

Graph Neural Network Model for Prediction of Non-Small Cell Lung Cancer Lymph Node Metastasis Using Protein–Protein Interaction Network and 18F-FDG PET/CT Radiomics


Abstract
1. Introduction
2. Results
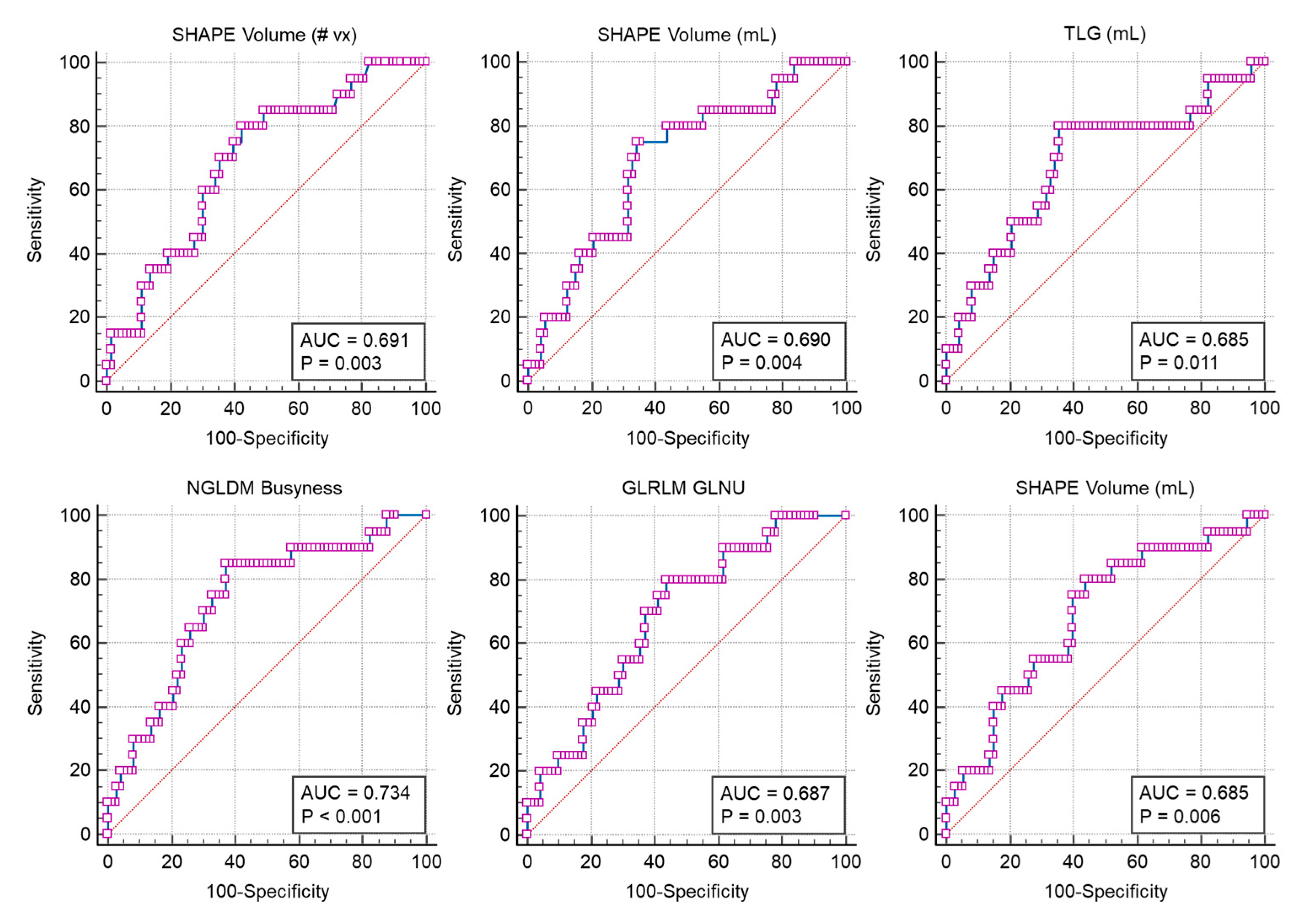
2.1. Gene and Image Texture Feature Selection
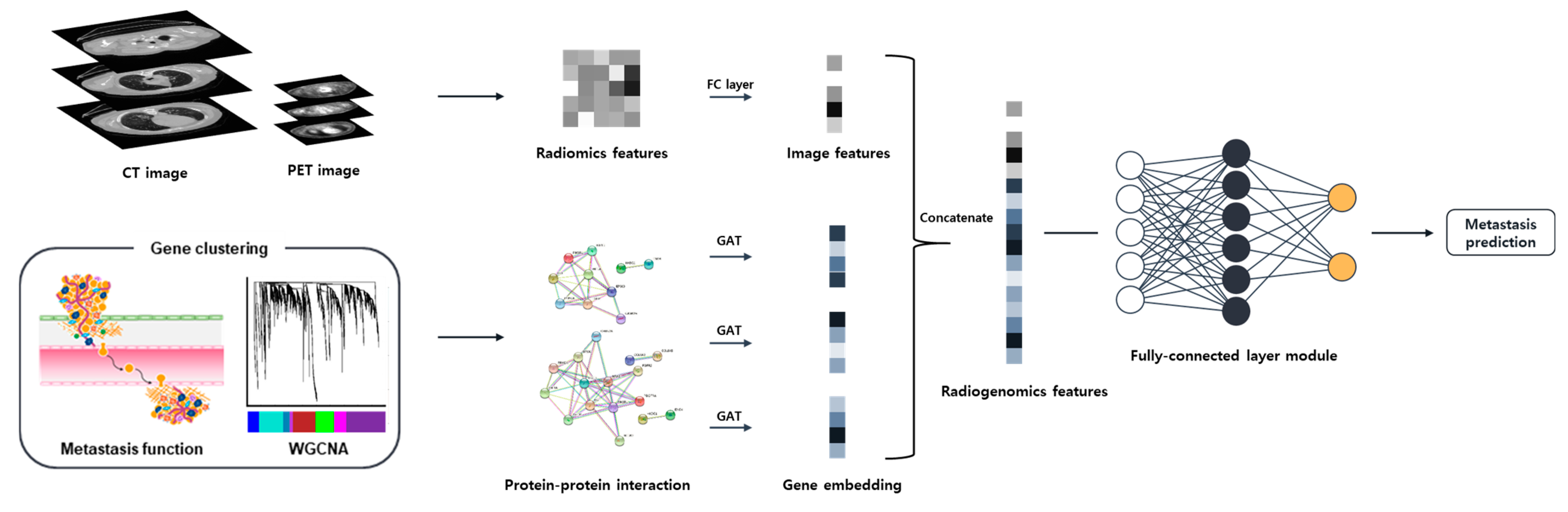
2.2. GNN Model
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Gene Analysis
4.3. Image Feature Extraction and Selection
4.4. Prediction Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meza, R.; Meernik, C.; Jeon, J.; Cote, M.L. Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS ONE 2015, 10, e0121323. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, A.; Matsuda, T.; Shibata, A.; Katanoda, K.; Sobue, T.; Nishimoto, H. Cancer incidence and incidence rates in Japan in 2008: A study of 25 population-based cancer registries for the Monitoring of Cancer Incidence in Japan (MCIJ) project. Jpn. J. Clin. Oncol. 2014, 44, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Little, A.G.; Gay, E.G.; Gaspar, L.E.; Stewart, A.K. National survey of non-small cell lung cancer in the United States: Epidemiology, pathology and patterns of care. Lung Cancer 2007, 57, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gu, Y.; Basu, S.; Berglund, A.; Eschrich, S.A.; Schabath, M.B.; Forster, K.; Aerts, H.J.; Dekker, A.; Fenstermacher, D.; et al. Radiomics: The process and the challenges. Magn. Reason. Imaging 2012, 30, 1234–1248. [Google Scholar] [CrossRef] [PubMed]
- Eloyan, A.; Yue, M.S.; Khachatryan, D. Tumor heterogeneity estimation for radiomics in cancer. Stat. Med. 2020, 39, 4704–4723. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.S.; Donington, J.S. Management of locally advanced non small cell lung cancer from a surgical perspective. Curr. Treat. Options Oncol. 2007, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lambin, P.; Rios-Velazquez, E.; Leijenaar, R.; Carvalho, S.; van Stiphout, R.G.P.M.; Granton, P.; Zegers, C.M.L.; Gillies, R.; Boellard, R.; Dekker, A.; et al. Radiomics: Extracting more information from medical images using advanced feature analysis. Eur. J. Cancer 2012, 48, 441–446. [Google Scholar] [CrossRef]
- Bowen, S.R.; Yuh, W.T.; Hippe, D.S.; Wu, W.; Partridge, S.C.; Elias, S.; Jia, G.; Huang, Z.; Sandison, G.A.; Nelson, D.; et al. Tumor radiomic heterogeneity: Multiparametric functional imaging to characterize variability and predict response following cervical cancer radiation therapy. J. Magn. Reason. Imaging 2018, 47, 1388–1396. [Google Scholar] [CrossRef]
