
Targeting Mitochondrial Dynamics during Lower-Limb Ischemia Reperfusion in Young and Old Mice: Effect of Mitochondrial Fission Inhibitor-1 (mDivi-1)

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
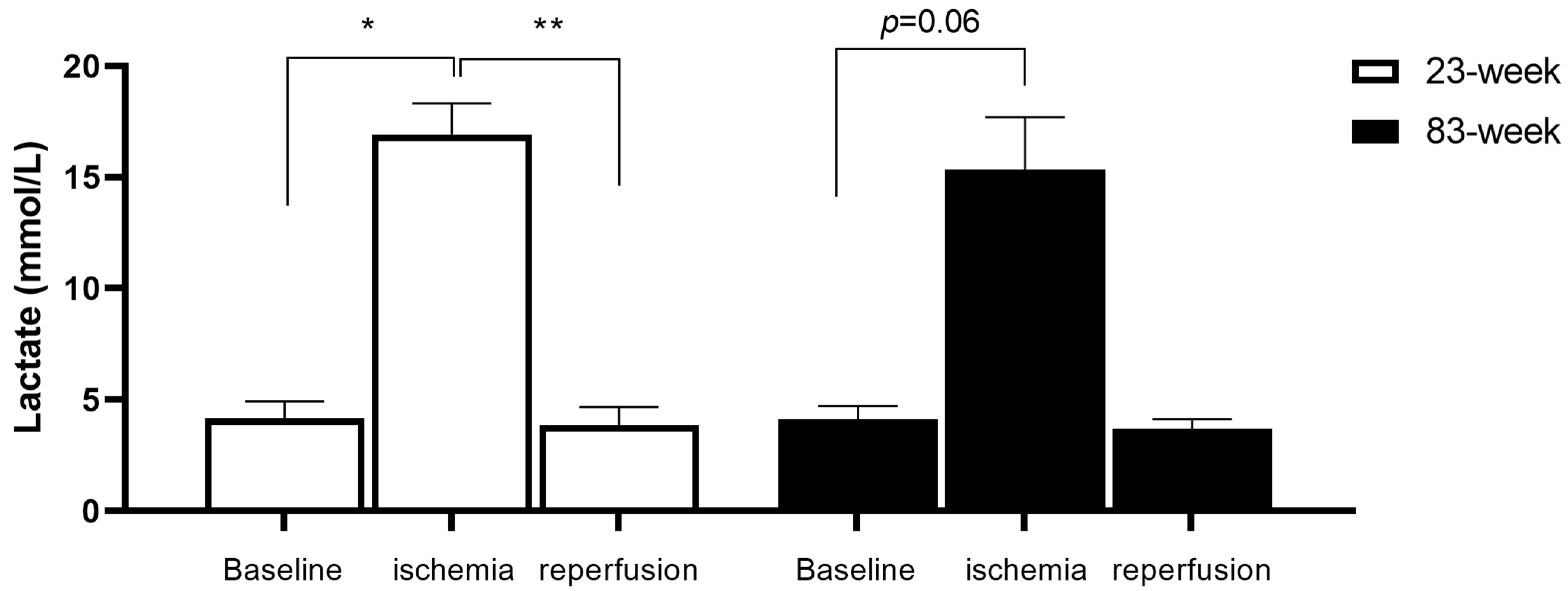
2.1. Effects of IR on Systemic Lactate in mDivi-1-Treated Young and Old Mice
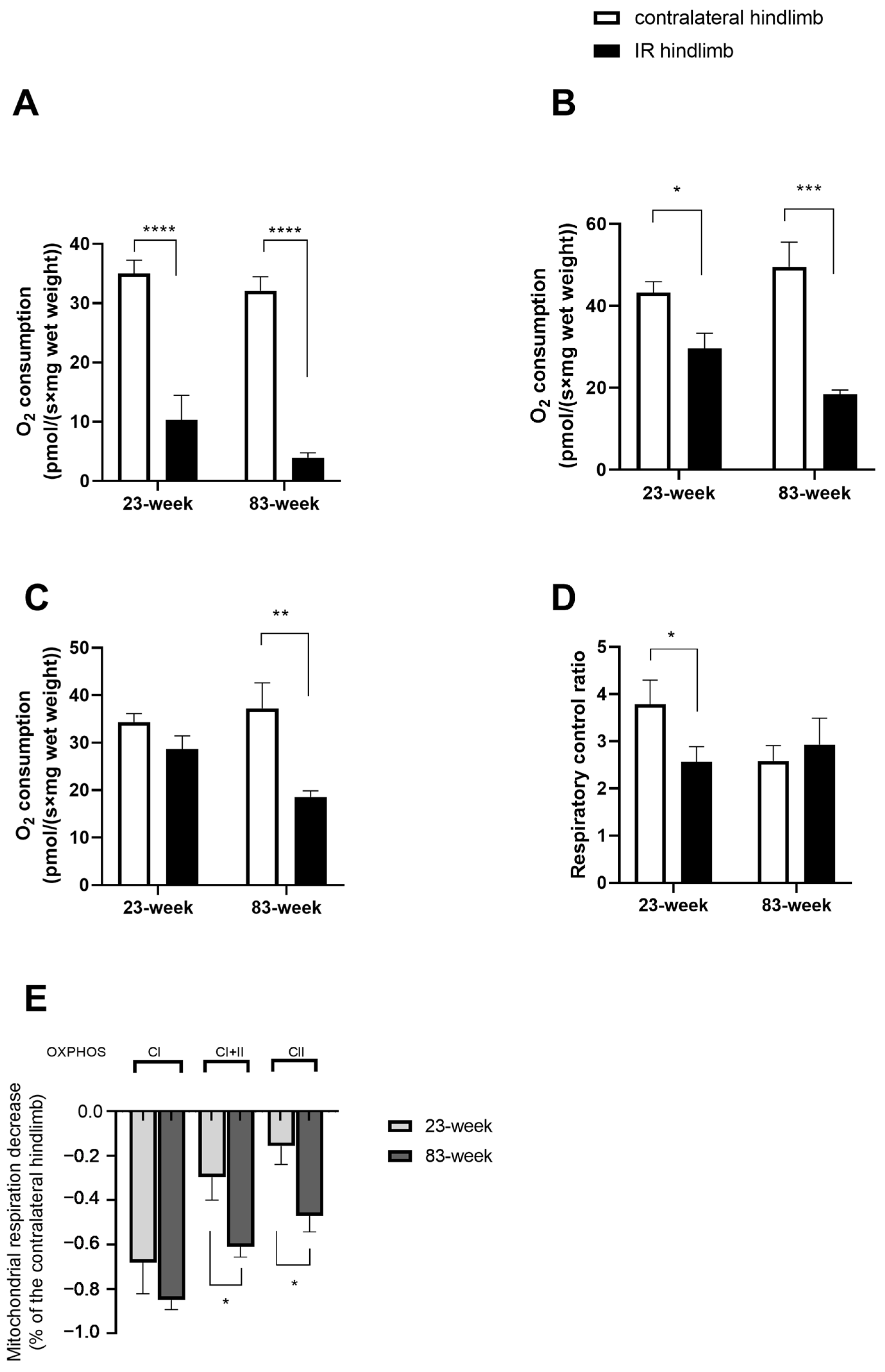
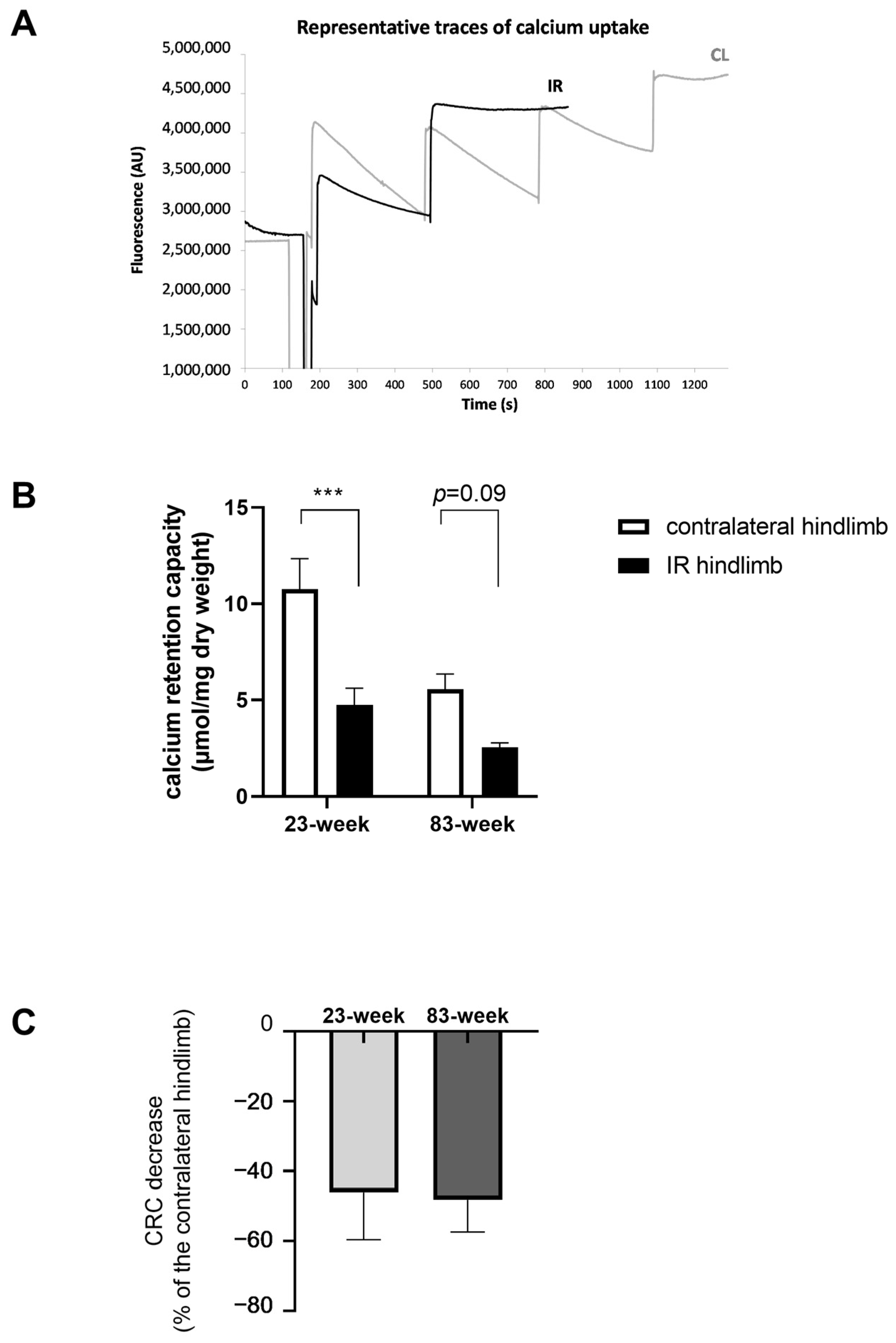
2.2. Effects of IR on Mitochondrial Respiration and Calcium Retention Capacity in mDivi-1-Treated Young and Old Mice
2.2.1. Mitochondrial Respiration
2.2.2. Mitochondrial Calcium Retention Capacity
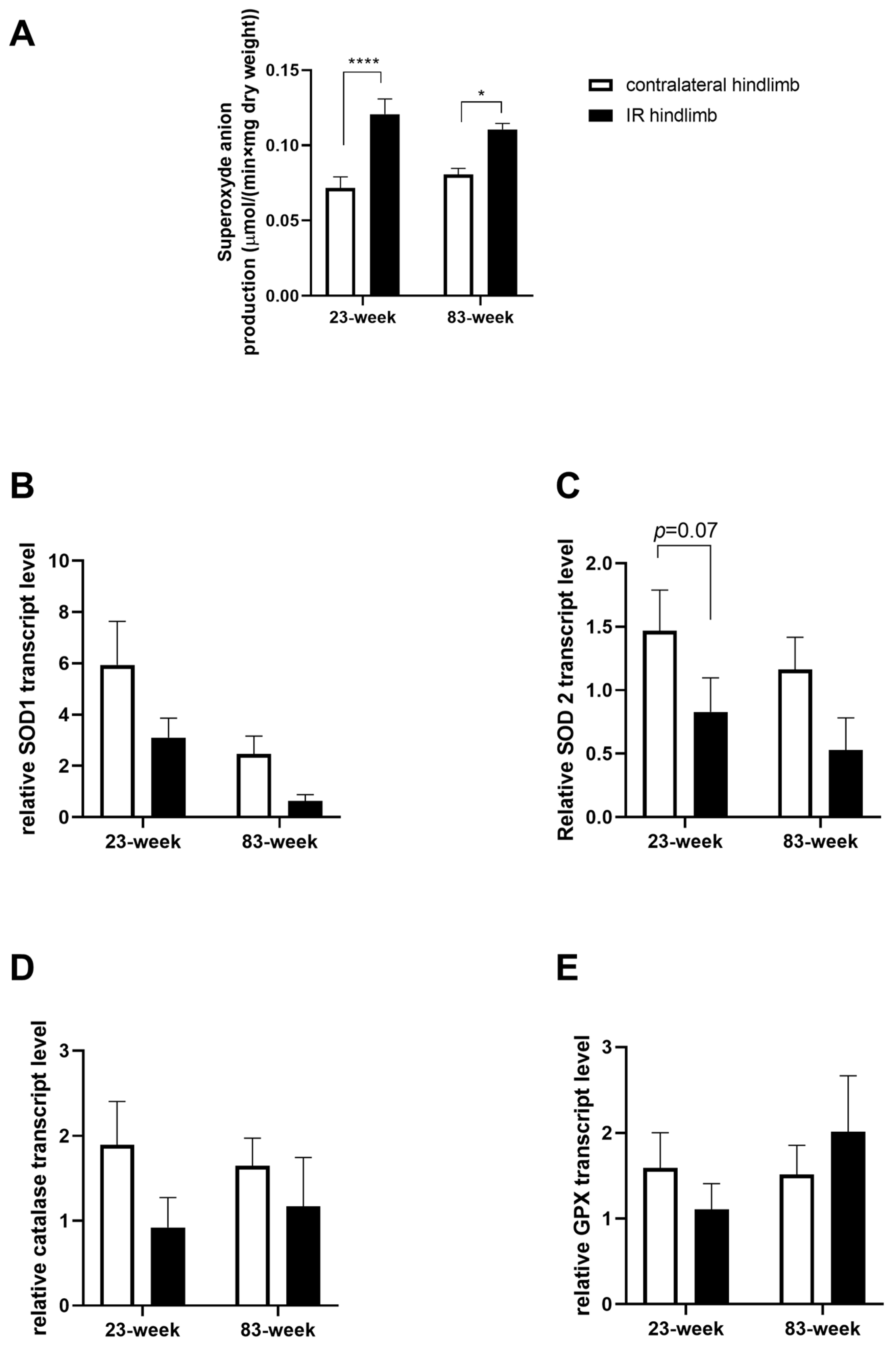
2.3. Effects of IR on Oxidative Balance in mDivi-1-Treated Young and Old Mice
2.3.1. Production of Superoxide Anion
2.3.2. Antioxidant System
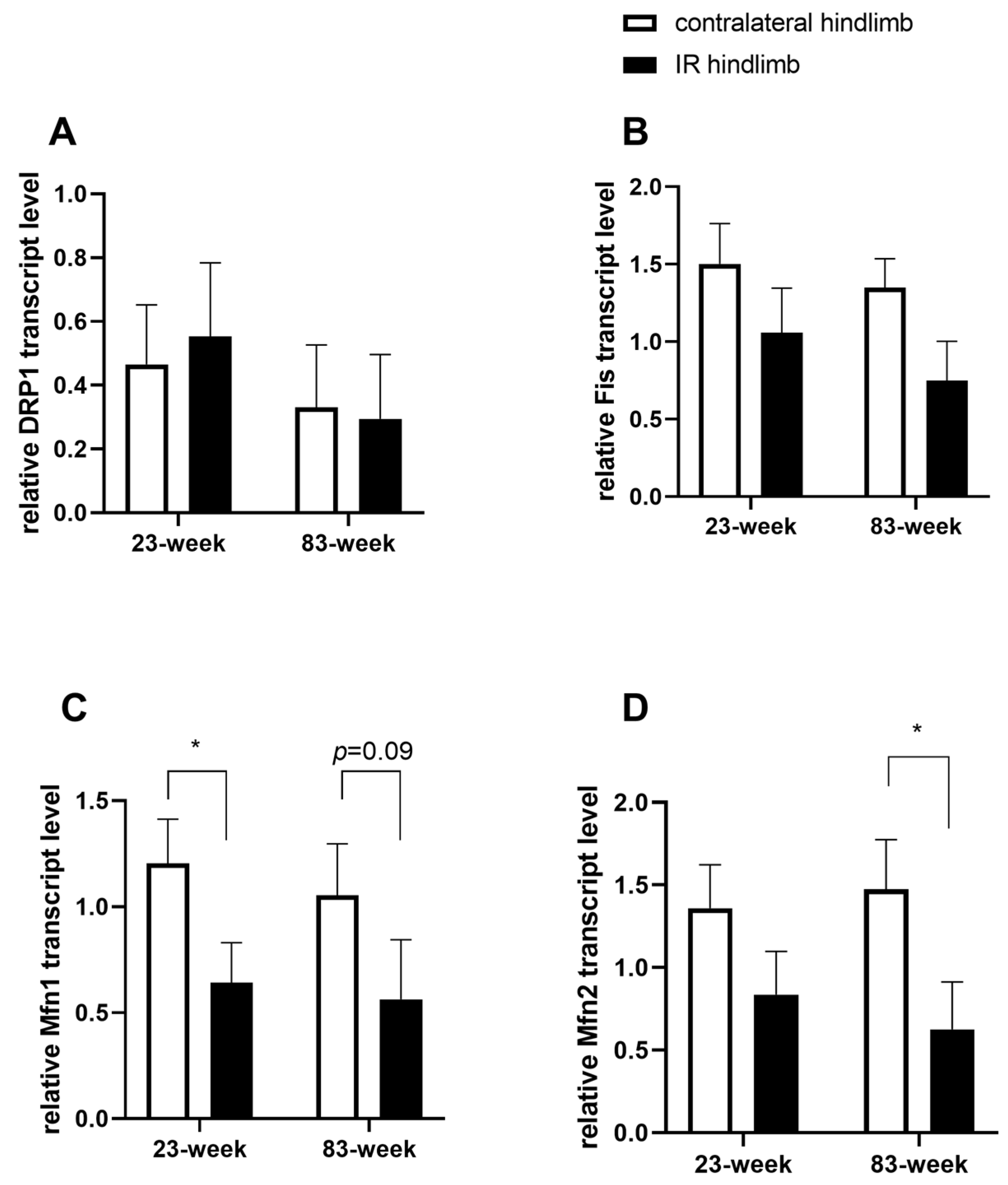
2.4. Effects of IR on Mitochondrial Dynamics in mDivi-1-Treated Young and Old Mice
3. Discussion
3.1. Effect of Age on IR-Induced Deleterious Effects
3.2. Inhibition of Mitochondrial Fission as a Therapeutic Option
3.3. No Protective Effect of mDivi-1 on Skeletal Muscle Ischemia Reperfusion-Induced Deleterious Effects
3.4. Effect of mDivi-1 on Mitochondrial Dynamics in the Setting of Lower-Limb Ischemia Reperfusion
3.5. Limitations of the Study
4. Materials and Methods
4.1. Animals
4.2. Experimental Procedure and Muscle Sampling
4.3. Permeabilization of Skeletal Muscle Fibers
4.4. Study of Mitochondrial Respiration by Oxymetry
4.5. Calcium Retention Capacity Evaluation in Ghost Fibers
4.6. Reactive Oxygen Species Production Measurement by Electron Paramagnetic Resonance Spectroscopy
4.7. RNA Transcripts Encoding for Antioxidant Defense and Mitochondrial Dynamic
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campia, U.; Gerhard-Herman, M.; Piazza, G.; Goldhaber, S.Z. Peripheral Artery Disease: Past, Present, and Future. Am. J. Med. 2019, 132, 1133–1141. [Google Scholar] [CrossRef]
- Criqui, M.H.; Aboyans, V. Epidemiology of Peripheral Artery Disease. Circ. Res. 2015, 116, 1509–1526. [Google Scholar] [CrossRef]
- Dua, A.; Lee, C.J. Epidemiology of Peripheral Arterial Disease and Critical Limb Ischemia. Tech. Vasc. Interv. Radiol. 2016, 19, 91–95. [Google Scholar] [CrossRef]
- Gardner, A.W.; Afaq, A. Management of Lower Extremity Peripheral Arterial Disease. J. Cardiopulm. Rehabil. Prev. 2008, 28, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, N.M.; Creager, M.A. Pathophysiology of Intermittent Claudication in Peripheral Artery Disease. Circ. J. 2017, 81, 281–289. [Google Scholar] [CrossRef]
- Pizzimenti, M.; Riou, M.; Charles, A.-L.; Talha, S.; Meyer, A.; Andres, E.; Chakfé, N.; Lejay, A.; Geny, B. The Rise of Mitochondria in Peripheral Arterial Disease Physiopathology: Experimental and Clinical Data. J. Clin. Med. 2019, 8, 2125. [Google Scholar] [CrossRef]
- Jongkind, V.; Earnshaw, J.J.; Bastos Gonçalves, F.; Cochennec, F.; Debus, E.S.; Hinchliffe, R.; Menyhei, G.; Svetlikov, A.V.; Tshomba, Y.; Van Den Berg, J.C.; et al. Editor’s Choice—Update of the European Society for Vascular Surgery (ESVS) 2020 Clinical Practice Guidelines on the Management of Acute Limb Ischaemia in Light of the COVID-19 Pandemic, Based on a Scoping Review of the Literature. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 80–89. [Google Scholar] [CrossRef]
