
Chicago/Turabian StyleBernardo, Pia, Claudia Cuccurullo, Marica Rubino, Gabriella De Vita, Gaetano Terrone, Leonilda Bilo, and Antonietta Coppola. 2024. "X-Linked Epilepsies: A Narrative Review" International Journal of Molecular Sciences 25, no. 7: 4110. https://doi.org/10.3390/ijms25074110
APA StyleBernardo, P., Cuccurullo, C., Rubino, M., De Vita, G., Terrone, G., Bilo, L., & Coppola, A. (2024). X-Linked Epilepsies: A Narrative Review. International Journal of Molecular Sciences, 25(7), 4110. https://doi.org/10.3390/ijms25074110