


Ischemia-Reperfusion Programming of Alzheimer’s Disease-Related Genes—A New Perspective on Brain Neurodegeneration after Cardiac Arrest



Abstract
1. Cardiac Arrest
2. Brain Neurodegeneration after Cardiac Arrest
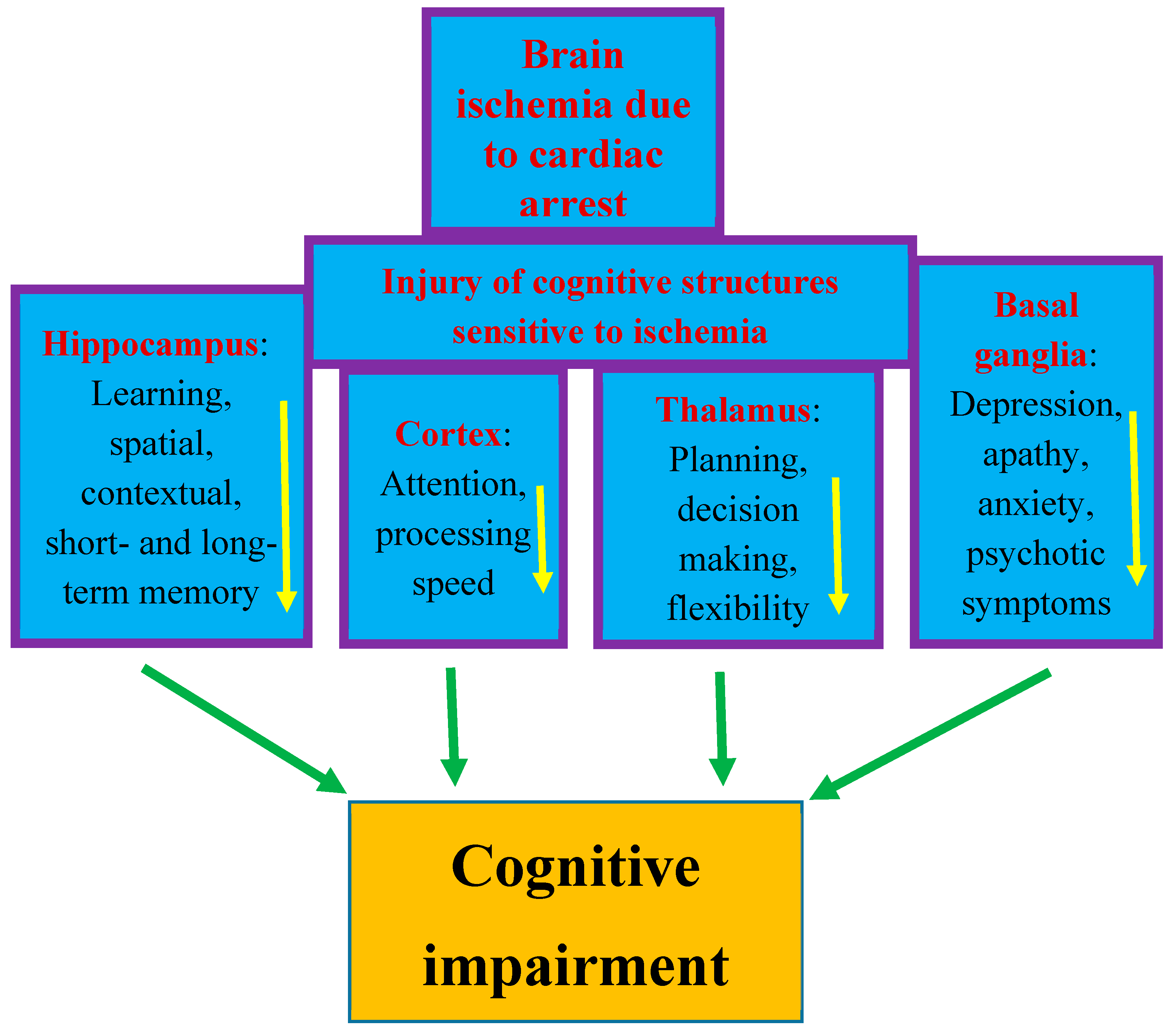
3. Cognitive Deficits after Cardiac Arrest
4. Alzheimer’s Disease Related Genes after Cardiac Arrest
5. Amyloid and Tau Protein in Humans after Cardiac Arrest
6. Amyloid and Tau Protein in Animals after Cardiac Arrest
7. Transport Genes of Amyloid and Tau Protein in the Brain after Cardiac Arrest
8. Genes Involved in Neuronal Death in Cardiac Arrest and Alzheimer’s Disease
9. Discussion
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stub, D.; Bernard, S.; Duffy, S.J.; Kaye, D.M. Post cardiac arrest syndrome: A review of therapeutic strategies. Circulation 2011, 123, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Sandroni, C.; Cronberg, T.; Sekhon, M. Brain injury after cardiac arrest: Pathophysiology, treatment, and prognosis. Intensiv. Care Med. 2021, 47, 1393–1414. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.W.; Holmberg, M.J.; Berg, K.M.; Donnino, M.W.; Granfeldt, A. In-hospital cardiac arrest: A review. JAMA 2019, 321, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Moseby-Knappe, M.; Benedet, A.L.; Grötschel, L.; Lantero-Rodriguez, J.; Karikari, T.K.; Hassager, C.; Wise, M.P.; Stammet, P.; Kjaergaard, J.; et al. Alzheimer Disease Blood Biomarkers in Patients with Out-of-Hospital Cardiac Arrest. JAMA Neurol. 2023, 80, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xing, J. A model for predicting return of spontaneous circulation and neurological outcomes in adults after in-hospital cardiac arrest: Development and evaluation. Front. Neurol. 2023, 14, 1323721. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Botha, J.A.; Tiruvoipati, R. Cognitive function, quality of life and mental health in survivors of our-of-hospital cardiac arrest: A review. Anaesth. Intensive Care 2015, 43, 568–576. [Google Scholar] [CrossRef]
- Glimmerveen, A.; Verhulst, M.; Verbunt, J.; Van Heugten, C.; Hofmeijer, J. Predicting Long-Term Cognitive Impairments in Survivors after Cardiac Arrest: A Systematic Review. J. Rehabil. Med. 2023, 55, jrm00368. [Google Scholar] [CrossRef]
- Mehra, R. Global public health problem of sudden cardiac death. J. Electrocardiol. 2007, 40, S118–S122. [Google Scholar] [CrossRef]
- Jung, E.; Park, J.H.; Ro, Y.S.; Ryu, H.H.; Cha, K.C.; Do Shin, S.; Hwang, S.O. Cardiac Arrest Pursuit Trial with Unique Registration, Epidemiologic Surveillance (CAPTURES) project investigators. Family history, socioeconomic factors, comorbidities, health behaviors, and the risk of sudden cardiac arrest. Sci. Rep. 2023, 13, 21341. [Google Scholar] [CrossRef]
- Wissenberg, M.; Lippert, F.K.; Folke, F.; Weeke, P.; Hansen, C.M.; Christensen, E.F. Association of national initiatives to improve cardiac arrest management with rates of bystander intervention and patient survival after out-of-hospital cardiac arrest. JAMA 2013, 310, 1377–1384. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, J.Y.; Teng, N.C.; Chao, T.T.; Tsai, S.L.; Chen, C.L.; Hsu, J.Y.; Wu, C.P.; Lai, C.C.; Chen, L. The secular trends in the incidence rate and outcomes of out-of-hospital cardiac arrest in Taiwan-A nationwide population-based study. PLoS ONE 2015, 10, e0122675. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Wilson, R.H.; Crouzet, C.; Amirhekmat, A.; Wei, K.S.; Akbari, Y. Resuscitating the Globally Ischemic Brain: TTM and Beyond. Neurotherapeutics 2020, 17, 539–562. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, G.; Ihle-Hansen, H.; Sandset, E.C.; Jacobsen, D.; Wimmer Hand Ihle-Hansen, H. Long Term Cognitive Function after Cardiac Arrest: A Mini-Review. Front. Aging Neurosci. 2022, 14, 885226. [Google Scholar] [CrossRef] [PubMed]