- Oti, M.; Snel, B.; Huynen, M.A.; Brunner, H.G. Predicting disease genes using protein–protein interactions. J. Med. Genet. 2006, 43, 691–698. [Google Scholar] [CrossRef]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N. Protein-protein interaction detection: Methods and analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef]
- Ni, D.; Lu, S.; Zhang, J. Emerging roles of allosteric modulators in the regulation of protein-protein interactions (PPIs): A new paradigm for PPI drug discovery. Med. Res. Rev. 2019, 39, 2314–2342. [Google Scholar] [CrossRef] [PubMed]
- Bebek, G.; Yang, J. PathFinder: Mining signal transduction pathway segments from protein-protein interaction networks. BMC Bioinform. 2007, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Thiel, P.; Kaiser, M.; Ottmann, C. Small-Molecule Stabilization of Protein–Protein Interactions: An Underestimated Concept in Drug Discovery? Angew. Chem. Int. Ed. 2012, 51, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Kar, G.; Gursoy, A.; Keskin, O. Human cancer protein-protein interaction network: A structural perspective. PLoS Comput Biol. 2009, 5, e1000601. [Google Scholar] [CrossRef] [PubMed]
- Engin, H.B.; Guney, E.; Keskin, O.; Oliva, B.; Gursoy, A. Integrating structure to protein-protein interaction networks that drive metastasis to brain and lung in breast cancer. PLoS ONE 2013, 8, e81035. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dai, Y.; Yu, K.; Zhang, Z.K. Enhancing cancer driver gene prediction by protein-protein interaction network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.D.; Zhang, Q.; Zhang, Y.H.; Chen, L.; Huang, T. Identification of genes associated with breast cancer metastasis to bone on a protein–protein interaction network with a shortest path algorithm. J. Proteome Res. 2017, 16, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S.Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef]
- Veyrin-Forrer, L.; Kamal, A.; Duffner, S.; Plantevit, M.; Robardet, C. On gnn explainability with activation rules. Data Min. Knowl. Discov. 2022, 1–35. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Sun, M. Graph neural networks: A review of methods and applications. AI Open 2020, 1, 57–81. [Google Scholar] [CrossRef]
- Liao, W.; Bak-Jensen, B.; Pillai, J.R.; Wang, Y.; Wang, Y. A review of graph neural networks and their applications in power systems. J. Mod. Power Syst. Clean Energy 2021, 10, 345–360. [Google Scholar] [CrossRef]
