

Immune-Related Molecules CD3G and FERMT3: Novel Biomarkers Associated with Sepsis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
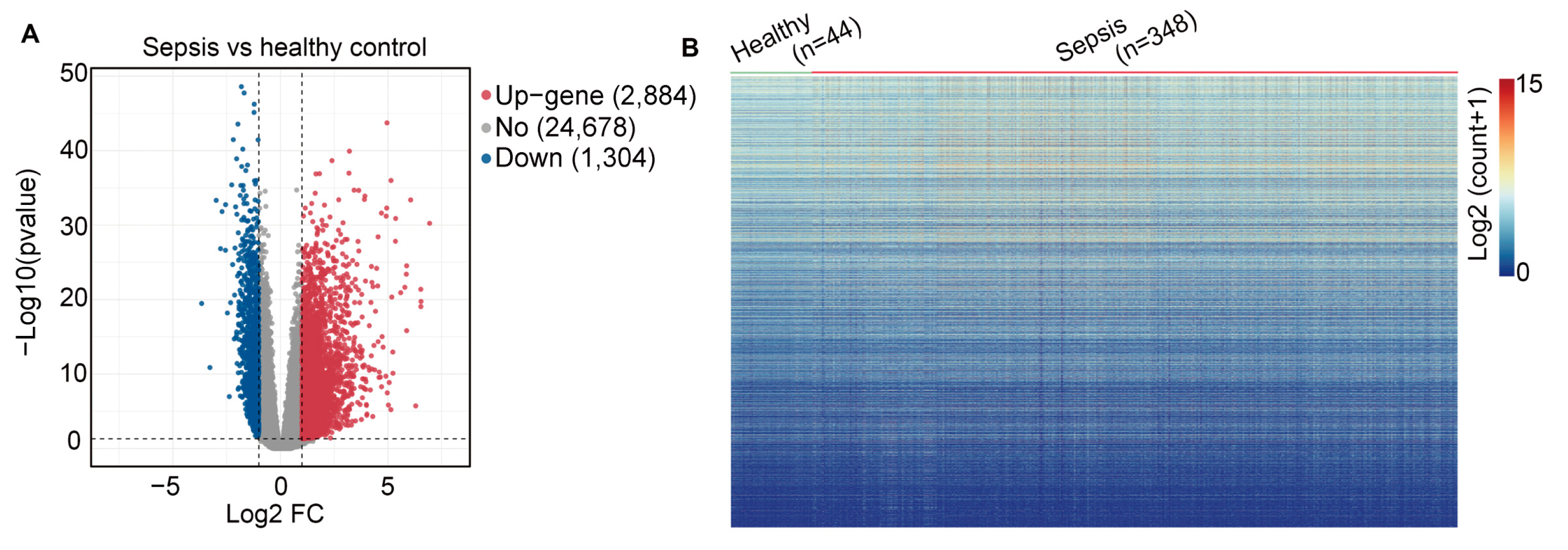
2.1. Data Processing and Identification of Differentially Expressed Genes (DEGs)
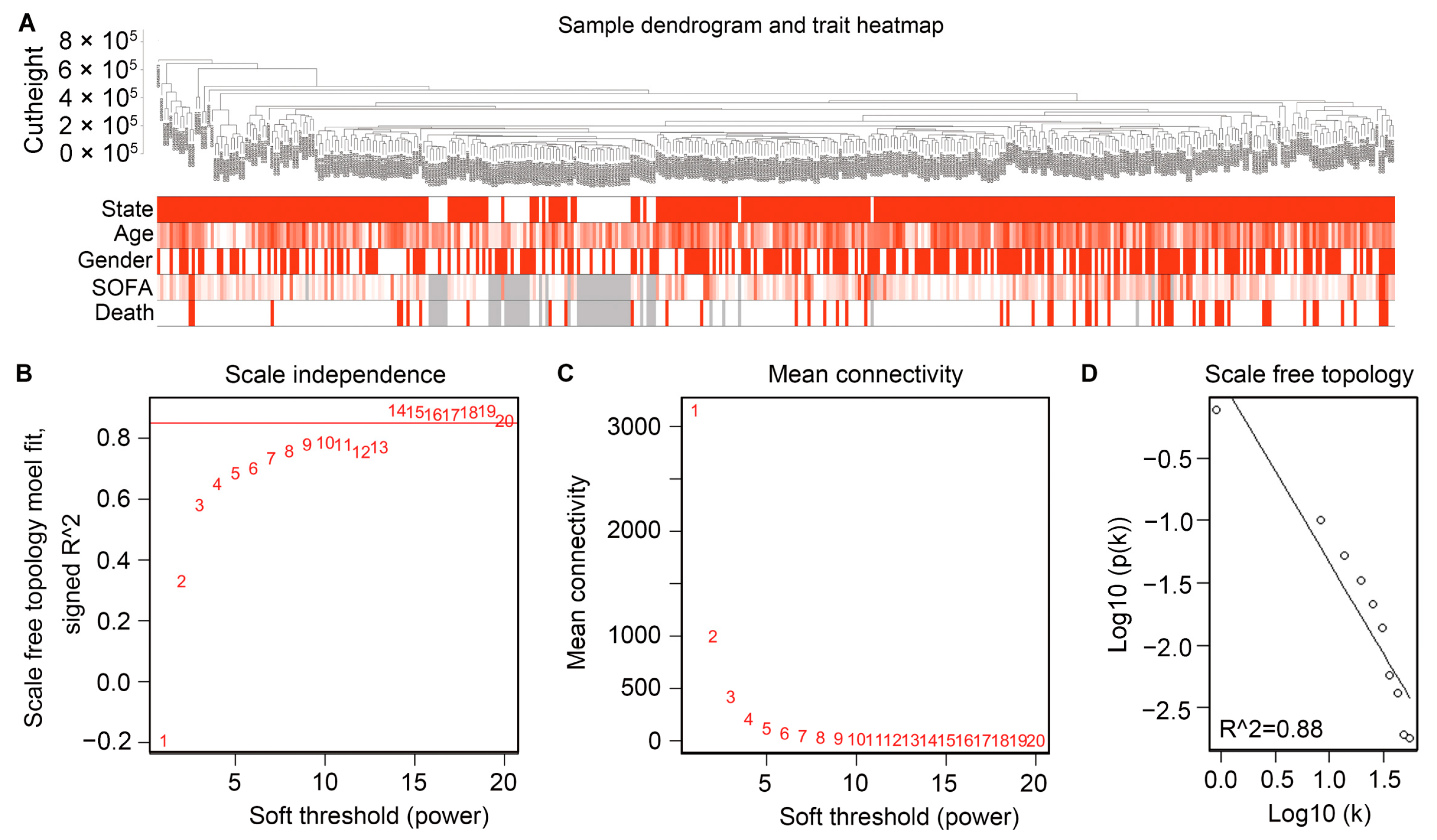
2.2. Filtering the Gene Modules by Constructing WGCNA
