1. Introduction
Glioblastoma (GBM), a highly aggressive primary brain tumor, presents a critical challenge in clinical management due to its significant heterogeneity, poor prognosis, and limited treatment options [
1,
2]. Management requires maximal surgical resection followed by concurrent chemoirradiation (CRT) with radiation therapy (RT) and temozolomide (TMZ), followed by the administration of adjuvant TMZ [
1,
2,
3,
4]. Identifying reliable prognostic markers, such as MGMT promoter methylation status, and harnessing MGMT protein expression levels are crucial for tailoring therapy and potentially improving patient outcomes [
5,
6].
MGMT (O6-methylguanine-DNA methyltransferase) [
7], is a gene that repairs alkylating agent-induced DNA damage [
8]. Patients with methylated MGMT promoters (mMGMT) have been found to exhibit significantly higher treatment sensitivity and improved survival [
9,
10], highlighting the importance of accurate status prediction for personalized treatment options [
7,
11]. Additionally, MGMT protein expression reflects the functional activity of the repair pathway, providing complementary information for treatment decisions [
3]. The capture of MGMT status has variability in technique [
12], but is based on tissue specimens. The connection between MGMT status and MGMT protein expression, which can be captured in serum, can be discordant, with no distinct threshold identified for MGMT protein expression [
13]. MGMT status is linked to the response to TMZ, which is the standard of care administered concurrently with RT, and adjuvantly; however, this mainstay approach still results in very poor outcomes, with a median survival of 14 months [
9]. The biological mechanisms that underlie treatment resistance to TMZ are multifaceted and poorly understood [
14]. The linkage of MGMT methylation status and MGMT protein expression has the potential to offer a more comprehensive picture of tumor biology, potentially leading to improved clinical outcomes and offering independent prognostic value [
5,
6]. Therefore, the accurate prediction of molecules and pathways that connect both aspects holds immense potential for personalizing treatment strategies. This can be achieved by studying new approaches to systemic management, including agents that act to potentially reverse or modify resistance by targeting GBM stem cells [
15,
16], or modify response to RT [
17] in an effort to link systemic management and RT to both MGMT-dependent and MGMT-independent mechanisms.
Proteomics represents the study of large-scale analyses of protein expression [
18] with immense potential for uncovering novel GBM biomarkers with high accuracy and clinical utility by providing complementary information to metabolic and genomic data [
18]. Compared to tissue acquisition, analyzing proteins in serum or plasma offers a minimally invasive approach [
2,
18]. By analyzing complex protein profiles, more profound insights are possible through the identification and monitoring of key players in treatment response and resistance mechanisms identified. Recent advancements in proteomics, whether they employed mass spectrometry and tumor tissue or large-scale panels and serum biospecimens, have uncovered crucial relationships between GBM tumor heterogeneity [
19], the proteome and the metabolome [
20], the proteome and outcomes [
21,
22], and the observable footprint of alterations in concurrent management in proteomic data [
23]. There is currently no serum biomarker in clinical use for GBM diagnosis or treatment monitoring. However, serum proteomic markers of promising importance are emerging in the literature, and several have applicability to GBM [
24].