- Dong, L.; Yao, H.; Li, D.; Wang, Y.; Li, S.; Liang, Q. Improving graph neural network via complex-network-based anchor structure. Knowl. Based Syst. 2012, 233, 107528. [Google Scholar] [CrossRef]
- Wang, S.; Tang, H.; Shan, P.; Wu, Z.; Zuo, L. ProS-GNN: Predicting effects of mutations on protein stability using graph neural networks. Comput. Biol. Chem. 2023, 107, 107952. [Google Scholar] [CrossRef]
- Burkhart, J.G.; Wu, G.; Song, X.; Raimondi, F.; McWeeney, S.; Wong, M.H.; Deng, Y. Biology-inspired graph neural network encodes reactome and reveals biochemical reactions of disease. Patterns Prejud. 2023, 4, 100758. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, M.; Fiannaca, A.; La Paglia, L.; Urso, A.A. Graph Neural Network Approach for the Analysis of siRNA-Target Biological Networks. Int. J. Mol. Sci. 2022, 23, 14211. [Google Scholar] [CrossRef]
- Shi, L.; He, Y.; Yuan, Z.; Benedict, S.; Valicenti, R.; Qiu, J.; Rong, Y. Radiomics for response and outcome assessment for non-small cell lung cancer. Technol. Cancer Res. Treat. 2018, 17, 1533033818782788. [Google Scholar] [CrossRef]
- Kirienko, M.; Sollini, M.; Corbetta, M.; Voulaz, E.; Gozzi, N.; Interlenghi, M.; Gallivanone, F.; Castiglioni, I.; Asselta, R.; Duga, S.; et al. Radiomics and gene expression profile to characterise the disease and predict outcome in patients with lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3643–3655. [Google Scholar] [CrossRef]
- Zhou, M.; Leung, A.; Echegaray, S.; Gentles, A.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Plevritis, S.K.; Rubin, D.L.; Napel, S.; et al. Non–small cell lung cancer radiogenomics map identifies relationships between molecular and imaging phenotypes with prognostic implications. Radiology 2018, 286, 307–315. [Google Scholar] [CrossRef]
- Li, R.; Zhou, L.; Wang, Y.; Shan, F.; Chen, X.; Liu, L. A graph neural network model for the diagnosis of lung adenocarcinoma based on multimodal features and an edge-generation network. Quant. Imaging Med. Surg. 2023, 13, 5333. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic Control of Metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Kim, S.J.; Rabbani, Z.N.; Dewhirst, M.W.; Vujaskovic, Z.; Vollmer, R.T.; Schreiber, E.-G.; Oosterwijk, E.; Kelley, M.J. Expression of HIF-1α, CA IX, VEGF, and MMP-9 in surgically resected non-small cell lung cancer. Lung Cancer 2005, 49, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; Van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Flamant, L.; Notte, A.; Ninane, N.; Raes, M.; Michiels, C. Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast cancer cells under hypoxia. Mol. Cancer 2010, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.D.; Yang, C.; Osterman, A.; Smith, J.W. Central carbon metabolism in the progression of mammary carcinoma. Breast Cancer Res. Treat. 2008, 110, 297–307. [Google Scholar] [CrossRef]