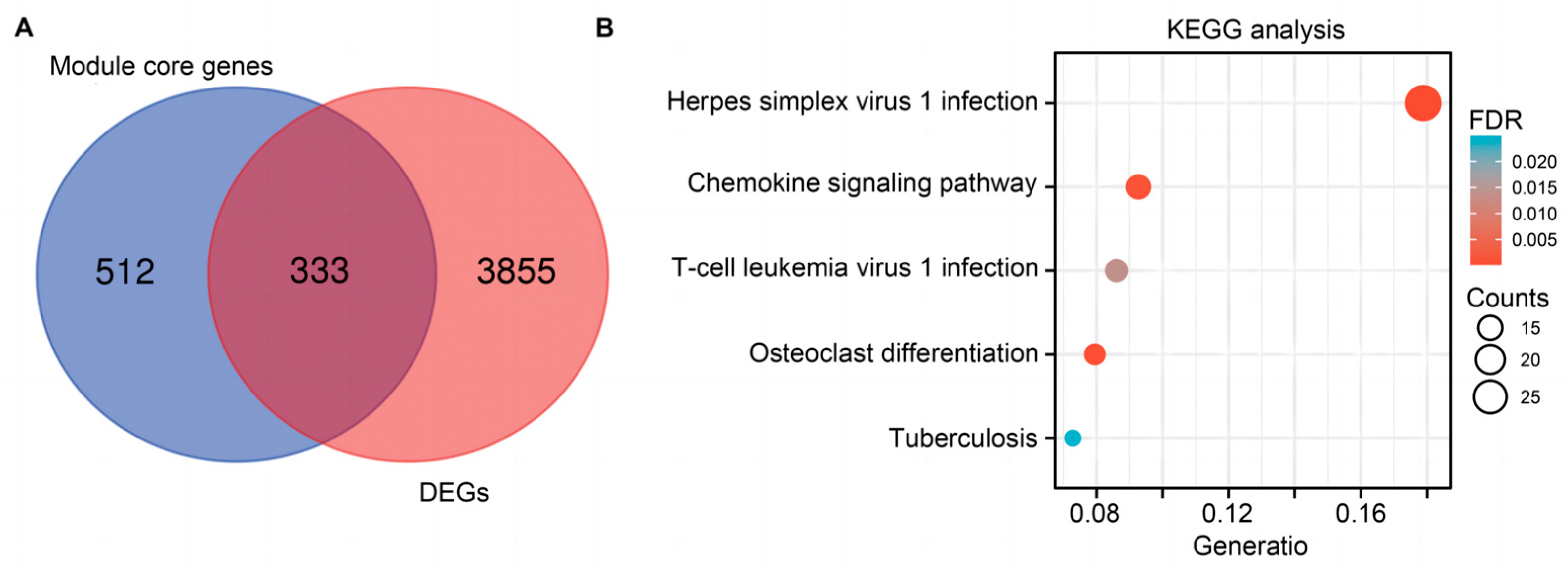
2.3. Functional and Pathway Enrichment Analysis for Hub Genes
2.4. PPI Network Construction
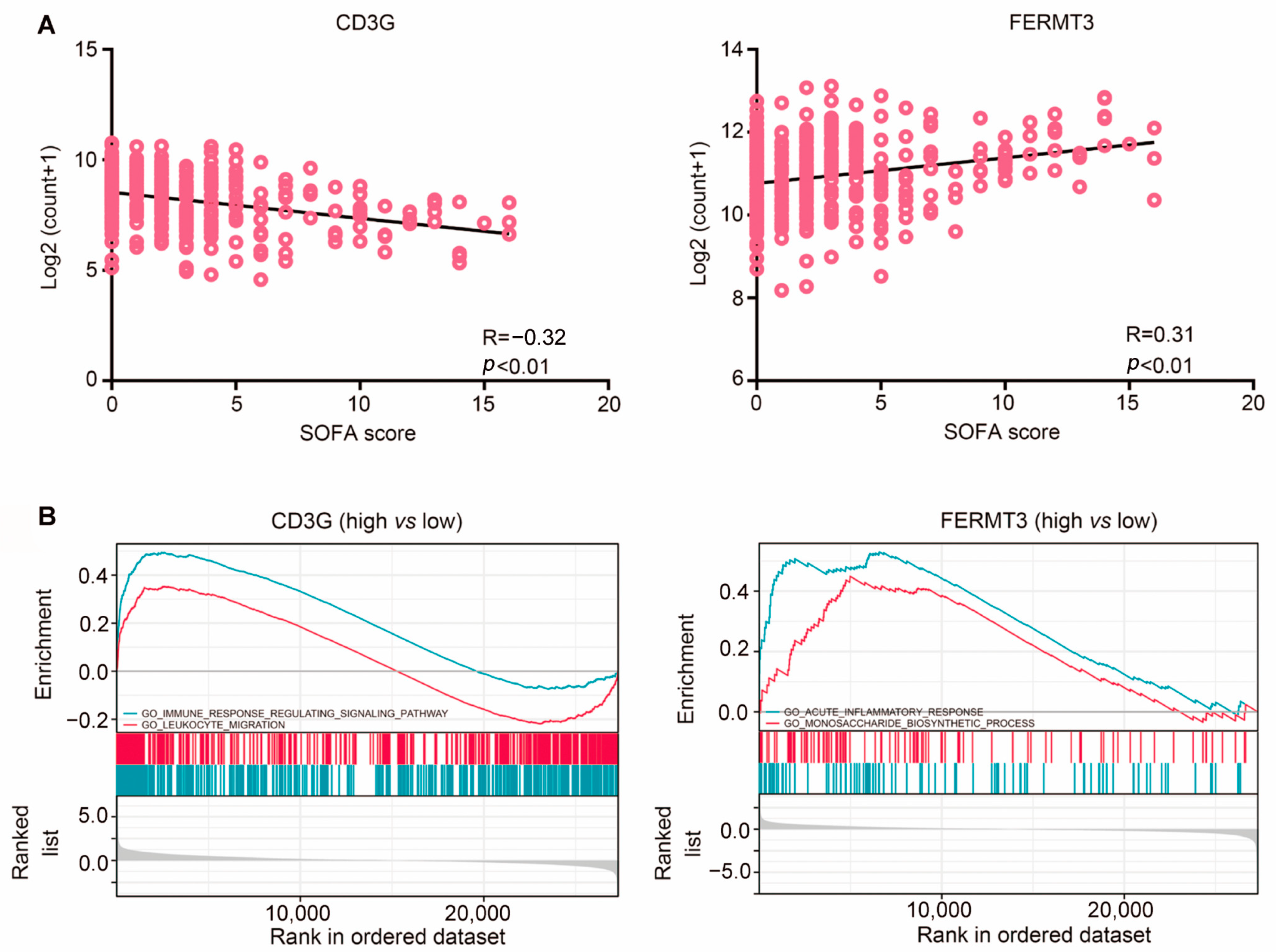
2.5. Gene Set Enrichment Analysis (GSEA) Analysis of Two Hub Genes Revealed Their Involvement of the Immune System