- Moras, E.; Khan, M.I.; Song, D.D.; Syed, M.; Prabhakaran, S.Y.; Gandhi, K.D.; Lavie, C.J.; Alam, M.; Sharma, R.; Krittanawong, C. Pharmacotherapy and Revascularization Strategies of Peripheral Artery Disease. Curr. Probl. Cardiol. 2024, 49, 102430. [Google Scholar] [CrossRef]
- Fitridge, R.; Chuter, V.; Mills, J.; Hinchliffe, R.; Azuma, N.; Behrendt, C.-A.; Boyko, E.J.; Conte, M.S.; Humphries, M.; Kirksey, L.; et al. The Intersocietal IWGDF, ESVS, SVS Guidelines on Peripheral Artery Disease in People with Diabetes Mellitus and a Foot Ulcer. J. Vasc. Surg. 2023, 78, 1101–1131. [Google Scholar] [CrossRef]
- Lejay, A.; Meyer, A.; Schlagowski, A.-I.; Charles, A.-L.; Singh, F.; Bouitbir, J.; Pottecher, J.; Chakfé, N.; Zoll, J.; Geny, B. Mitochondria: Mitochondrial Participation in Ischemia-Reperfusion Injury in Skeletal Muscle. Int. J. Biochem. Cell Biol. 2014, 50, 101–105. [Google Scholar] [CrossRef]
- Paradis, S.; Charles, A.-L.; Meyer, A.; Lejay, A.; Scholey, J.W.; Chakfé, N.; Zoll, J.; Geny, B. Chronology of Mitochondrial and Cellular Events during Skeletal Muscle Ischemia-Reperfusion. Am. J. Physiol.-Cell Physiol. 2016, 310, C968–C982. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Brown, D.A.; Neufer, P.D.; McClung, J.M. Mitochondrial Regulation of the Muscle Microenvironment in Critical Limb Ischemia. Front. Physiol. 2015, 6, 336. [Google Scholar] [CrossRef] [PubMed]
- Mansour, Z.; Bouitbir, J.; Charles, A.L.; Talha, S.; Kindo, M.; Pottecher, J.; Zoll, J.; Geny, B. Remote and Local Ischemic Preconditioning Equivalently Protects Rat Skeletal Muscle Mitochondrial Function during Experimental Aortic Cross-Clamping. J. Vasc. Surg. 2012, 55, 497–505.e1. [Google Scholar] [CrossRef][Green Version]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial Control of Cellular Life, Stress, and Death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef]
- Curcio, A.; Panarello, A.; Spaccarotella, C.; Indolfi, C. Cardiovascular Prognosis in Patients with Peripheral Artery Disease and Approach to Therapy. Biomedicines 2023, 11, 3131. [Google Scholar] [CrossRef] [PubMed]
- Rontoyanni, V.G.; Blears, E.; Nunez Lopez, O.; Ogunbileje, J.; Moro, T.; Bhattarai, N.; Randolph, A.C.; Fry, C.S.; Fankhauser, G.T.; Cheema, Z.F.; et al. Skeletal Muscle Bioenergetics in Critical Limb Ischemia and Diabetes. J. Surg. Res. 2023, 288, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Swanson, S.A.; Haynatzki, G.; Koutakis, P.; Johanning, J.M.; Reppert, P.R.; Papoutsi, E.; Miserlis, D.; Zhu, Z.; Casale, G.P.; et al. Protein Concentration and Mitochondrial Content in the Gastrocnemius Predicts Mortality Rates in Patients with Peripheral Arterial Disease. Ann. Surg. 2015, 261, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Pizzimenti, M.; Meyer, A.; Charles, A.-L.; Giannini, M.; Chakfé, N.; Lejay, A.; Geny, B. Sarcopenia and Peripheral Arterial Disease: A Systematic Review. J. Cachexia Sarcopenia Muscle 2020, 11, 866–886. [Google Scholar] [CrossRef] [PubMed]