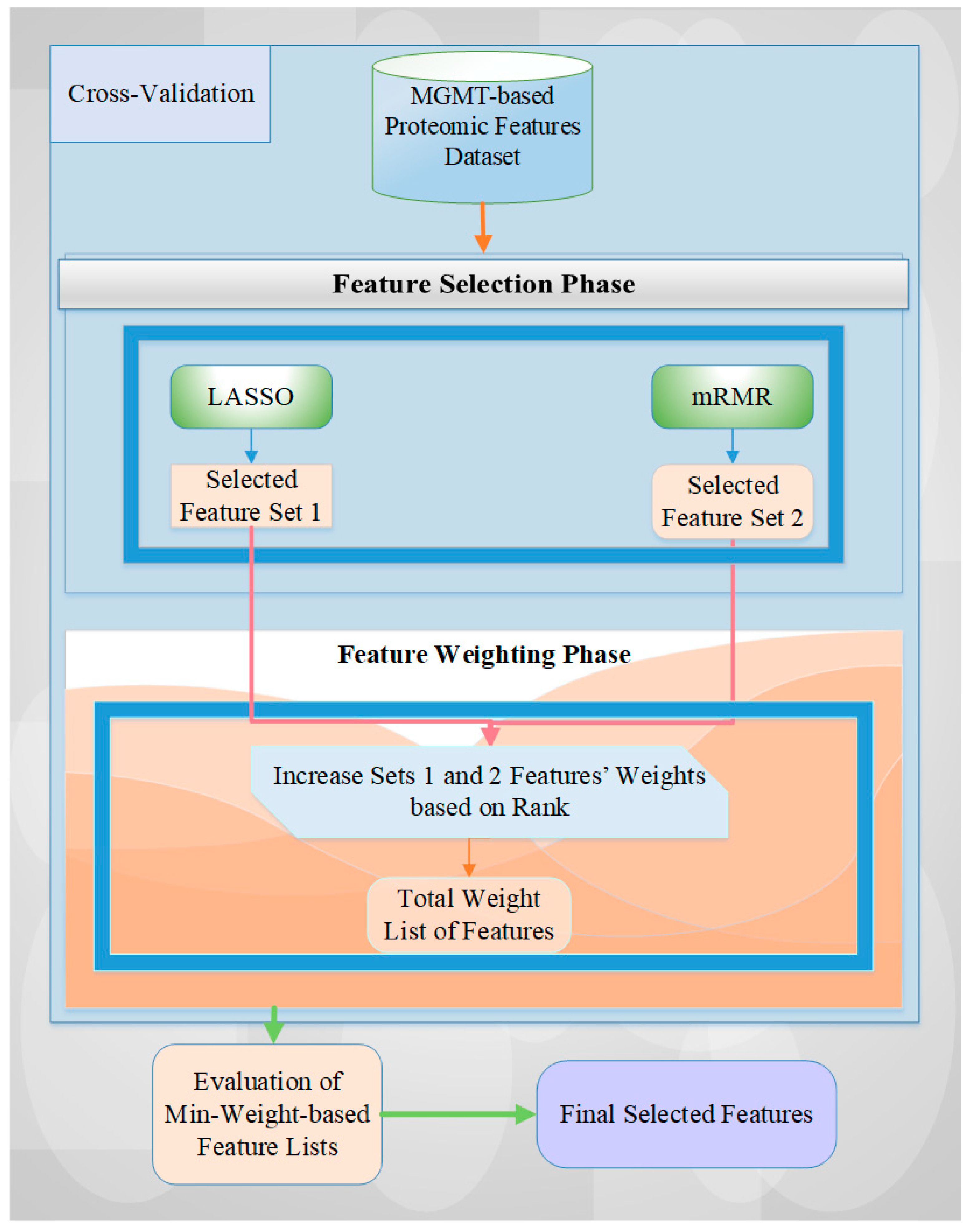
Utilizing feature selection methods and machine learning algorithms on proteomic data offers additional opportunities to refine biomarker discovery and prediction models. By using diverse datasets and effective dimensionality reduction techniques, hidden patterns can be extracted, and the prediction performance for MGMT status and protein expression can be improved. The feature selection process is a significant data pre-processing step and a dimensionality reduction technique to eliminate redundant and irrelevant predictors in high-dimensional data [
2,
25,
26]. This operation decreases the computational time, reduces complexity, extracts hidden data patterns, makes the related analysis more effective, and makes visualization easier [
2,
26,
27]. Feature selection methods can be categorized into filter (e.g., Minimum Redundancy Maximum Relevance (mRMR)), wrapper, and embedded (e.g., Least Absolute Shrinkage and Selection Operator (LASSO)) methods, according to evaluations of feature subsets [
28,
29,
30]. Apart from the feature selection process, feature weighting is also a crucial step in assigning suitable weights to the features for finding the most effective possible final feature subset, after applying each fold of the cross-validation operation task [
26,
31].