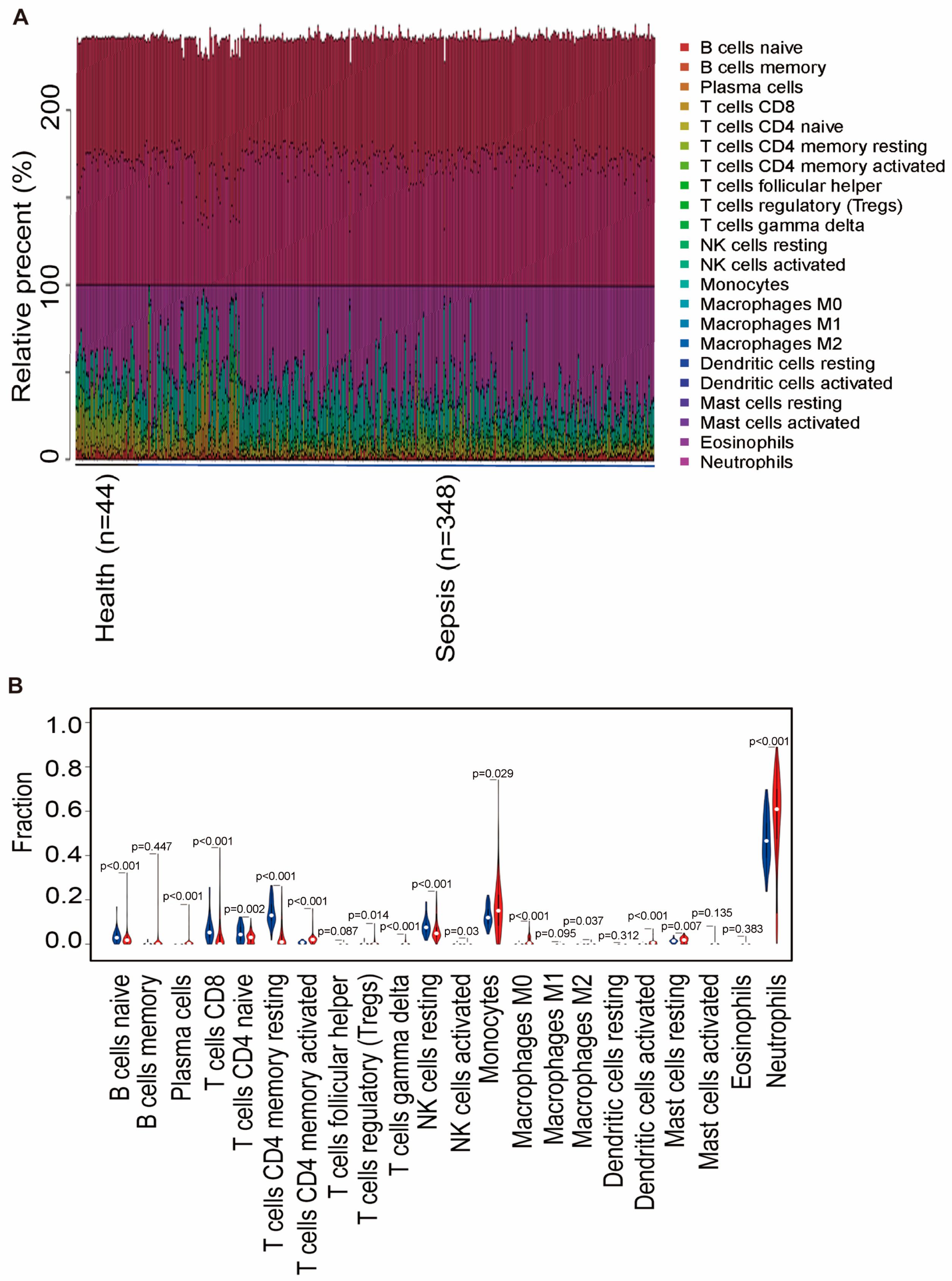
2.6. Two Hub Genes Involved in Immune Cell Infiltration
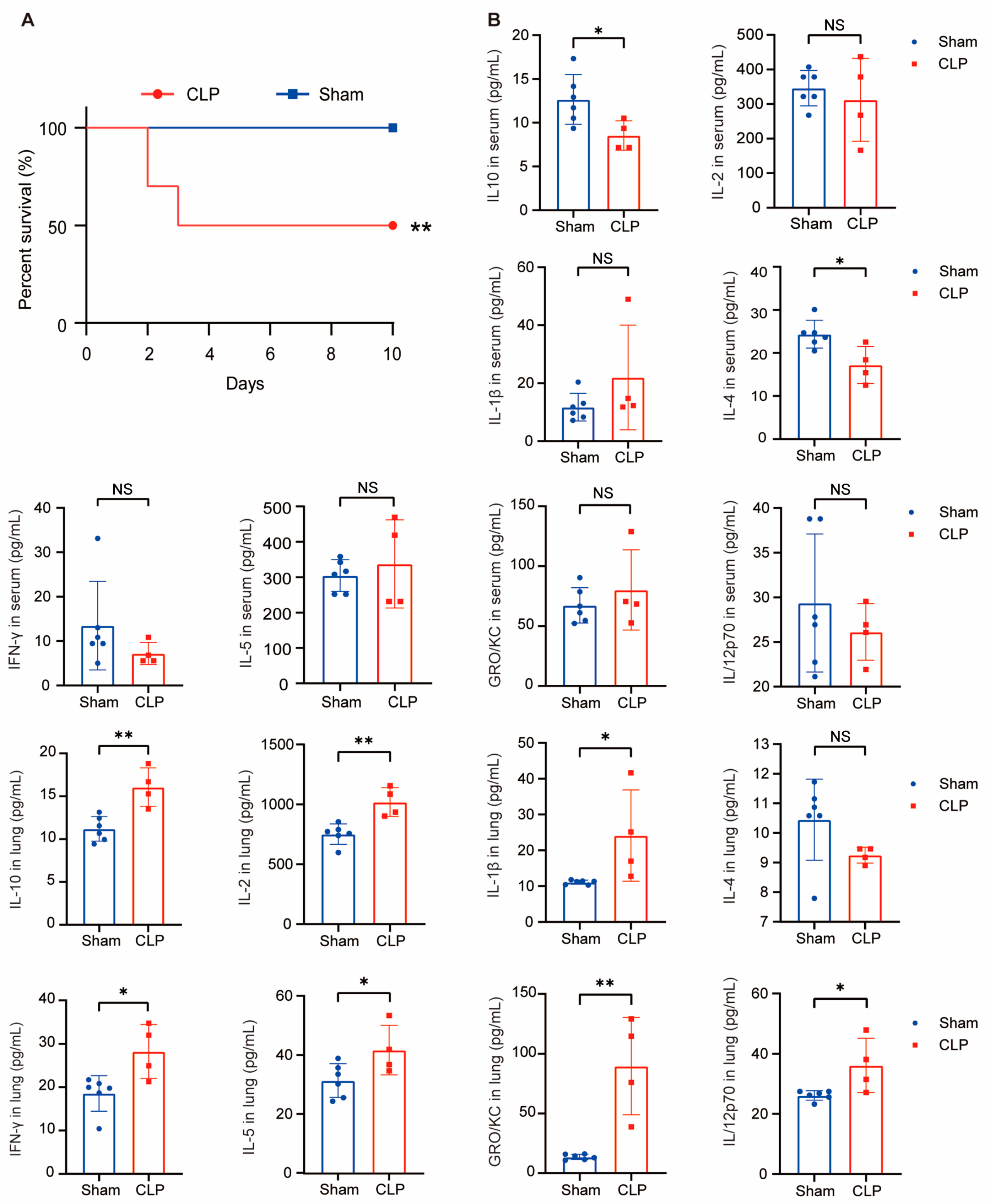
2.7. Sepsis Mice Model Construction and Cytokines Detecion In Vivo
2.8. The Expression of FERMT3 and CD3G in Sepsis Mice
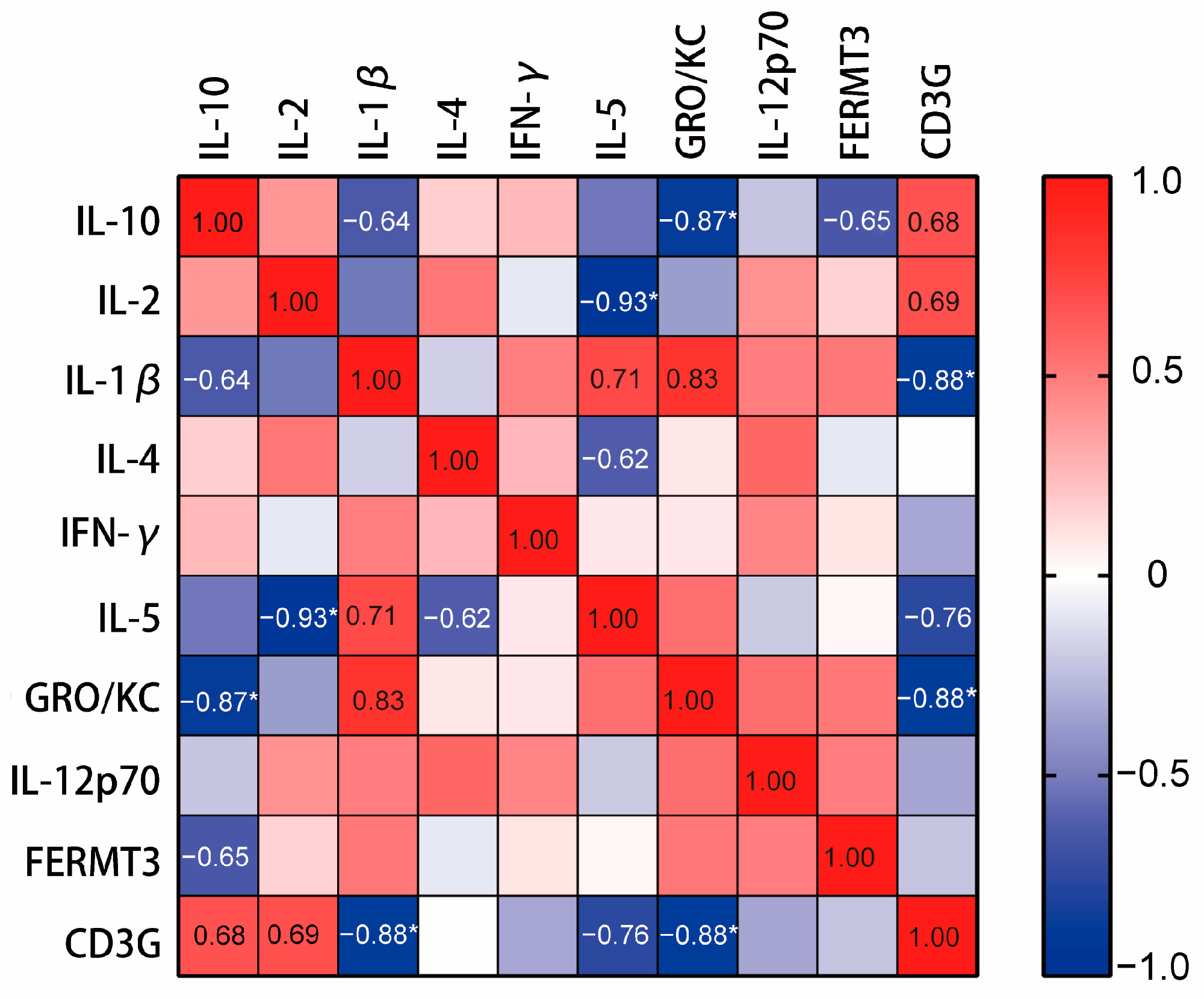
2.9. CD3Gregulated the Regulation of Cytokines In Vivo