- Pottecher, J.; Guillot, M.; Belaidi, E.; Charles, A.-L.; Lejay, A.; Gharib, A.; Diemunsch, P.; Geny, B. Cyclosporine A Normalizes Mitochondrial Coupling, Reactive Oxygen Species Production, and Inflammation and Partially Restores Skeletal Muscle Maximal Oxidative Capacity in Experimental Aortic Cross-Clamping. J. Vasc. Surg. 2013, 57, 1100–1108.e2. [Google Scholar] [CrossRef]
- Calo, L.; Dong, Y.; Kumar, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Dynamics: An Emerging Paradigm in Ischemia-Reperfusion Injury. Curr. Pharm. Des. 2013, 19, 6848–6857. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, Y. Mitochondrial Fission and Fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New Insights into the Function and Regulation of Mitochondrial Fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, X.; Li, H.; Jiang, H.; Zhou, J.; Zhang, S.; Guo, J.; Shen, L.; Yang, H.; Lin, J.; et al. The Role of Mitochondrial Dynamics in Disease. MedComm 2023, 4, e462. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, K.-Y.; Lindsey, J.D.; Dai, Y.; Heo, H.; Nguyen, D.H.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.-K. A Selective Inhibitor of Drp1, Mdivi-1, Increases Retinal Ganglion Cell Survival in Acute Ischemic Mouse Retina. Invest. Ophthalmol. Vis. Sci. 2011, 52, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Sumida, M.; Doi, K.; Ogasawara, E.; Yamashita, T.; Hamasaki, Y.; Kariya, T.; Takimoto, E.; Yahagi, N.; Nangaku, M.; Noiri, E. Regulation of Mitochondrial Dynamics by Dynamin-Related Protein-1 in Acute Cardiorenal Syndrome. J. Am. Soc. Nephrol. 2015, 26, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, P.; Li, S.; Wang, S.; Li, Y.; Liang, N.; Wang, M. Mdivi-1 Prevents Apoptosis Induced by Ischemia-Reperfusion Injury in Primary Hippocampal Cells via Inhibition of Reactive Oxygen Species-Activated Mitochondrial Pathway. J. Stroke Cerebrovasc. Dis. 2014, 23, 1491–1499. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, S.; Li, Y.; Che, L.; Zhao, Q. A Selective Inhibitor of Drp1, Mdivi-1, Acts against Cerebral Ischemia/Reperfusion Injury via an Anti-Apoptotic Pathway in Rats. Neurosci. Lett. 2013, 535, 104–109. [Google Scholar] [CrossRef]
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Pharmacological Inhibition of Mitochondrial Fission Attenuates Cardiac Ischemia-Reperfusion Injury in Pre-Diabetic Rats. Biochem. Pharmacol. 2020, 182, 114295. [Google Scholar] [CrossRef]
- Ong, S.-B.; Kwek, X.-Y.; Katwadi, K.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Ismail, N.I.; Lin, Y.-H.; Yap, E.P.; Lim, S.-Y.; Ja, K.P.M.M.; et al. Targeting Mitochondrial Fission Using Mdivi-1 in A Clinically Relevant Large Animal Model of Acute Myocardial Infarction: A Pilot Study. Int. J. Mol. Sci. 2019, 20, 3972. [Google Scholar] [CrossRef] [PubMed]
- Ueta, C.B.; Gomes, K.S.; Ribeiro, M.A.; Mochly-Rosen, D.; Ferreira, J.C.B. Disruption of Mitochondrial Quality Control in Peripheral Artery Disease: New Therapeutic Opportunities. Pharmacol. Res. 2017, 115, 96–106. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, H. Mitochondrial Quality Control Mechanisms as Molecular Targets in Cardiac Ischemia-Reperfusion Injury. Acta Pharm. Sin. B 2020, 10, 1866–1879. [Google Scholar] [CrossRef]