This study investigates the potential to employ a hybrid filter and embedded feature selection (i.e., mRMR and LASSO) and the rank-based weighting method to predict serum protein biomarkers associated with known molecular markers. To extract the most impactful signals from the high-dimensional serum proteomic data, we leverage a feature selection and weighting technique, ultimately yielding a focused possible minimal number of informative selected features for our large-scale oncologic dataset. By providing more accurate predictions of MGMT status and protein expression, this research can potentially improve clinical outcomes in patients with GBM by offering more tailored treatment approaches.
The primary contributions of this study, categorized as technical and clinical aspects, are summarized below.
1.1. Technical Aspects
This study stands out as the first to utilize the innovative and hybrid feature selection and rank-based weighting methodology (i.e., MGMT ProFWise) for both MGMT promoter methylation status classification and MGMT protein expression level regression tasks on proteomic data.
To validate the MGMT ProFWise method and identify shared biomarker features across diverse tasks and datasets, we apply it to two distinct proteomic datasets.
To compensate for the skewed class distribution in our dataset, we employed stratified cross-validation during machine learning training, ensuring each fold accurately reflected the overall class imbalance.
We also handle the identification of the names of the final feature subset after obtaining possible different feature subsets of each cross-validation fold for the feature selection process by employing a rank-based feature weighting procedure.
We explore the impact of the feature selection and rank-based weighting method on the performance of diverse learning models.
We aim to identify the best machine learning model with the minimum number of selected features providing the best performance (i.e., accuracy rate (ACC) or mean squared error (MSE)) for the relevant classification and regression tasks on large-scale proteomic datasets.
We give a general alteration analysis for the selected features of our local proteomic dataset regarding MGMT status.
1.2. Clinical Aspects
This study offers encouraging findings to advance GBM serum proteome biomarker research.
We investigate the associations between the identified features, GBM, relevant signaling pathways, and upstream regulatory proteins driving these pathways through STRING applications.
We also analyze shared selected features between MGMT promoter methylation classification and the MGMT protein expression feature selection processes towards ease of transferability for bioinformatics researchers in this domain.
The subsequent sections of this study are organized as follows:
Section 2 describes the results, including experiments, performance metrics, and comprehensive computational results.
Section 3 presents the discussion of the results.
Section 4 is comprised of the materials and methods, including the dataset description and characteristics, and an overview of the utilized feature selection and weighting methodology, and also explains the related feature selection methods and supervised learning models for both classification and regression tasks.
Section 5 concludes this study and proposes potential avenues for future research.
3. Discussion
MGMT promoter methylation remains the cardinal molecular feature associated with survival in GBM, rendering it both a prognostic and predictive biomarker [
9,
10]. The improvement in outcomes observed in MGMT promoter methylated patients has been attributed to its role as a repair enzyme and its activity in relationship to the administration of TMZ, specifically for GBM [
13]. However, its action in direct DNA repair, which constitutes less than 10% of the damage inflicted by TMZ [
14,
38], as well as its relevance to the presence of RT alone [
39] and to other malignancies, and in the prevention of cancer development [
40] suggest a broader role of the MGMT promoter. Further, the outcome is linked to several facets, including methylation patterns of CpGs associated with prognosis [
41].
In this study, we employed MGMT promoter status, serum MGMT expression measurements from a large-scale proteomic panel, and ML feature engineering to identify serum signals that capture the relationship between MGMT promoter status and MGMT protein expression. To our knowledge, this is the only study that has attempted this linkage through employing serum MGMT protein expression. Our hypothesis is that, with growing serum data availability and the interpretable use of ML and feature engineering, serum signals can be effectively harnessed in specific disease entities such as glioma.
