
Transcriptome and Metabolome Reveal Accumulation of Key Metabolites with Medicinal Properties of Phylloporia pulla


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome Features
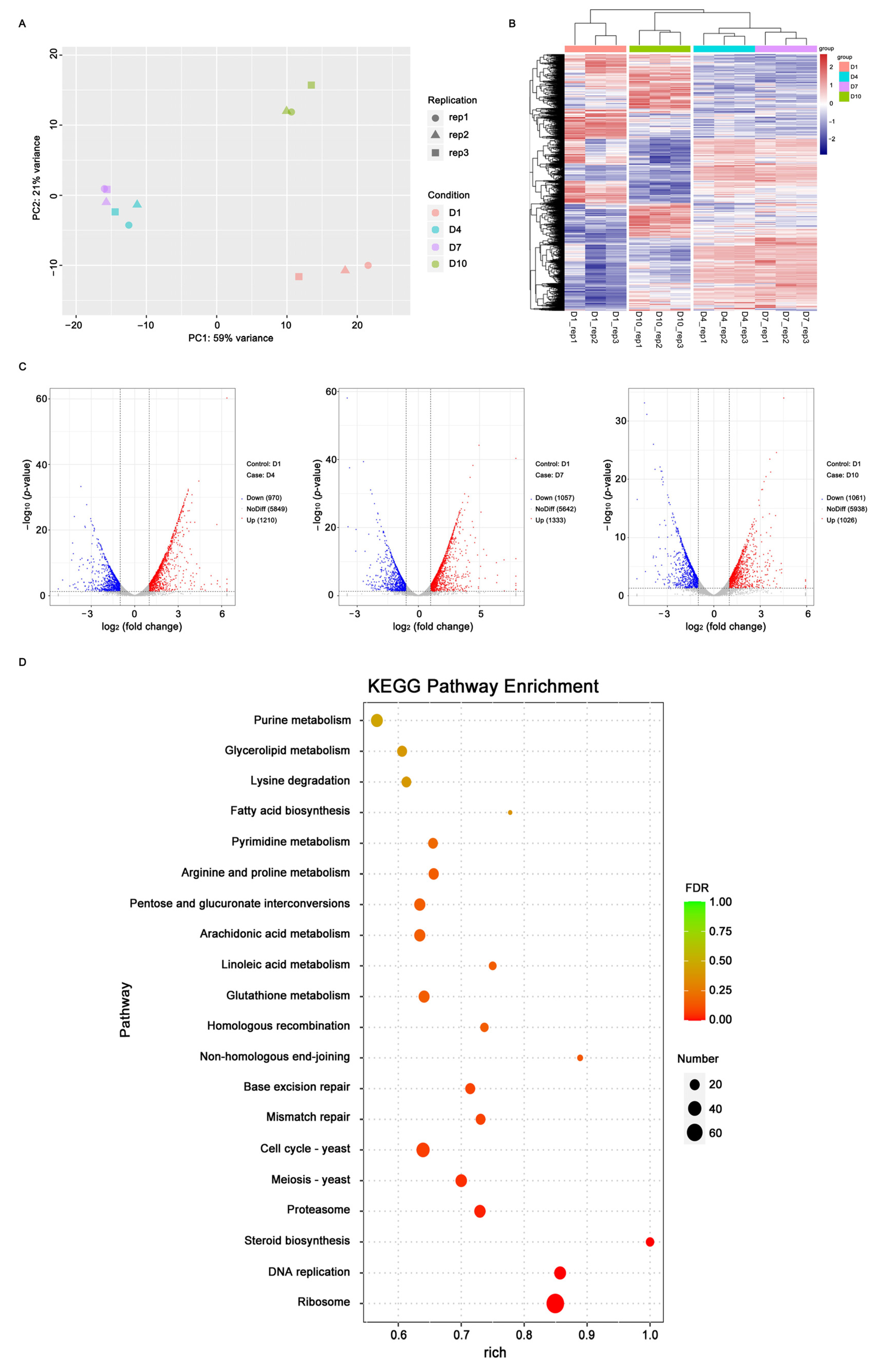
2.2. Gene Expression Profiles at Different Growth Times
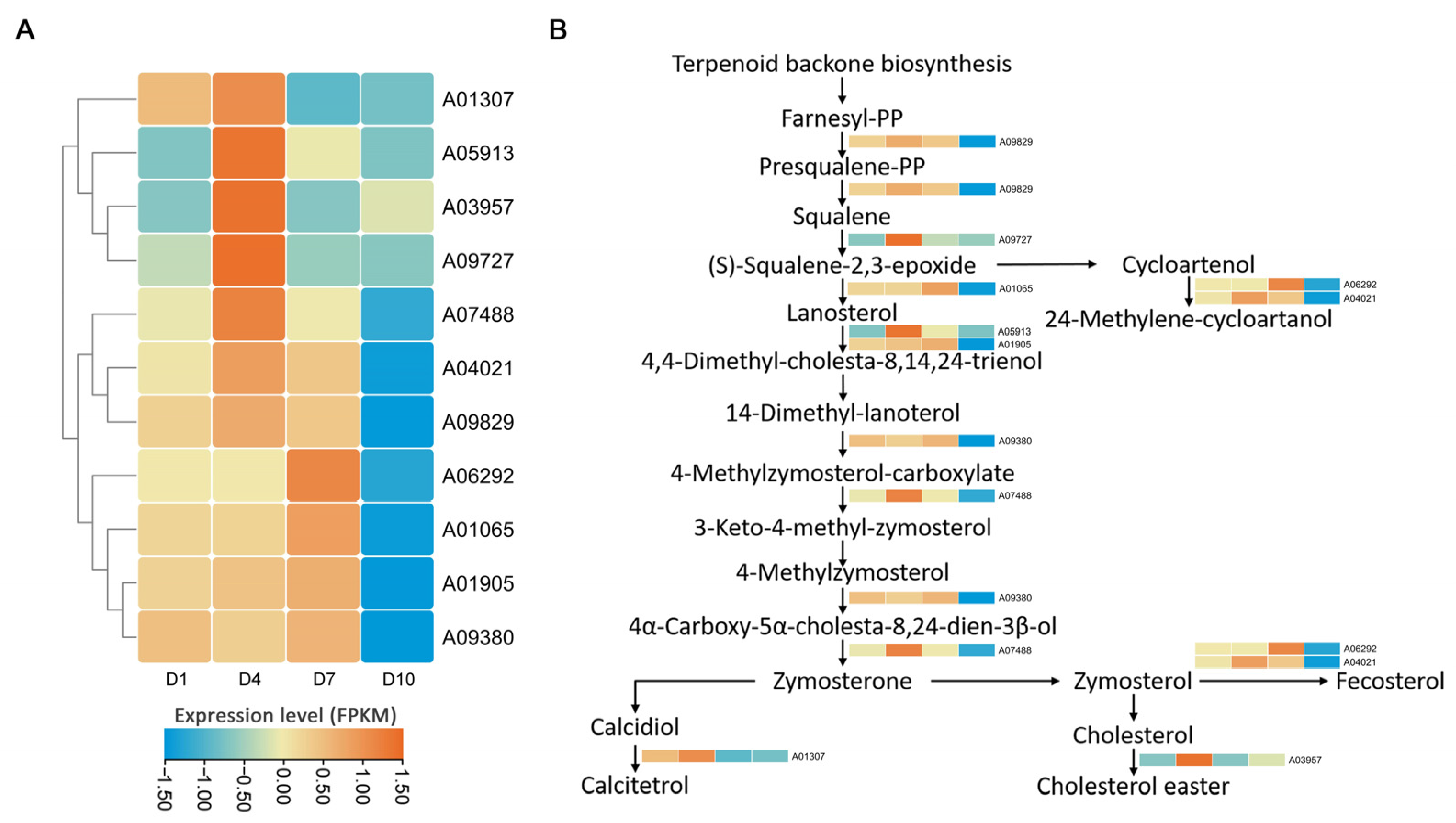
2.3. DEGs Involving the Steroid and Terpenoid Biosynthesis
2.4. Validation of RNA-Seq Data
2.5. Metabolome Profiles at Different Growth Times
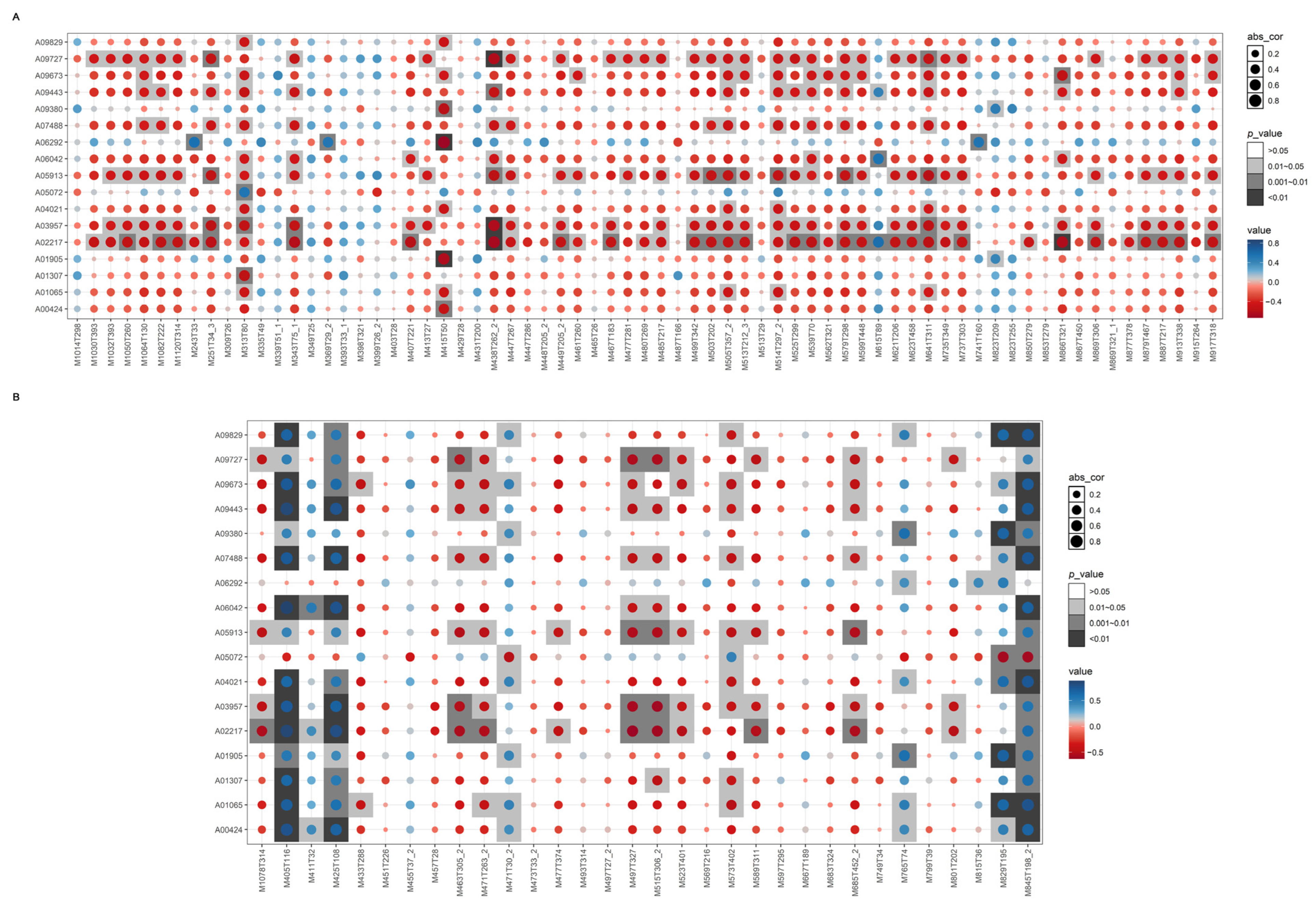
2.6. Integrative Analysis of Transcriptome and Metabolome
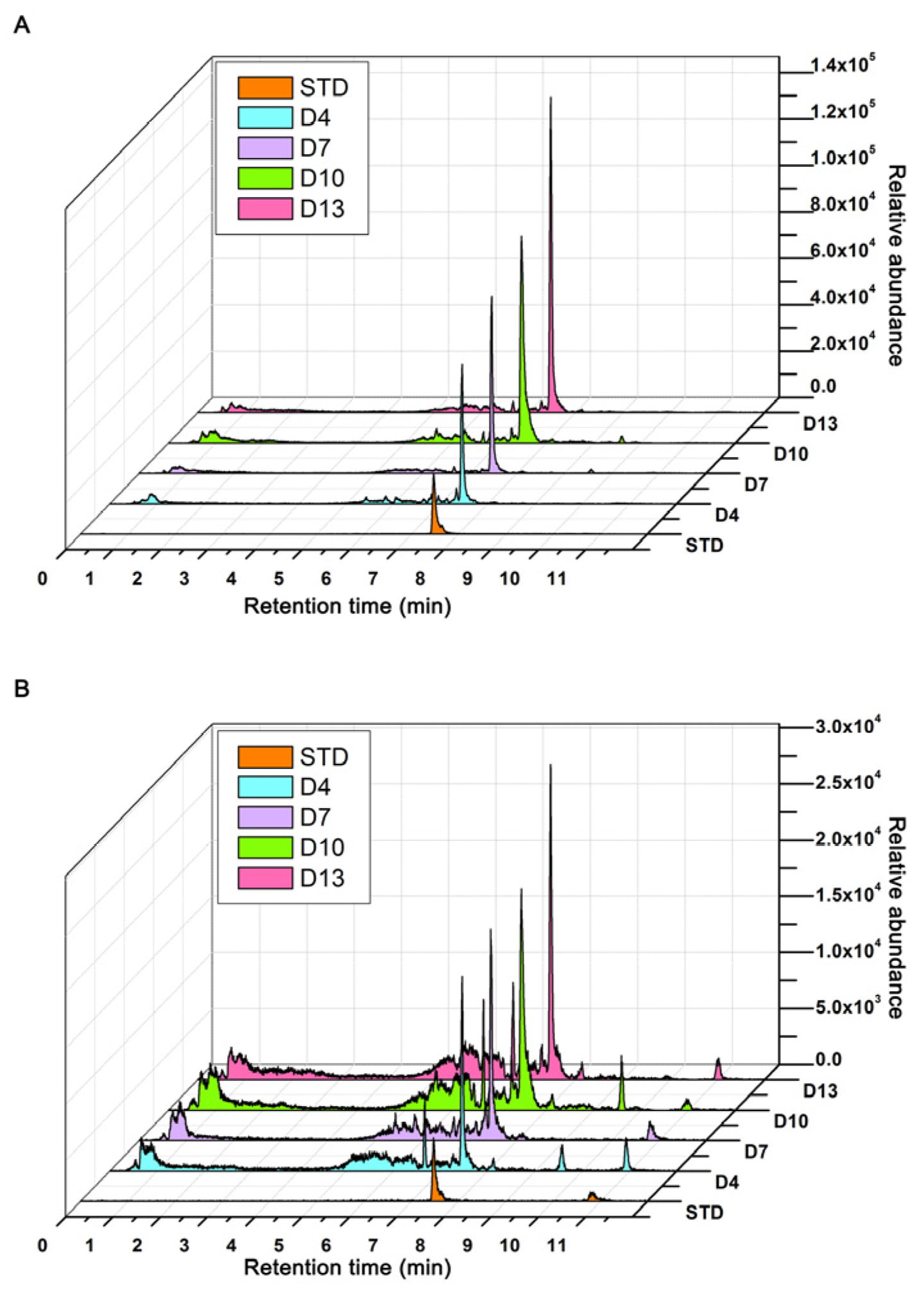
2.7. Targeted Detection of Celastrol
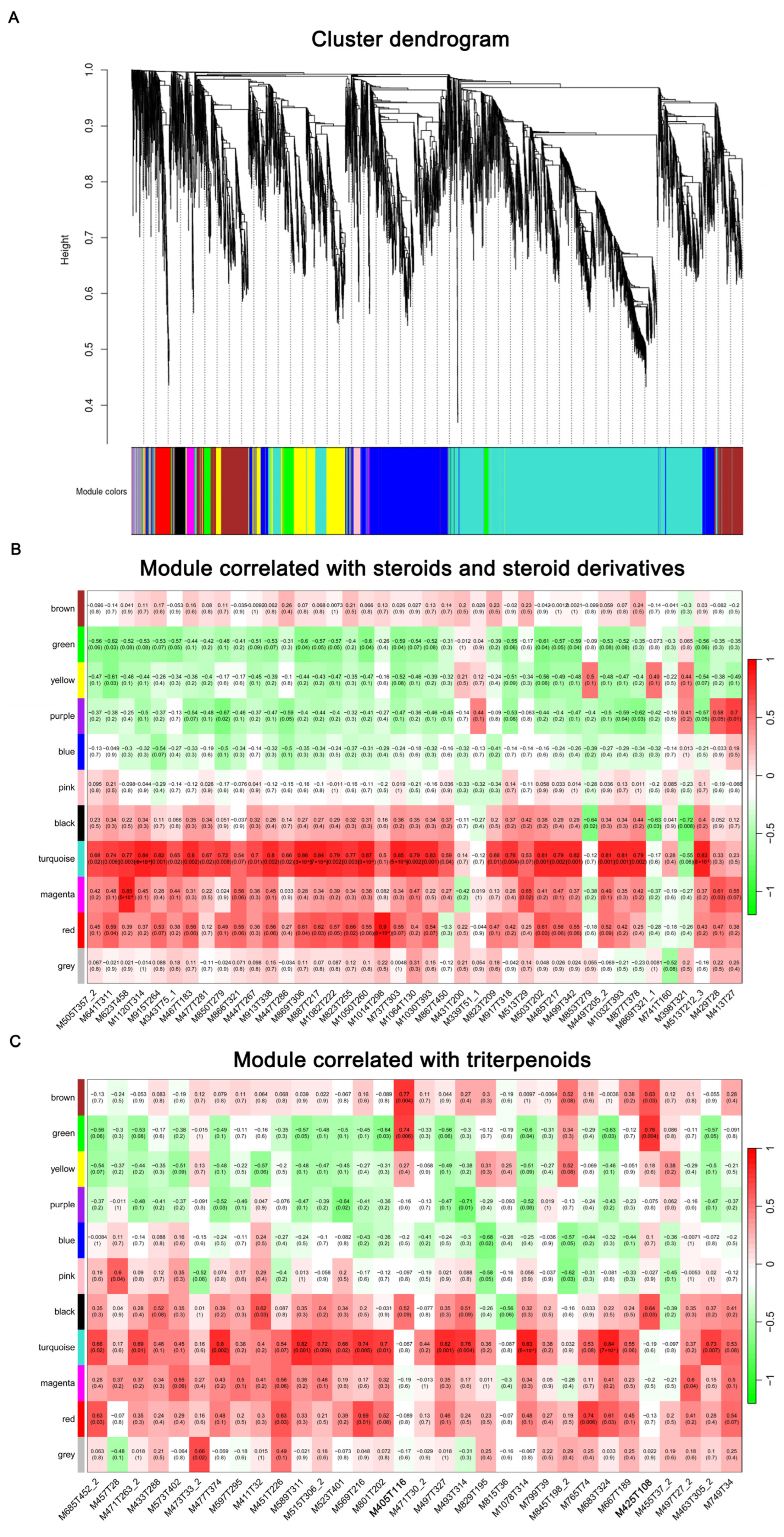
2.8. WGCNA
3. Discussion
4. Materials and Methods
4.1. Strain Cultivation and Genome Sequencing
4.2. RNA Extraction, Sequencing and Data Analyses
4.3. Real-Time Quantitative PCR (qPCR)
4.4. Metabolome Identification and Quantification
4.5. Determination of Triterpenes