- Paradis, S.; Charles, A.-L.; Georg, I.; Goupilleau, F.; Meyer, A.; Kindo, M.; Laverny, G.; Metzger, D.; Geny, B. Aging Exacerbates Ischemia-Reperfusion-Induced Mitochondrial Respiration Impairment in Skeletal Muscle. Antioxidants 2019, 8, 168. [Google Scholar] [CrossRef]
- Charles, A.-L.; Charloux, A.; Vogel, T.; Raul, J.-S.; Kindo, M.; Wolff, V.; Geny, B. Cumulative Deleterious Effects of Tetrahydrocannabinoid (THC) and Ethanol on Mitochondrial Respiration and Reactive Oxygen Species Production Are Enhanced in Old Isolated Cardiac Mitochondria. Int. J. Mol. Sci. 2024, 25, 1835. [Google Scholar] [CrossRef] [PubMed]
- Kugler, B.A.; Deng, W.; Duguay, A.L.; Garcia, J.P.; Anderson, M.C.; Nguyen, P.D.; Houmard, J.A.; Zou, K. Pharmacological Inhibition of Dynamin-Related Protein 1 Attenuates Skeletal Muscle Insulin Resistance in Obesity. Physiol. Rep. 2021, 9, e14808. [Google Scholar] [CrossRef]
- Charles, A.-L.; Meyer, A.; Dal-Ros, S.; Auger, C.; Keller, N.; Ramamoorthy, T.G.; Zoll, J.; Metzger, D.; Schini-Kerth, V.; Geny, B. Polyphenols Prevent Ageing-Related Impairment in Skeletal Muscle Mitochondrial Function through Decreased Reactive Oxygen Species Production. Exp. Physiol. 2013, 98, 536–545. [Google Scholar] [CrossRef]
- Avci, G.; Kadioglu, H.; Sehirli, A.O.; Bozkurt, S.; Guclu, O.; Arslan, E.; Muratli, S.K. Curcumin Protects against Ischemia/Reperfusion Injury in Rat Skeletal Muscle. J. Surg. Res. 2012, 172, e39–e46. [Google Scholar] [CrossRef] [PubMed]
- Bolcal, C.; Yildirim, V.; Doganci, S.; Sargin, M.; Aydin, A.; Eken, A.; Ozal, E.; Kuralay, E.; Demirkilic, U.; Tatar, H. Protective Effects of Antioxidant Medications on Limb Ischemia Reperfusion Injury. J. Surg. Res. 2007, 139, 274–279. [Google Scholar] [CrossRef]
- Hori, K.; Tsujii, M.; Iino, T.; Satonaka, H.; Uemura, T.; Akeda, K.; Hasegawa, M.; Uchida, A.; Sudo, A. Protective Effect of Edaravone for Tourniquet-Induced Ischemia-Reperfusion Injury on Skeletal Muscle in Murine Hindlimb. BMC Musculoskelet. Disord. 2013, 14, 113. [Google Scholar] [CrossRef]
- Lejay, A.; Paradis, S.; Lambert, A.; Charles, A.-L.; Talha, S.; Enache, I.; Thaveau, F.; Chakfe, N.; Geny, B. N-Acetyl Cysteine Restores Limb Function, Improves Mitochondrial Respiration, and Reduces Oxidative Stress in a Murine Model of Critical Limb Ischaemia. Eur. J. Vasc. Endovasc. Surg. 2018, 56, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-M.; Shih, J.-M.; Pai, M.-H.; Hou, Y.-C.; Yeh, C.-L.; Yeh, S.-L. Glutamine Administration After Sublethal Lower Limb Ischemia Reduces Inflammatory Reaction and Offers Organ Protection in Ischemia/Reperfusion Injury. JPEN J. Parenter. Enter. Nutr. 2016, 40, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Talha, S.; Bouitbir, J.; Charles, A.-L.; Zoll, J.; Goette-Di Marco, P.; Meziani, F.; Piquard, F.; Geny, B. Pretreatment with Brain Natriuretic Peptide Reduces Skeletal Muscle Mitochondrial Dysfunction and Oxidative Stress after Ischemia-Reperfusion. J. Appl. Physiol. 