3. Discussion
4. Materials and Methods
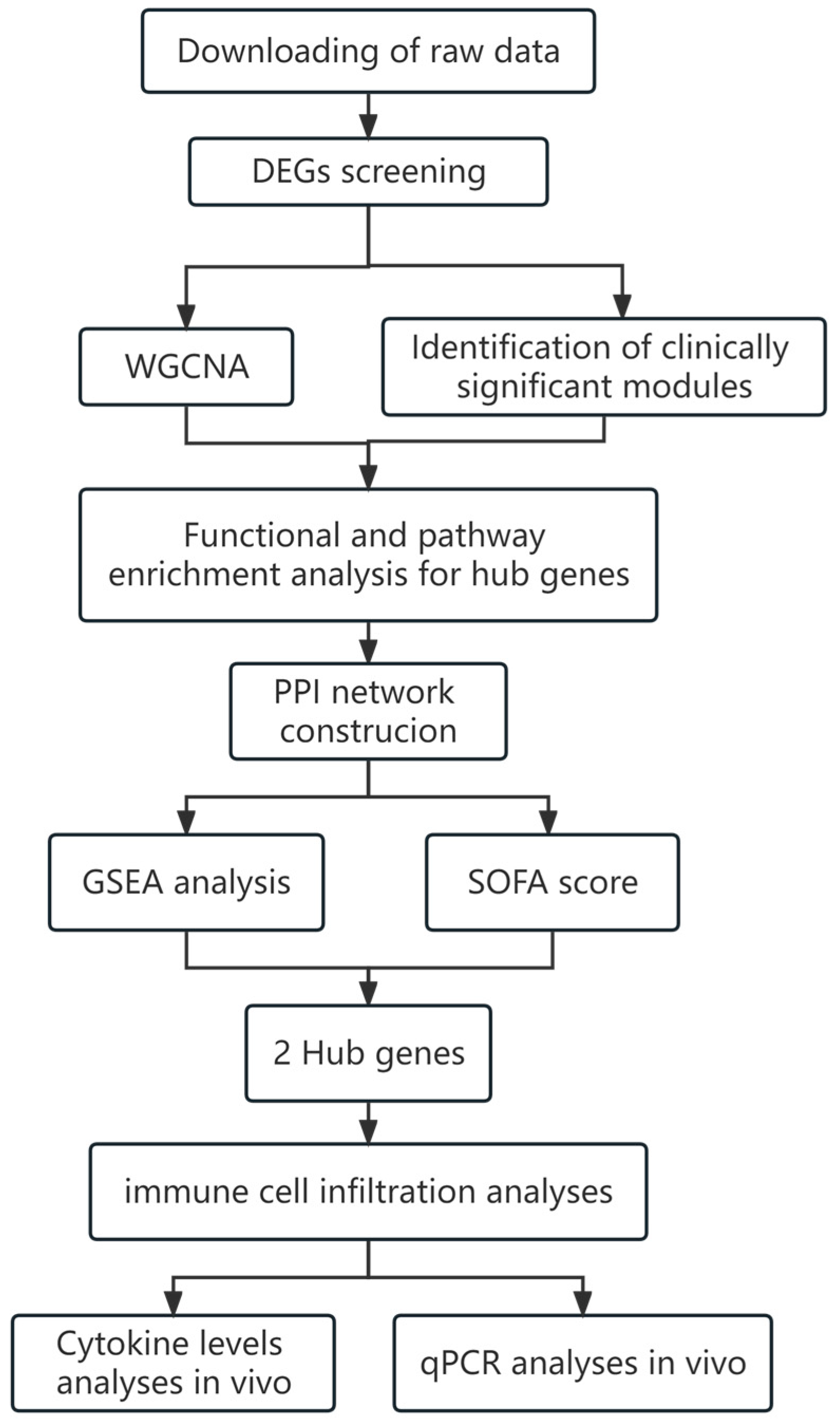
4.1. Experimental Design and Process
4.2. Data Processing and Identification of DEGs
4.3. Differentially Expressed Genes (DEGs) Screening
4.4. Weighted Gene Co-Expression Network Analysis
4.5. Identification of Clinically Significant Modules
4.6. Functional Enrichment Analyses
4.7. Construction of PPI Network and Identification of Hub Genes
4.8. Gene Set Enrichment Analysis Construction
4.9. Evaluating the Immune Cell Infiltration
4.10. In Vivo Validation Experiment of Animal Models
4.10.1. Sepsis Mice Model
4.10.2. qRT-PCR Detecion of FERMT3 and CD3G Gene Expression Levels
- 5′-CAGGCTGACCTCTGAAGGAA-3′ (FERMT3 forward primer),
- 5′-GGCTGTTTCTGCTAGCCTG-3′ (FERMT3 reverse primer),
- 5′-CTTCTGACTTGTGGCTTGACTGAC-3′ (CD3G forward primer),
- 5′-CCTTTGCTCCTTGACACTGATACG-3′ (CD3G reverse primer),
- 5′-ACGGCAAATTCAACGGCACAG-3′ (GAPDH forward primer),
- 5′-ACACCAGTAGACTCCACGACATAC-3′ (GAPDH reverse primer).