4.6. Targeted Detection of Celastrol by LC-MS/MS
4.7. Weighted Gene Co-Expression Network Analysis
4.8. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, L. Systematics is crucial for the traditional Chinese medicinal studies and industry of macrofungi. Fungal Biol. Rev. 2020, 34, 10–12. [Google Scholar] [CrossRef]
- Zhou, L.; Ghobad-Nejhad, M.; Tian, X.; Wang, Y.; Wu, F. Current Status of ‘Sanghuang’ as a Group of Medicinal Mushrooms and Their Perspective in Industry Development. Food Rev. Int. 2022, 38, 589–607. [Google Scholar] [CrossRef]
- Song, T.; Zhang, Z.; Jin, Q.; Feng, W.; Shen, Y.; Fan, L.; Cai, W. Nutrient profiles, functional compositions, and antioxidant activities of seven types of grain fermented with Sanghuangporus sanghuang fungus. J. Food Sci. Technol. 2021, 58, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Jiang, J.; Liu, S.; Gao, J.; Zhou, L. Metabolomics analysis of rice fermented by medicinal fungi providing insights into the preparation of functional food. Food Chem. 2024, 459, 140372. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhou, L.; Jiang, J.; Tian, X.; Zhou, L. Phylloporia (Hymenochaetales, Basidiomycota), a Medicinal Wood-inhabiting Fungal Genus with Much Potential for Commercial Development. Food Rev. Int. 2023, 39, 2776–2789. [Google Scholar] [CrossRef]
- Qin, W.; Wang, X.; Sawahata, T.; Zhou, L. Phylloporia lonicerae (Hymenochaetales, Basidiomycota), a new species on Lonicera japonica from Japan and an identification key to worldwide species of Phylloporia. MycoKeys 2018, 30, 17–30. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, L.; Vlasák, J.; Dai, Y. Global Diversity and Systematics of Hymenochaetaceae with Poroid Hymenophore. Fungal Divers. 2022, 113, 1–192. [Google Scholar] [CrossRef]