2013, 114, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.P.; Tu, H.; Liu, J.; Muelleman, R.L.; Li, Y.-L. Mitochondria-Derived Superoxide Links to Tourniquet-Induced Apoptosis in Mouse Skeletal Muscle. PLoS ONE 2012, 7, e43410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thaveau, F.; Zoll, J.; Rouyer, O.; Chafke, N.; Kretz, J.G.; Piquard, F.; Geny, B. Ischemic Preconditioning Specifically Restores Complexes I and II Activities of the Mitochondrial Respiratory Chain in Ischemic Skeletal Muscle. J. Vasc. Surg. 2007, 46, 541–547; discussion 547. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Disatnik, M.-H.; Ferreira, J.C.B.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.M.; Qi, X.; Mochly-Rosen, D. Acute Inhibition of Excessive Mitochondrial Fission after Myocardial Infarction Prevents Long-Term Cardiac Dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.-T.; Shaw, R.M.; Zhu, J. Dynasore Protects Mitochondria and Improves Cardiac Lusitropy in Langendorff Perfused Mouse Heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sesaki, H.; Qi, X. Drp1 Stabilizes P53 on the Mitochondria to Trigger Necrosis under Oxidative Stress Conditions in Vitro and in Vivo. Biochem. J. 2014, 461, 137–146. [Google Scholar] [CrossRef]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Reddy, P.H. Inhibitors of Mitochondrial Fission as a Therapeutic Strategy for Diseases with Oxidative Stress and Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef]
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 2547–2564. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.A.; Eguchi, S. Inhibition of Mitochondrial Fission as a Novel Therapeutic Strategy to Reduce Mortality upon Myocardial Infarction. Clin. Sci. 2018, 132, 2163–2167. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Youle, R.J. A Chemical Inhibitor of DRP1 Uncouples Mitochondrial Fission and Apoptosis. Mol. Cell 2008, 29, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Ding, H.; Chen, F.; Zhao, Y.; Yang, Q.; Dong, Q. Mdivi-1 Protects Against Ischemic Brain Injury via Elevating Extracellular Adenosine in a cAMP/CREB-CD39-Dependent Manner. Mol. Neurobiol. 2016, 53, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Grohm, J.; Kim, S.-W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 Provides Neuroprotection In Vitro and In Vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Differential Temporal Inhibition of Mitochondrial Fission by Mdivi-1 Exerts Effective Cardioprotection in Cardiac Ischemia/Reperfusion Injury. Clin. Sci. 2018, 132, 1669–1683. [Google Scholar] [CrossRef] [PubMed]
- Rosdah, A.A.; Bond, S.T.; Sivakumaran, P.; Hoque, A.; Oakhill, J.S.; Drew, B.G.; Delbridge, L.M.D.; Lim, S.Y. Mdivi-1 Protects Human W8B2+ Cardiac Stem Cells from Oxidative Stress and Simulated Ischemia-Reperfusion Injury. Stem Cells Dev. 2017, 26, 1771–1780. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-Related Protein 1 (Drp1)-Mediated Diastolic Dysfunction in Myocardial Ischemia-Reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Tian, Y.; Li, B.; Shi, W.-Z.; Chang, M.-Z.; Zhang, G.-J.; Di, Z.