- de Vreede-Swagemakers, J.J.; Gorgels, A.P.; Dubois-Arbouw, W.I.; van Ree, J.W.; Daemen, M.J.; Houben, L.G.; Wellens, H.J. Out-of-hospital cardiac arrest in the 1990’s: A population-based study in the Maastricht area on incidence, characteristics and survival. J. Am. Coll. Cardiol. 1997, 30, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Berdowski, J.; Berg, R.A.; Tijssen, J.G.; Koster, R.W. Global incidences of out-of-hospital cardiac arrest and survival rates: Systematic review of 67 prospective studies. Resuscitation 2010, 81, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.O.; Cha, K.C.; Jung, W.J.; Roh, Y.I.; Kim, T.Y.; Chung, S.P.; Kim, Y.M.; Park, J.D.; Kim, H.S.; Lee, M.J.; et al. Steering Committee of the 2020 Korean Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. 2020 Korean Guidelines for Cardiopulmonary Resuscitation. Part 2. Environment for cardiac arrest survival and the chain of survival. Clin. Exp. Emerg. Med. 2021, 8, S8–S14. [Google Scholar] [CrossRef]
- Gräsner, J.T.; Lefering, R.; Koster, R.W.; Masterson, S.; Böttiger, B.W.; Herlitz, J.; Wnent, J.; Tjelmeland, I.B.; Ortiz, F.R.; Maurer, H.; et al. EuReCa ONE-27 Nations, ONE Europe, ONE Registry: A prospective one month analysis of out-of-hospital cardiac arrest outcomes in 27 countries in Europe. Resuscitation 2016, 105, 188–195. [Google Scholar] [CrossRef]
- Gräsner, J.T.; Wnent, J.; Herlitz, J.; Perkins, G.D.; Lefering, R.; Tjelmeland, I.; Koster, R.W.; Masterson, S.; Rossell-Ortiz, F.; Maurer, H.; et al. Survival after out-of-hospital cardiac arrest in Europe –Results of the EuReCa TWO study. Resuscitation 2020, 148, 218–226. [Google Scholar] [CrossRef]
- Nichol, G.; Thomas, E.; Callaway, C.W.; Hedges, J.; Powell, J.L.; Aufderheide, T.P.; Rea, T.; Lowe, R.; Brown, T.; Dreyer, J.; et al. Resuscitation Outcomes Consortium Investigators. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA 2008, 300, 1423–1431. [Google Scholar] [CrossRef]
- Medina, J.A.; Quintero, J.A.; de Paz, D.A.; Scarpetta, D.F.; Castro, C.A.; Paker, N.A.; Carvajal, S.M. Cardiac arrest in an emergency department in Colombia during 2011–2020: A descriptive study. Int. J. Crit. Illn. Inj. Sci. 2023, 13, 132–137. [Google Scholar]
- Beck, B.; Bray, J.; Cameron, P.; Smith, K.; Walker, T.; Grantham, H.; Hein, C.; Thorrowgood, M.; Smith, A.; Inoue, M.; et al. Aus-ROC Steering Committee. Regional variation in the characteristics, incidence and outcomes of out-of-hospital cardiac arrest in Australia and New Zealand: Results from the Aus-ROC Epistry. Resuscitation 2018, 126, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Li, C.S.; Liang, L.R.; Qin, J.; Ding, N.; Fu, Y.; Yang, K.; Zhang, G.Q.; Zhao, L.; Zhao, B.; et al. Incidence and outcome of adult in-hospital cardiac arrest in Beijing, China. Resuscitation 2016, 102, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, M.S.; Ainslie, P.N.; Griesdale, D.E. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: A “two-hit” model. Crit. Care 2017, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- Lemiale, V.; Dumas, F.; Mongardon, N.; Giovanetti, O.; Charpentier, J.; Chiche, J.D.; Carli, P.; Mira, J.P.; Nolan, J.; Cariou, A. Intensive care unit mortality after cardiac arrest: The relative contribution of shock and brain injury in a large cohort. Intensive Care Med. 2013, 39, 1972–1980. [Google Scholar] [CrossRef]
- Geocadin, R.G.; Callaway, C.W.; Fink, E.L.; Golan, E.; Greer, D.M.; Ko, N.U.; Lang, E.; Licht, D.J.; Marino, B.S.; McNair, N.D.; et al. Standards for studies of neurological prognostication in comatose survivors of cardiac arrest: A scientific statement from the American Heart Association. Circulation 2019, 140, e517–e542. [Google Scholar] [CrossRef] [PubMed]
- Varon, J.; Marik, P.E.; Einav, S. Therapeutic hypothermia: A state-of-the-art emergency medicine perspective. Am. J. Emerg. Med. 2012, 30, 800–810. [Google Scholar] [CrossRef]