- Luo, M.; Guan, J.L. Focal adhesion kinase: A prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010, 289, 127–139. [Google Scholar] [CrossRef]
- Kornberg, L.J. Focal adhesion kinase and its potential involvement in tumor invasion and metastasis. Head Neck J. Sci. Spec. Head Neck 1998, 20, 745–752. [Google Scholar] [CrossRef]
- Schwock, J.; Dhani, N.; Hedley, D.W. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opin. Ther. Targets 2010, 14, 77–94. [Google Scholar] [CrossRef]
- Rahimi, N. The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer Ther. 2012, 11, 538–548. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions: Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Without Image Data | Image Features with AUC ≥ 0.68 | Whole Image Features | |||||
|---|---|---|---|---|---|---|---|---|
| PET (6) | CT (4) | PET/CT (10) | PET (55) | CT (55) | PET/CT (110) | |||
| 73 modules * | Accuracy | 0.4795 | 0.5375 | 0.5473 | 0.6420 | 0.7179 | 0.7429 | 0.8455 |
| Precision | 0.2679 | 0.4390 | 0.5448 | 0.7633 | 0.7039 | 0.7000 | 0.8392 | |
| Recall | 0.4518 | 0.7750 | 0.6321 | 0.6696 | 0.8268 | 0.9018 | 0.9054 | |
| F1 score | 0.3248 | 0.5496 | 0.5219 | 0.6354 | 0.7439 | 0.7820 | 0.8631 | |
| AUC | 0.5674 | 0.5744 | 0.6532 | 0.7430 | 0.7728 | 0.7907 | 0.8718 | |
| Without gene data | Accuracy | - | 0.5920 | 0.6857 | 0.7607 | 0.8357 | 0.8009 | 0.8286 |
| Precision | - | 0.5515 | 0.6627 | 0.7662 | 0.7875 | 0.7565 | 0.8026 | |
| Recall | - | 0.7625 | 0.8125 | 0.8000 | 0.9339 | 0.9161 | 0.8893 | |
| F1 score | - | 0.6363 | 0.7199 | 0.7630 | 0.8522 | 0.8236 | 0.8812 | |
| AUC | - | 0.6549 | 0.7415 | 0.8035 | 0.8530 | 0.8372 | 0.8812 | |
| Genes in 4 functions (34) ** | Accuracy | 0.5830 | 0.6509 | 0.6018 | 0.7330 | 0.8143 | 0.7955 | 0.8545 |
| Precision | 0.4538 | 0.6037 | 0.5858 | 0.7004 | 0.7928 | 0.7548 | 0.8213 | |
| Recall | 0.4036 | 0.6946 | 0.6018 | 0.8107 | 0.8768 | 0.9304 | 0.9143 | |
| F1 score | 0.3692 | 0.6377 | 0.5805 | 0.7477 | 0.8267 | 0.8238 | 0.8618 | |
| AUC | 0.6333 | 0.6776 | 0.7064 | 0.7709 | 0.8901 | 0.8849 | 0.9026 | |
| DEG results (19) *** | Accuracy | 0.5411 | 0.6321 | 0.6429 | 0.6973 | 0.8000 | 0.7768 | 0.8223 |
| Precision | 0.5239 | 0.6131 | 0.6146 | 0.6916 | 0.7553 | 0.7343 | 0.8006 | |