-L.; Liu, Y. Dynamin-Related Protein 1 Inhibitors Protect against Ischemic Toxicity through Attenuating Mitochondrial Ca2+ Uptake from Endoplasmic Reticulum Store in PC12 Cells. Int. J. Mol. Sci. 2014, 15, 3172–3185. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Cui, M.; Chen, S.-F.; Dong, Q.; Liu, X.-Y. Amelioration of Ischemic Mitochondrial Injury and Bax-Dependent Outer Membrane Permeabilization by Mdivi-1. CNS Neurosci. Ther. 2014, 20, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.K.; Austen, W.G.; Ibrahim, S.; Ding, G.Y.; Verna, N.; Hechtman, H.B.; Moore, F.D. Reperfusion Injury to Skeletal Muscle Affects Primarily Type II Muscle Fibers. J. Surg. Res. 2004, 122, 54–60. [Google Scholar] [CrossRef]
- Charles, A.-L.; Guilbert, A.-S.; Guillot, M.; Talha, S.; Lejay, A.; Meyer, A.; Kindo, M.; Wolff, V.; Bouitbir, J.; Zoll, J.; et al. Muscles Susceptibility to Ischemia-Reperfusion Injuries Depends on Fiber Type Specific Antioxidant Level. Front. Physiol. 2017, 8, 52. [Google Scholar] [CrossRef]
- Flück, M.; von Allmen, R.S.; Ferrié, C.; Tevaearai, H.; Dick, F. Protective Effect of Focal Adhesion Kinase against Skeletal Muscle Reperfusion Injury after Acute Limb Ischemia. Eur. J. Vasc. Endovasc. Surg. 2015, 49, 306–313. [Google Scholar] [CrossRef][Green Version]
- Woitaske, M.D.; McCarter, R.J. Effects of Fiber Type on Ischemia-Reperfusion Injury in Mouse Skeletal Muscle. Plast. Reconstr. Surg. 1998, 102, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Davidson, M.M.; Zhou, H.; Wang, C.; Walker, W.F.; Hei, T.K. Cytoplasmic Irradiation Results in Mitochondrial Dysfunction and DRP1-Dependent Mitochondrial Fission. Cancer Res. 2013, 73, 6700–6710. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.-R.; Wang, X.; Hu, W.-W.; et al. Cerebral Ischemia-Reperfusion-Induced Autophagy Protects against Neuronal Injury by Mitochondrial Clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal Ischemia/Reperfusion-Induced Mitophagy Protects against Renal Dysfunction via Drp1-Dependent-Pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Ishikita, A.; Matoba, T.; Ikeda, G.; Koga, J.-I.; Mao, Y.; Nakano, K.; Takeuchi, O.; Sadoshima, J.; Egashira, K. Nanoparticle-Mediated Delivery of Mitochondrial Division Inhibitor 1 to the Myocardium Protects the Heart From Ischemia-Reperfusion Injury Through Inhibition of Mitochondria Outer Membrane Permeabilization: A New Therapeutic Modality for Acute Myocardial Infarction. J. Am. Heart Assoc. 2016, 5, e003872. [Google Scholar] [CrossRef]
- Yu, J.; Maimaitili, Y.; Xie, P.; Wu, J.J.; Wang, J.; Yang, Y.N.; Ma, H.P.; Zheng, H. High Glucose Concentration Abrogates Sevoflurane Post-Conditioning Cardioprotection by Advancing Mitochondrial Fission but Dynamin-Related Protein 1 Inhibitor Restores These Effects. Acta Physiol. 2017, 220, 83–98. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor Mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor That Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Hood, D.A. Oxidative Stress-Induced Mitochondrial Fragmentation and Movement in Skeletal Muscle Myoblasts. Am. J. Physiol. Cell Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Liu, S.; Iida, T.; Yi, H.; Huang, W.; Levitt, R.C.; Lubarsky, D.A.; Candiotti, K.A.; Hao, S. Inhibition of Mitochondrial Fission Protein Reduced Mechanical Allodynia and Suppressed Spinal Mitochondrial Superoxide Induced by Perineural Human Immunodeficiency Virus Gp120 in Rats. Anesth. Analg. 