- Lilja, G.; Nielsen, N.; Friberg, H.; Horn, J.; Kjaergaard, J.; Nilsson, F.; Pellis, T.; Wetterslev, J.; Wise, M.P.; Bosch, F.; et al. Cognitive function in survivors of out-of-hospital cardiac arrest after target temperature management at 33 °C versus 36 °C. Circulation 2015, 131, 1340–1349. [Google Scholar] [CrossRef]
- Ahn, J.H.; Lee, T.K.; Tae, H.J.; Kim, B.; Sim, H.; Lee, J.C.; Kim, D.W.; Kim, Y.S.; Shin, M.C.; Park, Y.; et al. Neuronal Death in the CNS Autonomic Control Center Comes Very Early after Cardiac Arrest and Is Not Significantly Attenuated by Prompt Hypothermic Treatment in Rats. Cells 2021, 10, 60. [Google Scholar] [CrossRef]
- Weng, Y.C.; Huang, Y.T.; Chiang, I.C.; Chuang, H.C.; Lee, T.H.; Tan, T.H.; Chou, W.H. DUSP6 deficiency attenuates neurodegeneration after global cerebral ischemia. Int. J. Mol. Sci. 2023, 24, 7690. [Google Scholar] [CrossRef]
- Kagstrom, E.; Smith, M.L.; Siesjo, B.K. Local cerebral blood flow in the recovery period following complete cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 1983, 3, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, P.; Wieloch, T. Ischemic brain damage in rats following cardiac arrest using a long-term recovery model. J. Cereb. Blood Flow Metab. 1985, 5, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Nitecka, L.; Ruetzler, C.A.; Nagashima, G.; Joó, F.; Mies, G.; Nowak TSJr Saito, N.; Lohr, J.M.; Klatzo, I. Global cerebral ischemia associated with cardiac arrest in the rat: I. Dynamics of early neuronal changes. J. Cereb. Blood Flow Metab. 1992, 12, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Mai, J.K.; Krajewska, M.; Sikorska, M.; Mossakowski, M.J.; Reed, J.C. Upregulation of bax protein levels in neurons following cerebral ischemia. J. Neurosci. 1995, 15, 6364–6376. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Nowak, T.S., Jr.; Klatzo, I. Loss of parvalbumin immunoreactivity defines selectively vulnerable thalamic reticular nucleus neurons following cardiac arrest in the rat. Acta Neuropathol. 1995, 89, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Böttiger, B.W.; Schmitz, B.; Wiessner, C.; Vogel, P.; Hossmann, K.A. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J. Cereb. Blood Flow Metab. 1998, 18, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K.; Feldmann, E.; Pulsinelli, W.A.; Plum, F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 1987, 37, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.R.; Liu, C.L.; Park, D.J. Alteration of MAP kinase pathways after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2000, 20, 1089–1095. [Google Scholar] [CrossRef]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef]
- Sadowski, M.; Wisniewski, H.M.; Jakubowska-Sadowska, K.; Tarnawski, M.; Lazarewicz, J.W.; Mossakowski, M.J. Pattern of neuronal loss in the rat hippocampus following experimental cardiac arrest-induced ischemia. J. Neurol. Sci. 1999, 168, 13–20. [Google Scholar] [CrossRef]
- Sadowski, M.; Lazarewicz, J.W.; Jakubowska-Sadowska, K.; Wisniewski, H.M.; Mossakowski, M.J.; Brown, W.T. Long-term changes in calbindin D(28K) immunoreactivity in the rat hippocampus after cardiac arrest. Neurosci. Lett. 2002, 321, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek, M.; Jabłoński, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. 2009, 292, 1863–1881. [Google Scholar] [CrossRef] [PubMed]
- Warenits, A.M.; Hatami, J.; Müllebner, A.; Ettl, F.; Teubenbacher, U.; Magnet, I.A.M.; Bauder, B.; Janata, A.; Miller, I.; Moldzio, R.; et al. Motor Cortex and Hippocampus Display Decreased Heme Oxygenase Activity 2 Weeks after Ventricular Fibrillation Cardiac Arrest in Rats. Front. Med. 2020, 7, 513. [Google Scholar] [CrossRef] [PubMed]
- Sekeljic, V.; Bataveljic, D.; Stamenkovic, S.; Ułamek, M.; Jabłoński, M.; Radenovic, L.; Pluta, R.; Andjus, P.R. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 2012, 217, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging 2020, 12, 12251–12267. [Google Scholar] [CrossRef] [PubMed]