We showcase several technical innovations employed for the purpose of linking MGMT status and MGMT protein expression to molecules measured in the serum of patients, post-surgical resection but prior to upfront management with CRT. The technical approaches presented here carry broad transferability to other molecular domains and have wide dimensionality for large-scale data. In our previous work, we identified features associated with the administration of CRT [
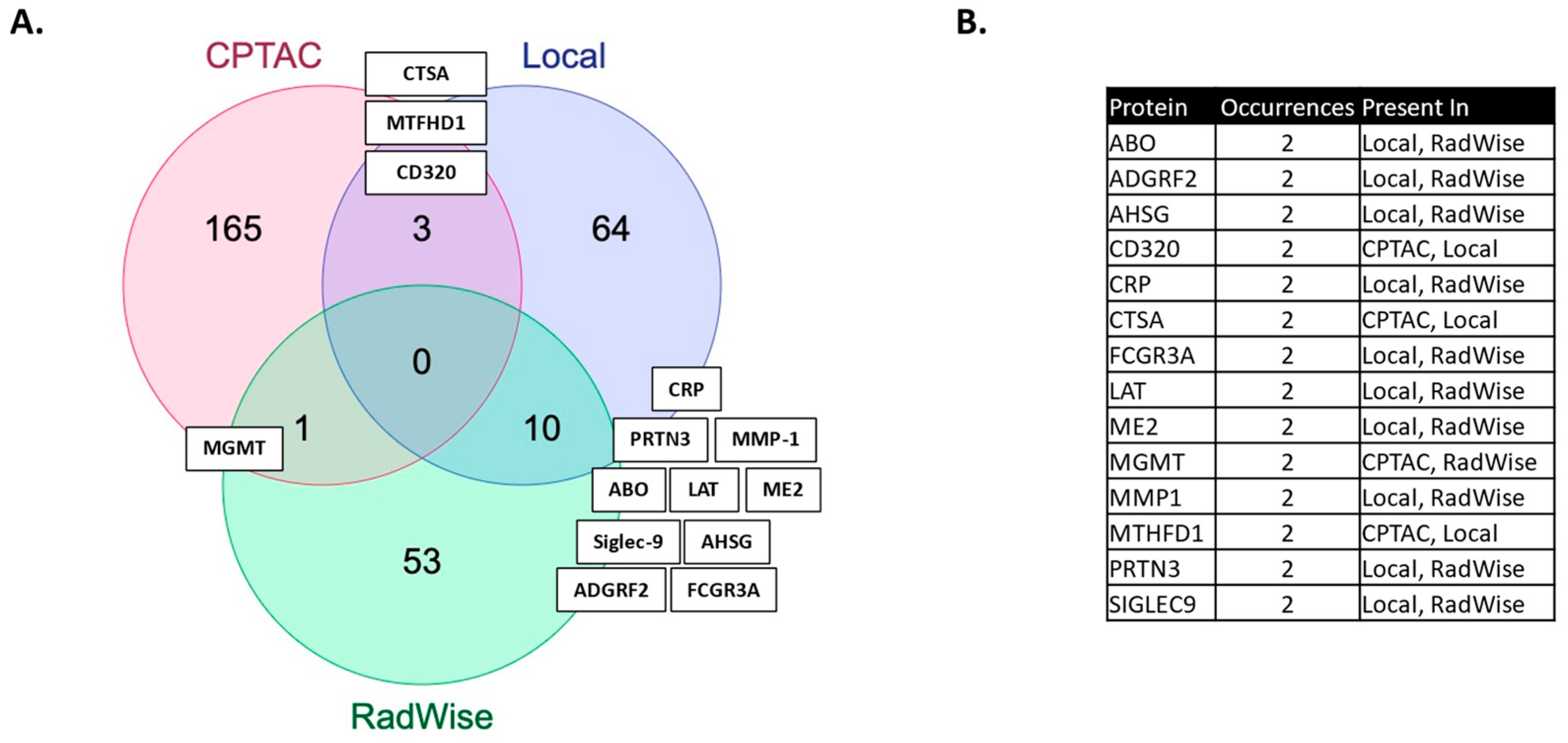
2]. More features were shared between MGMT status and CRT administration than by MGMT protein expression and CRT administration in this study (
Figure 2,
Supplementary Table S1).