2016, 122, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, B.; Sun, F.; Li, Y.; Li, Q.; Lang, H.; Zhao, Z.; Gao, P.; Zhao, Y.; Shang, Q.; et al. Mitochondrial Respiratory Dysfunctions of Blood Mononuclear Cells Link with Cardiac Disturbance in Patients with Early-Stage Heart Failure. Sci. Rep. 2015, 5, 10229. [Google Scholar] [CrossRef]
- Troncoso, R.; Paredes, F.; Parra, V.; Gatica, D.; Vásquez-Trincado, C.; Quiroga, C.; Bravo-Sagua, R.; López-Crisosto, C.; Rodriguez, A.E.; Oyarzún, A.P.; et al. Dexamethasone-Induced Autophagy Mediates Muscle Atrophy through Mitochondrial Clearance. Cell Cycle 2014, 13, 2281–2295. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Luo, C.; Zhang, Z.; Hu, J.; Gao, X.; Zuo, Y.; Wang, Y.; Zhu, S. Mdivi-1 Attenuates Sodium Azide-induced Apoptosis in H9c2 Cardiac Muscle Cells. Mol. Med. Rep. 2017, 16, 5972–5978. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Ferdjallah, I.; Elenberg, F.; Chen, S.K.; Deuster, P.; Chen, Y. Mitochondrial Fission Contributes to Heat-Induced Oxidative Stress in Skeletal Muscle but Not Hyperthermia in Mice. Life Sci. 2018, 200, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Dong, Q.; Liu, Z.; Liu, Z.; Qu, Y.; Li, X.; Huo, C.; Jia, X.; Fu, F.; Wang, X. Inhibition of Dynamin-Related Protein 1 Protects against Myocardial Ischemia-Reperfusion Injury in Diabetic Mice. Cardiovasc. Diabetol. 2017, 16, 19. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, P.; Bisetto, S.; Yoon, Y.; Chen, Q.; Sheu, S.-S.; Wang, W. A Novel Fission-Independent Role of Dynamin-Related Protein 1 in Cardiac Mitochondrial Respiration. Cardiovasc. Res. 2017, 113, 160–170. [Google Scholar] [CrossRef]
- Kim, J.-H.; Park, S.-J.; Kim, B.; Choe, Y.-G.; Lee, D.-S. Insulin-Stimulated Lipid Accumulation Is Inhibited by ROS-Scavenging Chemicals, but Not by the Drp1 Inhibitor Mdivi-1. PLoS ONE 2017, 12, e0185764. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.-S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.-Y. Inhibition of Drp1-Dependent Mitochondrial Division Impairs Myogenic Differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef]
- Rosdah, A.A.; K Holien, J.; Delbridge, L.M.D.; Dusting, G.J.; Lim, S.Y. Mitochondrial Fission—A Drug Target for Cytoprotection or Cytodestruction? Pharmacol. Res. Perspect. 2016, 4, e00235. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxid. Redox Signal 2011, 14, 439–457. [Google Scholar] [CrossRef]
- Ali, S.; McStay, G. Regulation of Mitochondrial Dynamics by Proteolytic Processing and Protein Turnover. Antioxidants 2018, 7, 15. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial Fission and Fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef]
- Ruiz, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 (Mdivi-1) Protects Neurons against Excitotoxicity through the Modulation of Mitochondrial Function and Intracellular Ca2+ Signaling. Front. Mol. Neurosci. 2018, 11, 3. [Google Scholar] [CrossRef]