- Traub, J.; Frey, A.; Störk, S. Chronic Neuroinflammation and Cognitive Decline in Patients with Cardiac Disease: Evidence, Relevance, and Therapeutic Implications. Life 2023, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia–reperfusion brain injury. A model of early Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 903, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, E.; Tam, E.; Allan, L.; Hall, R.; Khundakar, A.; Oakley, A.E.; Thomas, A.; Deramecourt, V.; Kalaria, R.N. Neuron volumes in hippocampal subfields in delayed poststroke and aging-related dementias. J. Neuropathol. Exp. Neurol. 2014, 73, 305–311. [Google Scholar] [CrossRef]
- Goulay, R.; Mena Romo, L.; Hol, E.M.; Dijkhuizen, R.M. From stroke to dementia: A Comprehensive review exposing tight interactions between stroke and amyloid-β formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef]
- Rost, N.S.; Brodtmann, A.; Pase, M.P.; van Veluw, S.J.; Biffi, A.; Duering, M.; Hinman, J.D.; Dichgans, M. Post-stroke cognitive impairment and dementia. Circ. Res. 2022, 130, 1252–1271. [Google Scholar] [CrossRef]
- Kiryk, A.; Pluta, R.; Figiel, I.; Mikosz, M.; Ulamek, M.; Niewiadomska, G.; Jablonski, M.; Kaczmarek, L. Transient brain ische-mia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav. Brain Res. 2011, 219, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cohan, C.H.; Neumann, J.T.; Dave, K.R.; Alekseyenko, A.; Binkert, M.; Stransky, K.; Lin, H.W.; Barnes, C.A.; Wright, C.B.; Perez Pinzon, M.A. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS ONE 2015, 10, e0124918. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; You, Y.; Min, J.H.; Yoo, I.; Jeong, W.; Cho, Y.; Ryu, S.; Lee, J.; Kim, S.W.; Cho, S.U.; et al. Study on the timing of severe blood-brain barrier disruption using cerebrospinal fluid-serum albumin quotient in post cardiac arrest patients treated with targeted temperature management. Resuscitation 2019, 135, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wisniewski, H.M.; Mossakowski, M.J. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Hirnforsch. 1994, 35, 463–471. [Google Scholar] [PubMed]
- Pluta, R.; Barcikowska, M.; Januszewski, S.; Misicka, A.; Lipkowski, A.W. Evidence of blood-brain barrier permeability/leakage for circulating human Alzheimer’s beta-amyloid-(1-42)-peptide. Neuroreport 1996, 7, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Miziak, B.; Czuczwar, S.J. Post-Ischemic Permeability of the Blood-Brain Barrier to Amyloid and Platelets as a Factor in the Maturation of Alzheimer’s Disease-Type Brain Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 10739. [Google Scholar] [CrossRef] [PubMed]
- Ousta, A.; Piao, L.; Fang, Y.H.; Vera, A.; Nallamothu, T.; Garcia, A.J., 3rd; Sharp, W.W. Microglial Activation and Neurological Outcomes in a Murine Model of Cardiac Arrest. Neurocrit. Care 2022, 36, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Mörtberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 2011, 6, e28263. [Google Scholar] [CrossRef]
- Peeters-Scholte, C.; Meilin, S.; Berckovich, Y.; Westers, P. 2-iminobiotin, a selective inhibitor of nitric oxide synthase, improves memory and learning in a rat model after four vessel occlusion, mimicking cardiac arrest. PLoS ONE 2023, 18, e0291915. [Google Scholar] [CrossRef]
- Moulaert, V.R.; Verbunt, J.A.; van Heugten, C.M.; Wade, D.T. Cognitive impairments in survivors of out-of-hospital cardiac arrest: A systematic review. Resuscitation 2009, 80, 297–305. [Google Scholar] [CrossRef]
- Sabedra, A.R.; Kristan, J.; Raina, K.; Holm, M.B.; Callaway, C.W.; Guyette, F.X.; Dezfulian, C.; Doshi, A.A.; Rittenberger, J.C.; Post Cardiac Arrest Service. Neurocognitive outcomes following successful resuscitation from cardiac arrest. Resuscitation 2015, 90, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Samudra, N.; Aiyagari, V. Cognitive and Functional Consequence of Cardiac Arrest. Curr. Neurol. Neurosci. Rep. 2016, 16, 70. [Google Scholar] [CrossRef]