The proteins identified have relevance to GBM. PLA2G12B, a phospholipase A2 variant, is of interest, given recent evidence for the role of phospholipases in cancer and documented high expression levels in glioma, as well as its emergence as an oncogene associated with glioma progression [
20,
42]. ABO, histo-blood group ABO system transferase, poses an interesting link between MGMT status and blood group. The blood group has been examined in connection with several conditions, including GBM, but there is currently no conclusive evidence [
43]. The most shared features (10) (
Figure 2) between the MGMT analysis and the CRT analysis in the local dataset have reported relevance to GBM: ADGRF2 (overexpressed in glioma, regulating proliferation and migration) [
44], AHSG (serum level predicts survival in GBM) [
45], CRP [
46], ME2 (promotes proneural mesenchymal transition) [
47], MMP1 (expression upregulated in GBM and associated with decreased survival) [
48], Sigle9 (immune evasion, reduced survival) [
49], and others which are not yet fully defined: FCGR3A, LAT, and PRTN3 [
20]. We have previously identified these signals as associated with CRT [
2] and MGMT expression, and associated with lower survival [
21].
Serum MGMT expression and CPTAC notably overlapped with respect to CTSA (upregulated in glioma and associated with immune infiltration) [
50], MTFHD1 (one-carbon metabolism association in GBM) [
51], and CD320 (implicated in a cobalmin-mediated metabolism) [
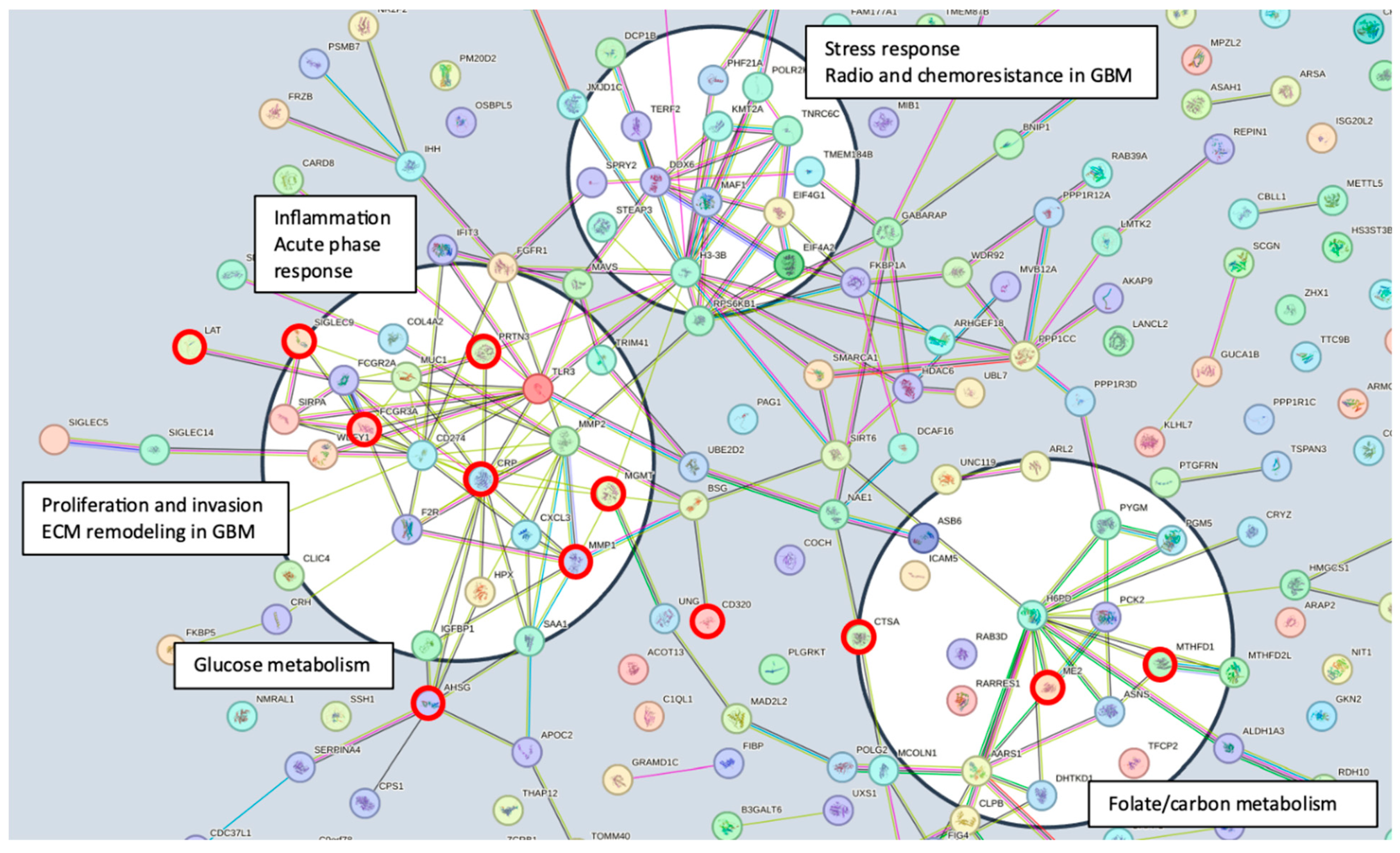
52]. The shared features are linked in STRING (
Figure 3) with MGMT protein expression, in nodes that are connected to glucose, folate, and a one-carbon metabolism, as well as stress response, radio, and chemoresistance in GBM [
53]. MGMT protein expression, however, only emerged as a shared feature between CPTAC and our previous study through examining features associated with CRT in the local dataset [
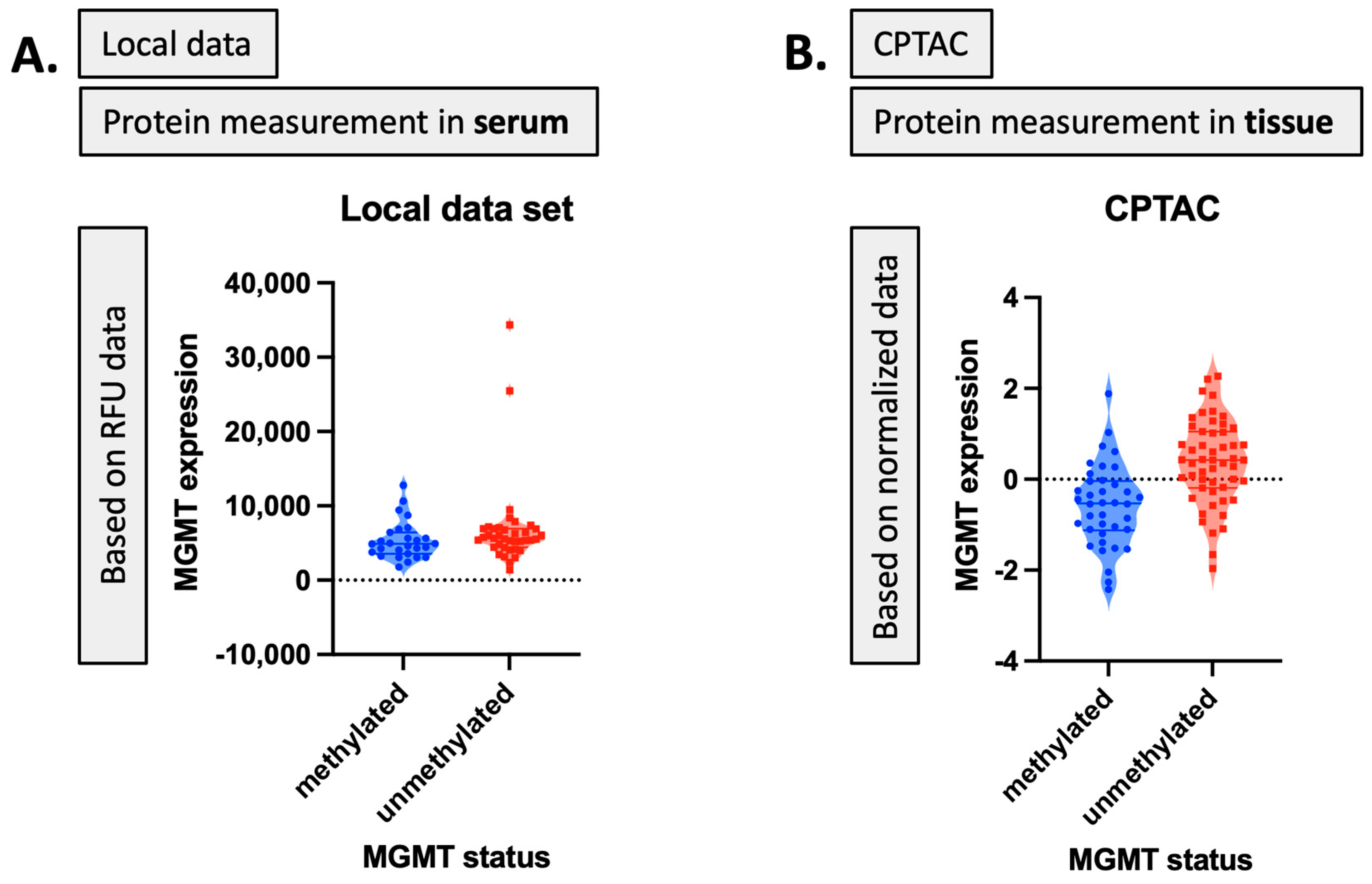
2]. This may reflect the inconsistent or low capture of MGMT protein expression levels in serum. While mean MGMT protein expression was higher in unmethylated patients vs. methylated patients, overall MGMT expression levels carried significant overlap between MGMT methylated and unmethylated patients in both the local dataset and in CPTAC (
Figure 4), which was similar to previously reported trends in the literature [
20]. This study, however, is encouraging, since MGMT protein expression in serum can be linked to MGMT protein expression in tissue via mechanistic connections wherein each method of measurement (MGMT promoter status, MGMT protein expression) more optimally captures a specific