| Recall | 0.5875 | 0.6464 | 0.7357 | 0.7250 | 0.9179 | 0.9179 | 0.8893 | |
| F1 score | 0.4910 | 0.6153 | 0.6421 | 0.6963 | 0.8238 | 0.8098 | 0.8345 | |
| AUC | 0.5231 | 0.6879 | 0.7092 | 0.7458 | 0.8585 | 0.8245 | 0.8948 | |
| Univariate analysis (12) **** | Accuracy | 0.5036 | 0.6045 | 0.7268 | 0.7580 | 0.8134 | 0.7580 | 0.8420 |
| Precision | 0.3998 | 0.6098 | 0.6861 | 0.7443 | 0.7660 | 0.7256 | 0.7893 | |
| Recall | 0.5893 | 0.6750 | 0.8625 | 0.7982 | 0.9179 | 0.8464 | 0.9607 | |
| F1 score | 0.4627 | 0.5929 | 0.7602 | 0.7668 | 0.8325 | 0.7643 | 0.8610 | |
| AUC | 0.4784 | 0.7229 | 0.7627 | 0.7765 | 0.8606 | 0.8397 | 0.8793 | |
| Characteristics | Non Metastasis (n = 73) | Lymph Node Metastasis (n = 20) |
|---|---|---|
| Age (%) | ||
| <65 | 22 (30) | 3 (15) |
| ≥65 | 51 (70) | 17 (85) |
| Mean age (y) | 68.82 | 69.1 |
| Sex, n (%) | ||
| Male | 56 (77) | 15 (75) |
| Female | 17 (23) | 5 (25) |
| Pathological stage, n (%) | ||
| T1a | 15 (21) | 1 (5) |
| T1b | 15 (21) | 7 (35) |
| T2a | 26 (36) | 5 (25) |
| T2b | 6 (8) | 3 (15) |
| T3 | 7 (10) | 2 (10) |
| T4 | 4 (5) | 2 (10) |
| Pathological stage (%) | ||
| N0 | 73 | |
| N1 | 7 (35) | |
| N2 | 13 (65) | |
| Pathological stage (%) | ||
| I | 56 (77) | |
| II | 13 (18) | 7 (35) |
| III | 4 (5.48) | 12 (60) |
| IV | 1 (5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, H.; Kim, K.; Kim, B.I.; Woo, S.-K. Graph Neural Network Model for Prediction of Non-Small Cell Lung Cancer Lymph Node Metastasis Using Protein–Protein Interaction Network and 18F-FDG PET/CT Radiomics. Int. J. Mol. Sci. 2024, 25, 698. https://doi.org/10.3390/ijms25020698
Ju H, Kim K, Kim BI, Woo S-K. Graph Neural Network Model for Prediction of Non-Small Cell Lung Cancer Lymph Node Metastasis Using Protein–Protein Interaction Network and 18F-FDG PET/CT Radiomics. International Journal of Molecular Sciences. 2024; 25(2):698. https://doi.org/10.3390/ijms25020698
Chicago/Turabian StyleJu, Hyemin, Kangsan Kim, Byung Il Kim, and Sang-Keun Woo. 2024. "Graph Neural Network Model for Prediction of Non-Small Cell Lung Cancer Lymph Node Metastasis Using Protein–Protein Interaction Network and 18F-FDG PET/CT Radiomics" International Journal of Molecular Sciences 25, no. 2: 698. https://doi.org/10.3390/ijms25020698
APA StyleJu, H., Kim, K., Kim, B. I., & Woo, S.-K. (2024). Graph Neural Network Model for Prediction of Non-Small Cell Lung Cancer Lymph Node Metastasis Using Protein–Protein Interaction Network and 18F-FDG PET/CT Radiomics. International Journal of Molecular Sciences, 25(2), 698. https://doi.org/10.3390/ijms25020698