- Steinbusch, C.V.M.; van Heugten, C.M.; Rasquin, S.M.C.; Verbunt, J.A.; Moulaert, V.R.M. Cognitive impairments and subjective cognitive complaints after survival of cardiac arrest: A prospective longitudinal cohort study. Resuscitation 2017, 120, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.K.; Berg, S.K.; Hassager, C.; Armand, S.; Møller, J.E.; Ekholm, O.; Rasmussen, T.B.; Fisher, P.M.; Knudsen, G.M.; Stenbæk, D.S. Cognitive impairment and psychopathology in out-of-hospital cardiac arrest survivors in Denmark: The REVIVAL cohort study protocol. BMJ Open 2020, 10, e038633. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.L.; Tang, L.H.; Borregaard, B.; Zinckernagel, L.; Mikkelsen, T.B.; Taylor, R.S.; Christiansen, S.R.; Nielsen, J.F.; Zwisler, A.D. Long-term physical and psychological outcomes after out-of-hospital cardiac arrest-protocol for a national cross-sectional survey of survivors and their relatives (the DANCAS survey). BMJ Open 2021, 11, e045668. [Google Scholar] [CrossRef] [PubMed]
- Amacher, S.A.; Bohren, C.; Blatter, R.; Becker, C.; Beck, K.; Mueller, J.; Loretz, N.; Gross, S.; Tisljar, K.; Sutter, R.; et al. Long-term survival after out-of-hospital cardiac arrest: A systematic review and meta-analysis. JAMA Cardiol. 2022, 7, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Huebschmann, N.A.; Cook, N.E.; Murphy, S.; Iverson, G.L. Cognitive and Psychological Outcomes Following Pediatric Cardiac Arrest. Front. Pediatr. 2022, 10, 780251. [Google Scholar] [CrossRef] [PubMed]
- Lilja, G.; Nilsson, G.; Nielsen, N.; Friberg, H.; Hassager, C.; Koopmans, M.; Kuiper, M.; Martini, A.; Mellinghoff, J.; Pelosi, P.; et al. Anxiety and depression among out-of-hospital cardiac arrest survivors. Resuscitation 2015, 97, 68–75. [Google Scholar] [CrossRef]
- Mion, M.; Simpson, R.; Johnson, T.; Oriolo, V.; Gudde, E.; Rees, P.; Quinn, T.; Vopelius-Feldt, V.J.; Gallagher, S.; Mozid, A.; et al. British cardiovascular intervention society consensus position statement on out-of-hospital cardiac arrest 2: Post-discharge rehabilitation. Interv. Cardiol. 2022, 17, e19. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, Y.J.; Ryoo, S.M.; Ahn, S.; Kim, W.Y. Telephone-based evaluation of cognitive impairment and mood disorders in cardiac arrest survivors with good neurologic outcomes: A retrospective cohort study. Sci. Rep. 2023, 13, 18065. [Google Scholar] [CrossRef]
- Andersson, A.E.; Rosén, H.; Sunnerhagen, K.S. Life after cardiac arrest: A very long-term follow-up. Resuscitation 2015, 91, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Verfaellie, M.; Schnyer, D.; Lafleche, G.; Alexander, M.P. Recovery, long-term cognitive outcome and quality of life following out-of-hospital cardiac arrest. J. Rehabil. Med. 2014, 46, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Fertl, E.; Vass, K.; Sterz, F.; Gabriel, H.; Auff, E. Neurological rehabilitation of severely disabled cardiac arrest survivors. Part I. Course of post-acute inpatient treatment. Resuscitation 2000, 47, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Lundgren-Nilsson, Å.; Rosén, H.; Hofgren, C.; Sunnerhagen, K.S. The first year after successful cardiac resuscitation: Function, activity, participation and quality of life. Resuscitation 2005, 66, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Mateen, F.J.; Josephs, K.A.; Trenerry, M.R.; Felmlee-Devine, M.D.; Weaver, A.L.; Carone, M.; White, R.D. Long-term cognitive outcomes following out-of-hospital cardiac arrest. Neurology 2011, 77, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Czuczwar, M. Cognitive impairment and dementia following ischemic stroke and cardiac arrest in humans. In Brain Ischemia: Alzheimer’s Disease Mechanisms; Pluta, R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2019; pp. 1–10. [Google Scholar]