mechanistic aspect vs. another. This study also importantly illustrated that features associated with the administration of CRT for GBM are also associated with MGMT status. This can potentially extend our understanding of how MGMT promoter methylation may be exerting wide-ranging effects to result in improved outcomes in patients with MGMT methylated disease. Large-scale proteomic panels are actively evolving to include larger repertoires of proteins; thus, as datasets grow and validation of findings improves, ranges can be generated for various GBM clinical feature sets, and markers displayed in
Figure 2B can be potentially employed to predict MGMT status. Currently, MGMT status is determined based on tissue sample obtained at the time of surgical resection, the analysis of which can be cost prohibitive and delay results which are often not available at the time of initial consult (2–3 weeks post resection). Serum markers can be measured with results returned within hours, as is currently the case with markers such as CRP, which is one of the markers identified in the current analysis. For treatment, monitoring panels can be finetuned further to home in on patterns that reflect the likelihood of response or treatment failure, rendering the patient a candidate for clinical trial management. For example, serum markers that phenotypically reflect tumor behavior that mirrors the unmethylated patients in
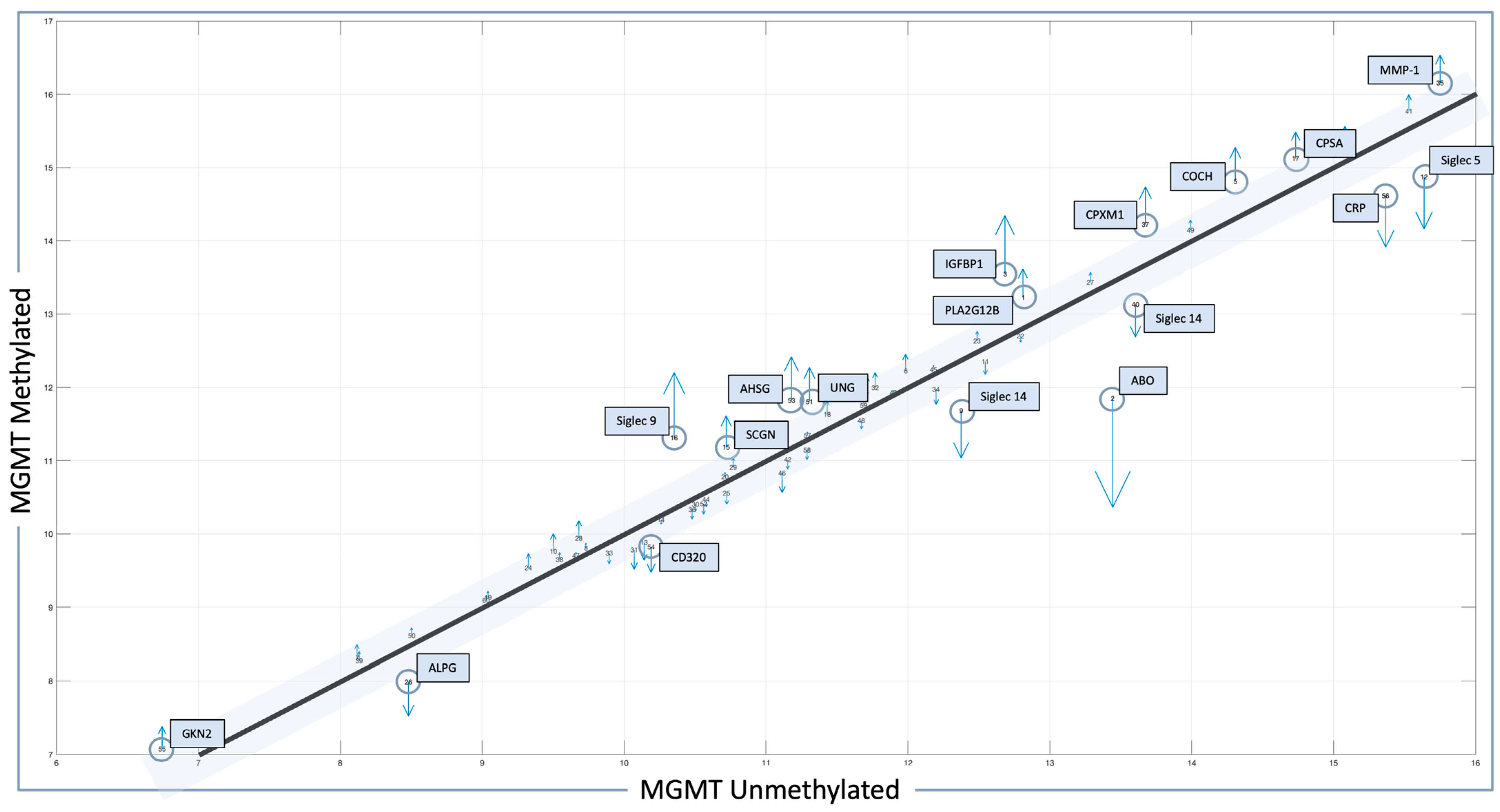
Figure 1 could be considered more optimal candidates for treatment with agents other than TMZ [
15,
54]. Limitations of this study include the large period over which patients were diagnosed and treated, and the difficulty in comparing protein signal originating in serum to protein signal measured in tissue. MGMT status was established through different methods in this study vs. in CPTAC data [
20]. Future directions of our research include the validation and clinical translation of serum biomarkers into clinical trials. Validation is subject to comparative analyses with serum and plasma proteomic datasets, several of which are currently evolving [
34]. Clinical translations are contingent on the implementation of serum markers in GBM trials. While over 30 trials are ongoing that aim to leverage biomarkers for the diagnosis or management of GBM [
55], most involve tissue as the biospecimen of origin. A shift to liquid biopsies is needed; however, a template for the real-time measurement of novel serum markers needs to be established for analysis to occur in the clinic to direct management in real-time, and this is the subject of future directions.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}