4.10.3. Inflammatory Cytokine Measurements
4.10.4. Correlation Analysis on the Hub Genes and Cytokines
4.10.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Gauer, R.; Forbes, D.; Boyer, N. Sepsis: Diagnosis and Management. Am. Fam. Physician 2020, 101, 409–418. [Google Scholar] [PubMed]
- Ferreira, F.L.; Bota, D.P.; Bross, A.; Mélot, C.; Vincent, J.L. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA 2001, 286, 1754. [Google Scholar] [CrossRef] [PubMed]
- Pierrakos, C.; Vincent, J.L. Sepsis biomarkers: A review. Crit. Care 2010, 14, R15. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.-M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight 2020, 5, e140289. [Google Scholar] [CrossRef]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Qi, X.; Yu, Y.; Sun, R.; Huang, J.; Liu, L.; Yang, Y.; Rui, T.; Sun, B. Identification and characterization of neutrophil heterogeneity in sepsis. Crit. Care 2021, 25, 50. [Google Scholar] [CrossRef]
- Lerman, Y.V.; Kim, M. Neutrophil Migration Under Normal and Sepsis Conditions. Cardiovasc. Hematol. Disord. Drug Targets 2015, 15, 19–28. [Google Scholar] [CrossRef]
- Chaturvedi, V.; Marsh, R.A.; Zoref-Lorenz, A.; Owsley, E.; Chaturvedi, V.; Nguyen, T.C.; Goldman, J.R.; Henry, M.M.; Greenberg, J.N.; Ladisch, S.; et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood 2021, 137, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.M.; Carlet, J.M.; Bion, J.; Parker, M.M.; Jaeschke, R.; Reinhart, K.; Angus, D.C.; Brun-Buisson, C.; Beale, R.; et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2007, 34, 17–60. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Wang, J.; Li, L.; Lin, X.; Wu, G.; Chen, J.; Zeng, Z.; Zhang, H. Identification of key genes involved in the recurrence of glioblastoma multiforme using weighted gene co-expression network analysis and differential expression analysis. Bioengineered 2021, 12, 3188–3200. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zheng, P.; Luo, D.; Xie, Q.; Liu, F.; Shao, Q.; Zhao, N.; Qian, K. LPIN1 Is a Regulatory Factor Associated with Immune Response and Inflammation in Sepsis. Front. Immunol. 2022, 13, 820164. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, J.; Lu, L.; Tian, W.; Ma, J.; Wu, M. Identification of key genes and novel immune infiltration-associated biomarkers of sepsis. Innate Immunol. 2020, 26, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Godini, R.; Fallahi, H.; Ebrahimie, E. Network analysis of inflammatory responses to sepsis by neutrophils and peripheral blood mononuclear cells. PLoS ONE 2018, 13, e0201674. [Google Scholar]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Toronen, P. Selection of informative clusters from hierarchical cluster tree with gene classes. BMC Bioinform. 2004, 5, 32. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 155545. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Faix, J.D. Biomarkers of sepsis. Crit. Rev. Clin. Lab. Sci. 2013, 50, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, R.; Martin-Loeches, I.; Phillips, G.; Osborn, T.M.; Townsend, S.; Dellinger, R.P.; Artigas, A.; Schorr, C.; Levy, M.M. Empiric Antibiotic Treatment Reduces Mortality in Severe Sepsis and Septic Shock From the First Hour. Crit. Care Med. 2014, 42, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; Generoso, J.S.; Singer, M.; Dal-Pizzol, F. Biomarkers for sepsis: More than just fever and leukocytosis—A narrative review. Crit. Care 2022, 26, 14. [Google Scholar] [CrossRef] [PubMed]
- Kambli, P.M.; Bargir, U.A.; Yadav, R.M.; Gupta, M.R.; Dalvi, A.D.; Hule, G.; Kelkar, M.; Sawant-Desai, S.; Setia, P.; Jodhawat, N.; et al. Clinical and Genetic Spectrum of a Large Cohort of Patients With Leukocyte Adhesion Deficiency Type 1 and 3: A Multicentric Study from India. Front. Immunol. 2020, 11, 612703. [Google Scholar] [CrossRef] [PubMed]
- Manukjan, G.; Wiegering, V.A.; Reindl, T.; Strauß, G.; Klopocki, E.; Schulze, H.; Andres, O. Novel variants in FERMT3 and RASGRP2—Genetic linkage in Glanzmann-like bleeding disorders. Pediatr. Blood Cancer 2019, 67, e28078. [Google Scholar] [CrossRef] [PubMed]
- Essa, M.F.; Elbashir, E.; Alroqi, F.; Mohammed, R.; Alsultan, A. Successful hematopoietic stem cell transplant in leukocyte adhesion deficiency type III presenting primarily as malignant infantile osteopetrosis. Clin. Immunol. 2020, 213, 108365. [Google Scholar] [CrossRef]
- Levi, M.; Poll, T. Coagulation in Patients with Severe Sepsis. Semin. Thromb. Hemost. 2015, 41, 009–015. [Google Scholar] [CrossRef]
- Ogura, H.; Gando, S.; Saitoh, D.; Takeyama, N.; Kushimoto, S.; Fujishima, S.; Mayumi, T.; Araki, T.; Ikeda, H.; Kotani, J.; et al. Epidemiology of severe sepsis in Japanese intensive care units: A prospective multicenter study. J. Infect. Chemother. 2014, 20, 157–162. [Google Scholar] [CrossRef]