- Wolters, F.J.; Segufa, R.A.; Darweesh, S.K.L.; Bos, D.; Ikram, M.A.; Sabayan, B.; Hofman, A.; Sedaghat, S. Coronary heart disease, heart failure, and the risk of dementia: A systematic review and meta-analysis. Alzheimers Dement. 2018, 14, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Luo, P. Myocardial Infarction Predisposes Neurodegenerative Diseases. J. Alzheimers Dis. 2020, 74, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Buanes, E.A.; Gramstad, A.; Sovig, K.K.; Hufthammer, K.O.; Flaatten, H.; Husby, T.; Langorgen, J.; Heltne, J.K. Cognitive function and health-related quality of life four years after cardiac arrest. Resuscitation 2015, 89, 13–18. [Google Scholar] [CrossRef]
- Caro-Codon, J.; Rey, J.R.; Lopez-de-Sa, E.; Gonzalez Fernandez, O.; Rosillo, S.O.; Armada, E.; Iniesta, A.M.; Fernandez de Bobadilla, J.; Ruiz Cantador, J.; Rodriguez Sotelo, L.; et al. Long-term neurological outcomes in out-of-hospital cardiac arrest patients treated with targeted-temperature management. Resuscitation 2018, 133, 33–39. [Google Scholar] [CrossRef]
- Byron-Alhassan, A.; Collins, B.; Bedard, M.; Quinlan, B.; Le May, M.; Duchesne, L.; Osborne, C.; Wells, G.; Smith, A.M.; Tulloch, H.E. Cognitive dysfunction after out-of-hospital cardiac arrest: Rate of impairment and clinical predictors. Resuscitation 2021, 165, 154–160. [Google Scholar] [CrossRef]
- Pluta, R. Alzheimer’s disease connected genes in the post-ischemic hippocampus and temporal cortex. Genes 2022, 13, 1059. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kida, E.; Lossinsky, A.S.; Golabek, A.A.; Mossakowski, M.J.; Wisniewski, H.M. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest: I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994, 649, 323–328. [Google Scholar] [CrossRef] [PubMed]
- van Groen, T.; Puurunen, K.; Mäki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse β-amyloid precursor protein and β-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Wu, H.; Yang, Y.; Wang, D.D.; Chen, Y.X.; Gu, Y.H.; Liu, T. Cerebral ischemia and Alzheimer’s disease: The expression of amyloid and apolipoprotein E in human hippocampus. J. Alzheimer’s Dis. 2007, 12, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, H.M.; Maślińska, D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996, 34, 65–71. [Google Scholar] [PubMed]
- Mörtberg, E.; Zetterberg, H.; Nordmark, J.; Blennow, K.; Catry, C.; Decraemer, H.; Vanmechelen, E.; Rubertsson, S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 2011, 55, 1132–1138. [Google Scholar] [CrossRef]
- Randall, J.; Mörtberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2013, 84, 351–356. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Nielsen, N.; Blennow, K.; Dankiewicz, J.; Friberg, H.; Lilja, G.; Insel, P.S.; Rylander, C.; Stammet, P.; et al. Serum tau and neurological outcome in cardiac arrest. Ann. Neurol. 2017, 82, 665–675. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, C.; Liu, C.; Zhan, H.; Li, B.; Lu, Y.; Wei, H.; Cheng, J.; Li, S.; Wang, C.; et al. Identification and Validation of Novel Potential Pathogenesis and Biomarkers to Predict the Neurological Outcome after Cardiac Arrest. Brain Sci. 2022, 12, 928. [Google Scholar] [CrossRef]
- Majd, S.; Power, J.H.; Koblar, S.A.; Grantham, H.J. Early glycogen synthase kinase-3 and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur. J. Neurosci. 2016, 44, 1987–1997. [Google Scholar] [CrossRef]
- Majd, S.; Power, J.H.; Koblar, S.A.; Grantham, H.J.M. Introducing a developed model of reversible cardiac arrest to produce global brain ischemia and its impact on microtubule-associated protein tau phosphorylation at Ser396. Int. J. Neurol. Neurother. 2016, 3, 040. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Brain ischemia as a prelude to Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 636653. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Yuldasheva, N.Y.; Batten, T.F.C.; Pickles, A.R.; Kellett, K.A.B.; Saha, S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia—A potential model for vascular dementia? Neurochem. Int. 2018, 118, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yang, S.H.; Liu, R.; Perez, E.J.; Brun-Ziukemagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta 2007, 1772, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hirano, A.; Katagiri, T.; Sasaki, H.; Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 1988, 23, 620–623. [Google Scholar] [CrossRef]