- Smith, G.; Gallo, G. To Mdivi-1 or Not to Mdivi-1: Is That the Question? Dev. Neurobiol. 2017, 77, 1260–1268. [Google Scholar] [CrossRef]
- Mao, X.; Gu, Y.; Sui, X.; Shen, L.; Han, J.; Wang, H.; Xi, Q.; Zhuang, Q.; Meng, Q.; Wu, G. Phosphorylation of Dynamin-Related Protein 1 (DRP1) Regulates Mitochondrial Dynamics and Skeletal Muscle Wasting in Cancer Cachexia. Front. Cell Dev. Biol. 2021, 9, 673618. [Google Scholar] [CrossRef]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883.e5. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Hepple, R.T.; Burelle, Y. Mitochondrial Functional Specialization in Glycolytic and Oxidative Muscle Fibers: Tailoring the Organelle for Optimal Function. Am. J. Physiol.-Cell Physiol. 2012, 302, C629–C641. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial Structure and Function Are Disrupted by Standard Isolation Methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer 5′ → 3′ | Reverse Primer 5′ → 3′ |
|---|---|---|
| SOD1 | CCAGTGCAGGACCTCATTTT | TTGTTTCTCATGGACCACCA |
| SOD2 | ACCCAAAGTCACGCTTGATAG | GGACAAACCTGAGCCCTAAG |
| Catalase | CACTGACGAGATGGCACACT | TGTGGAGAATCGAACGGCAA |
| Glutathione peroxidase | TGCAATCAGTTCGGACACCA | AAGGTAAAGAGCGGGTGAGC |
| Drp1 | AGAAAACTGTCTGCCCGAGA | GCTGCCCTACCAGTTCACTC |
| Fis1 | CCGGCTCAAGGAATATGAAA | ACAGCCAGTCCAATGAGTCC |
| Mfn1 | CCTCCATGGGCATCATCGTT | TGCAGCTTCTCGGTTGCATA |
| Mfn2 | CTCAGGAGCAGCGGGTTTAT | GAGAGGCGCCTGATCTCTTC |
| 18S | CGCGGTTCTATTTTGTTGGT | TCGTCTTCGAAACTCCGACT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paradis, S.; Charles, A.-L.; Giannini, M.; Meyer, A.; Lejay, A.; Talha, S.; Laverny, G.; Charloux, A.; Geny, B. Targeting Mitochondrial Dynamics during Lower-Limb Ischemia Reperfusion in Young and Old Mice: Effect of Mitochondrial Fission Inhibitor-1 (mDivi-1). Int. J. Mol. Sci. 2024, 25, 4025. https://doi.org/10.3390/ijms25074025
Paradis S, Charles A-L, Giannini M, Meyer A, Lejay A, Talha S, Laverny G, Charloux A, Geny B. Targeting Mitochondrial Dynamics during Lower-Limb Ischemia Reperfusion in Young and Old Mice: Effect of Mitochondrial Fission Inhibitor-1 (mDivi-1). International Journal of Molecular Sciences. 2024; 25(7):4025. https://doi.org/10.3390/ijms25074025
Chicago/Turabian StyleParadis, Stéphanie, Anne-Laure Charles, Margherita Giannini, Alain Meyer, Anne Lejay, Samy Talha, Gilles Laverny, Anne Charloux, and Bernard Geny. 2024. "Targeting Mitochondrial Dynamics during Lower-Limb Ischemia Reperfusion in Young and Old Mice: Effect of Mitochondrial Fission Inhibitor-1 (mDivi-1)" International Journal of Molecular Sciences 25, no. 7: 4025. https://doi.org/10.3390/ijms25074025
APA StyleParadis, S., Charles, A.-L., Giannini, M., Meyer, A., Lejay, A., Talha, S., Laverny, G., Charloux, A., & Geny, B. (2024). Targeting Mitochondrial Dynamics during Lower-Limb Ischemia Reperfusion in Young and Old Mice: Effect of Mitochondrial Fission Inhibitor-1 (mDivi-1). International Journal of Molecular Sciences, 25(7), 4025. https://doi.org/10.3390/ijms25074025