- Almansa, R.; Heredia-Rodríguez, M.; Gomez-Sanchez, E.; Andaluz-Ojeda, D.; Iglesias, V.; Rico, L.; Ortega, A.; Gomez-Pesquera, E.; Liu, P.; Aragón, M.; et al. Transcriptomic correlates of organ failure extent in sepsis. J. Infect. 2015, 70, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Frimpong, A.; Owusu, E.D.A.; Amponsah, J.A.; Obeng-Aboagye, E.; Puije, W.V.D.; Frempong, A.F.; Kusi, K.A.; Ofori, M.F. Cytokines as Potential Biomarkers for Differential Diagnosis of Sepsis and Other Non-Septic Disease Conditions. Front. Cell. Infect. Microbiol. 2022, 12, 901433. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor. Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Mera, S.; Tatulescu, D.; Cismaru, C.; Bondor, C.; Slavcovici, A.; Zanc, V.; Carstina, D.; Oltean, M. Multiplex cytokine profiling in patients with sepsis. Apmis 2010, 119, 155–163. [Google Scholar]
- Wang, Y.; Huang, Q.; He, F. Aberrant blood MALT1 and its relevance with multiple organic dysfunctions, T helper cells, inflammation, and mortality risk of sepsis patients. J. Clin. Lab. Anal. 2022, 36, e24331. [Google Scholar] [CrossRef]
- Simonsen, K.A.; Anderson-Berry, A.L.; Delair, S.F.; Davies, H.D. Early-Onset Neonatal Sepsis. Clin. Microbiol. Rev. 2014, 27, 21–47. [Google Scholar] [CrossRef]
- Gharamti, A.A.; Samara, O.; Monzon, A.; Montalbano, G.; Scherger, S.; DeSanto, K.; Chastain, D.B.; Sillau, S.; Montoya, J.G.; Franco-Paredes, C.; et al. Proinflammatory cytokines levels in sepsis and healthy volunteers, and tumor necrosis factor-alpha associated sepsis mortality: A systematic review and meta-analysis. Cytokine 2022, 158, 156006. [Google Scholar] [CrossRef]
- Chimenti, L.; Morales-Quinteros, L.; Puig, F.; Camprubi-Rimblas, M.; Guillamat-Prats, R.; Gómez, M.N.; Tijero, J.; Blanch, L.; Matute-Bello, G.; Artigas, A. Comparison of direct and indirect models of early induced acute lung injury. Intensive Care Med. Exp. 2020, 8, 62. [Google Scholar]
- Pollard, B.S.; Blancol, J.C.; Pollard, J.R. Classical Drug Digitoxin Inhibits Influenza Cytokine Storm, With Implications for COVID-19 Therapy. In Vivo 2020, 34, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Sadikot, R.T.; Blackwell, T.S.; Christman, J.W.; Prince, A.S. Pathogen–Host Interactions inPseudomonas aeruginosaPneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Ertel, W.; Kremer, J.P.; Kenney, J.; Steckholzer, U.; Jarrar, D.; Trentz, O.; Schildberg, F.W. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood 1995, 85, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Grätz, C.; Bui, M.L.U.; Thaqi, G.; Kirchner, B.; Loewe, R.P.; Pfaffl, M.W. Obtaining Reliable RT-qPCR Results in Molecular Diagnostics—MIQE Goals and Pitfalls for Transcriptional Biomarker Discovery. Life 2022, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Baghela, A.; Pena, O.M.; Lee, A.H.; Baquir, B.; Falsafi, R.; An, A.; Farmer, S.W.; Hurlburt, A.; Mondragon-Cardona, A.; Rivera, J.D.; et al. Predicting sepsis severity at first clinical presentation: The role of endotypes and mechanistic signatures. eBioMedicine 2022, 75, 103776. [Google Scholar] [CrossRef]
- Ren, P.; Wang, J.-Y.; Chen, H.-L.; Lin, X.-W.; Zhao, Y.-Q.; Guo, W.-Z.; Zeng, Z.-R.; Li, Y.-F. Diagnostic model constructed by nine inflammation-related genes for diagnosing ischemic stroke and reflecting the condition of immune-related cells. Front. Immunol. 2022, 13, 1046966. [Google Scholar] [CrossRef]
- Albert, R.K. Scale-free networks in cell biology. J. Cell Sci. 2005, 118, 4947–4957. [Google Scholar] [CrossRef]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2008, 4, 31–36. [Google Scholar] [CrossRef]
- Seymour, C.W.; Kerti, S.J.; Lewis, A.J.; Kennedy, J.; Brant, E.; Griepentrog, J.E.; Zhang, X.; Angus, D.C.; Chang, C.-C.H.; Rosengart, M.R. Murine sepsis phenotypes and differential treatment effects in a randomized trial of prompt antibiotics and fluids. Crit. Care 2019, 23, 384. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Ren, P.; Wang, J.; Zhu, X.; Qiao, X.; Zeng, Z.; Ye, T.; Wang, S.; Meng, Z.; Gan, H.; et al. Immune-Related Molecules CD3G and FERMT3: Novel Biomarkers Associated with Sepsis. Int. J. Mol. Sci. 2024, 25, 749. https://doi.org/10.3390/ijms25020749
Li N, Ren P, Wang J, Zhu X, Qiao X, Zeng Z, Ye T, Wang S, Meng Z, Gan H, et al. Immune-Related Molecules CD3G and FERMT3: Novel Biomarkers Associated with Sepsis. International Journal of Molecular Sciences. 2024; 25(2):749. https://doi.org/10.3390/ijms25020749
Chicago/Turabian StyleLi, Nanxi, Peng Ren, Jingya Wang, Xiaohui Zhu, Xuan Qiao, Zhirui Zeng, Tong Ye, Shanshan Wang, Zhiyun Meng, Hui Gan, and et al. 2024. "Immune-Related Molecules CD3G and FERMT3: Novel Biomarkers Associated with Sepsis" International Journal of Molecular Sciences 25, no. 2: 749. https://doi.org/10.3390/ijms25020749
APA StyleLi, N., Ren, P., Wang, J., Zhu, X., Qiao, X., Zeng, Z., Ye, T., Wang, S., Meng, Z., Gan, H., Liu, S., Sun, Y., Zhu, X., Dou, G., & Gu, R. (2024). Immune-Related Molecules CD3G and FERMT3: Novel Biomarkers Associated with Sepsis. International Journal of Molecular Sciences, 25(2), 749. https://doi.org/10.3390/ijms25020749