- Hatsuta, H.; Takao, M.; Nogami, A.; Uchino, A.; Sumikura, H.; Takata, T.; Morimoto, S.; Kanemaru, K.; Adachi, T.; Arai, T.; et al. Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol. Commun. 2019, 7, 49. [Google Scholar] [CrossRef]
- Pluta, R.; Czuczwar, S.J.; Januszewski, S.; Jabłoński, M. The many faces of post-ischemic tau protein in brain neurodegeneration of the Alzheimer’s disease type. Cells 2021, 10, 2213. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Bogucki, J.; Bogucka-Kocka, A.; Czuczwar, S.J. LRP1 and RAGE Genes Transporting Amyloid and Tau Protein in the Hippocampal CA3 Area in an Ischemic Model of Alzheimer’s Disease with 2-Year Survival. Cells 2023, 12, 2763. [Google Scholar] [CrossRef]
- Maślińska, D.; Laure-Kamionowska, M.; Taraszewska, A.; Deręgowski, K.; Maśliński, S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011, 49, 295–300. [Google Scholar]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of autophagy, mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s dis-ease in the rat. J. Alzheimers Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Furmaga-Jabłońska, W.; Brzozowska, J.; et al. Dysregulation of Autophagy, Mitophagy, and Apoptotic Genes in the Medial Temporal Lobe Cortex in an Ischemic Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 54, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Kriska, J.; Hermanova, Z.; Knotek, T.; Tureckova, J.; Anderova, M. On the Common Journey of Neural Cells through Ischemic Brain Injury and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9689. [Google Scholar] [CrossRef] [PubMed]
- Elman-Shina, K.; Efrati, S. Ischemia as a common trigger for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1012779. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Miners, J.S.; Love, S. Pathological changes within the cerebral vasculature in Alzheimer’s disease: New perspectives. Brain Pathol. 2022, 32, e13061. [Google Scholar] [CrossRef]
- Lecordier, S.; Pons, V.; Rivest, S.; ElAli, A. Multifocal Cerebral Microinfarcts Modulate Early Alzheimer’s Disease Pathology in a Sex-Dependent Manner. Front. Immunol. 2022, 12, 813536. [Google Scholar] [CrossRef]
- Das, T.K.; Ganesh, B.P.; Fatima-Shad, K. Common signaling pathways involved in Alzheimer’s disease and stroke: Two Faces of the Same Coin. J. Alzheimers Dis. Rep. 2023, 7, 381–398. [Google Scholar] [CrossRef]
- Eisenmenger, L.B.; Peret, A.; Famakin, B.M.; Spahic, A.; Roberts, G.S.; Bockholt, J.H.; Johnson, K.M.; Paulsen, J.S. Vascular contributions to Alzheimer’s disease. Transl. Res. 2023, 254, 41–53. [Google Scholar] [CrossRef]
- Chen, C.; Liu, C.; Niu, Z.; Li, M.; Zhang, Y.; Gao, R.; Chen, H.; Wang, Q.; Zhang, S.; Zhou, R.; et al. RNA-seq analysis of the key long noncoding RNAs and mRNAs related to cognitive impairment after cardiac arrest and cardiopulmonary resuscitation. Aging 2020, 12, 14490–14505. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Tuxiu, X.; Xiaobing, L.; Guijun, J.; Lulu, K.; Jie, J.; Lu, Y.; Liying, Z.; Xiaoxing, X.; Jingjun, L. LncRNA GAS5/miR-137 Is a Hypoxia-Responsive Axis Involved in Cardiac Arrest and Cardiopulmonary Cerebral Resuscitation. Front. Immunol. 2022, 12, 790750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Peng, F.; Zhang, L.; Kang, K.; Yang, M.; Chen, C.; Yu, H. Long noncoding RNA upregulates adapter ShcA protein expression to promote cognitive impairment after cardiac arrest and resuscitation. Shock 2022, 58, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.J.; Xu, J.; Lahousse, S.A.; Caggiano, N.L.; de la Monte, S.M. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: Potential strategies for neuroprotection. J. Alzheimers Dis. 2003, 5, 209–228. [Google Scholar] [CrossRef]
- Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Januszewski, S.; Jabłoński, M.; Gil-Kulik, P.; Brzozowska, J.; Petniak, A.; Furmaga-Jabłońska, W.; Bogucki, J.; et al. Dysregulation of Amyloid-β Protein Precursor, β-Secretase, Presenilin 1 and 2 Genes in the Rat Selectively Vulnerable CA1 Subfield of Hippocampus Following Transient Global Brain Ischemia. J. Alzheimers Dis. 2015, 47, 1047–1056. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Brzozowska, J.; Furmaga-Jabłońska, W.; et al. Discrepancy in Expression of β-Secretase and Amyloid-β Protein Precursor in Alzheimer-Related Genes in the Rat Medial Temporal Lobe Cortex Following Transient Global Brain Ischemia. J. Alzheimers Dis. 2016, 51, 1023–1031. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Gil-Kulik, P.; Januszewski, S.; Jabłoński, M.; Petniak, A.; Brzozowska, J.; Bogucki, J.; et al. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol. Rep. 2016, 68, 155–161. [Google Scholar] [CrossRef]
- Pluta, R.; Bogucka-Kocka, A.; Ułamek-Kozioł, M.; Bogucki, J.; Januszewski, S.; Kocki, J.; Czuczwar, S.J. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2018, 70, 881–884. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Kocki, J.; Bogucki, J.; Januszewski, S.; Bogucka-Kocka, A.; Czuczwar, S.J. Expression of the tau protein and amyloid protein precursor processing genes in the CA3 Area of the hippocampus in the ischemic model of Alzheimer’s Disease in the Rat. Mol. Neurobiol. 2020, 57, 1281–1290. [Google Scholar] [CrossRef]

{kind=link}
| Genes Survival | APP | ADAM10 | BACE1 | PSEN1 | PSEN2 | MAPT |
|---|---|---|---|---|---|---|
| CA1 subfield of hippocampus | ||||||
| 2 days | ↑ | N.A. | ↑↑ | ↑ | ↑↑ | ↑↑ |
| 7 days | ↑ | N.A. | ↑ | ↑ | ↑ | ↔ |
| 30 days | ↑ | N.A. | ↓ | ↓ | ↓ | ↔ |
| CA3 subfield of hippocampus | ||||||
| 2 days | ↔ | ↓ | ↓ | ↑ | ↔ | ↔ |
| 7 days | ↑ | ↓ | ↓ | ↑ | ↓ | ↑ |
| 30 days | ↔ | ↓ | ↑ | ↔ | ↑ | ↑ |
| Medial temporal cortex | ||||||
| 2 days | ↓ | N.A. | ↑↑ | ↔ | ↑↑ | N.A. |
| 7 days | ↑ | N.A. | ↔ | ↔ | ↔ | N.A. |
| 30 days | ↑ | N.A. | ↔ | ↔ | ↔ | N.A. |
| Survival Genes | 2 Days | 7 Days | 30 Days | 12 Months | 18 Months | 24 Months |
|---|---|---|---|---|---|---|
| LRP1 | ↓ | ↓ | ↔ | ↑ | ↑↑ | ↑ |
| RAGE | ↓ | ↑ | ↑ | ↓↓ | ↓ | ↓ |
| Genes Survival | BECN1 | BNIP3 | CASP3 |
|---|---|---|---|
| CA1 subfield of hippocampus | |||
| 2 days | ↔ | ↑ | ↑↑↑ |
| 7 days | ↔ | ↔ | ↑ |
| 30 days | ↔ | ↔ | ↓ |
| CA3 subfield of hippocampus | |||
| 2 days | ↔ | ↓ | ↓ |
| 7 days | ↓ | ↓ | ↑ |
| 30 days | ↑ | ↓ | ↑ |
| Medial temporal cortex | |||
| 2 days | ↑ | ↓↓ | ↓ |
| 7 days | ↔ | ↑↑↑ | ↑ |
| 30 days | ↔ | ↑ | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pluta, R.; Czuczwar, S.J. Ischemia-Reperfusion Programming of Alzheimer’s Disease-Related Genes—A New Perspective on Brain Neurodegeneration after Cardiac Arrest. Int. J. Mol. Sci. 2024, 25, 1291. https://doi.org/10.3390/ijms25021291
Pluta R, Czuczwar SJ. Ischemia-Reperfusion Programming of Alzheimer’s Disease-Related Genes—A New Perspective on Brain Neurodegeneration after Cardiac Arrest. International Journal of Molecular Sciences. 2024; 25(2):1291. https://doi.org/10.3390/ijms25021291
Chicago/Turabian StylePluta, Ryszard, and Stanisław J. Czuczwar. 2024. "Ischemia-Reperfusion Programming of Alzheimer’s Disease-Related Genes—A New Perspective on Brain Neurodegeneration after Cardiac Arrest" International Journal of Molecular Sciences 25, no. 2: 1291. https://doi.org/10.3390/ijms25021291
APA StylePluta, R., & Czuczwar, S. J. (2024). Ischemia-Reperfusion Programming of Alzheimer’s Disease-Related Genes—A New Perspective on Brain Neurodegeneration after Cardiac Arrest. International Journal of Molecular Sciences, 25(2), 1291. https://doi.org/10.3390/ijms25021291