- Lu, L.; Fan, Y.; Yao, W.; Xie, W.; Guo, J.; Yan, Y.; Yang, F.; Xu, L. Safety assessment of the fermented Phylloporia ribis (Lonicera japonica Thunb.) mycelia by oral acute toxicity study in mice and 90-day feeding study in rats. Food Chem. Toxicol. 2014, 69, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Huang, X.; Cheng, L.; Bu, Y.; Liu, G.; Yi, F.; Yang, Z.; Song, F. Evaluation of antioxidant and immune activity of Phellinus ribis glucan in mice. Food Chem. 2009, 115, 581–584. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Jiang, H.; Xu, L.; Cheng, Y.; Wang, P.G.; Wang, F. Preparation, antiangiogenic and antitumoral activities of the chemically sulfated glucan from Phellinus ribis. Carbohydr. Polym. 2014, 106, 42–48. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, C.; Jiang, H.; Zhou, H.; Li, P.; Wang, F. Isolation, structural characterization and neurotrophic activity of a polysaccharide from Phellinus ribis. Carbohydr. Polym. 2015, 127, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, J.; Zong, A.; Wang, J.; Liu, Y.; Jia, W.; Jin, J.; Yan, G.; Zhang, Y. Anti-angiogenic activity and mechanism of a chemically sulfated natural glucan from Phellinus ribis. Int. J. Biol. Macromol. 2018, 107, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhou, R.; Lin, J.; Zang, X.; Wang, Q.; Wang, P.; Wang, L.; Li, Z.; Wang, W. Transcriptome and metabolome analyses reveal transcription factors regulating ganoderic acid biosynthesis in Ganoderma lucidum development. Front. Microbiol. 2022, 13, 956421. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, Y.; Zhang, X.; Wang, T.; Wang, G.; Zhou, L.; Yu, H.; Tian, X. Transcriptome and Metabolome Integration Reveals the Impact of Fungal Elicitors on Triterpene Accumulation in Sanghuangporus sanghuang. J. Fungi 2023, 9, 604. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.; Taghizadeh, M.S.; Haghi, R.; Tahmasebi, A.; Niazi, A.; Ebrahimie, E. Exploring novel insights: Methyl jasmonate treatment reveals novel lncRNA-mediated regulation of secondary metabolite biosynthesis pathways in Echinacea purpurea. Food Biosci. 2024, 57, 103457. [Google Scholar] [CrossRef]
- Moghadam, A.; Foroozan, E.; Tahmasebi, A.; Taghizadeh, M.S.; Bolhassani, M.; Jafari, M. System network analysis of Rosmarinus officinalis transcriptome and metabolome—Key genes in biosynthesis of secondary metabolites. PLoS ONE 2023, 18, e0282316. [Google Scholar] [CrossRef]
- Shao, Y.; Guo, H.; Zhang, J.; Liu, H.; Wang, K.; Zuo, S.; Xu, P.; Xia, Z.; Zhou, Q.; Zhang, H.; et al. The Genome of the Medicinal Macrofungus Sanghuang Provides Insights Into the Synthesis of Diverse Secondary Metabolites. Front. Microbiol. 2019, 10, 3035. [Google Scholar] [CrossRef]
- Yang, B.; He, S.; Liu, Y.; Liu, B.; Ju, Y.; Kang, D.; Sun, X.; Fang, Y. Transcriptomics integrated with metabolomics reveals the effect of regulated deficit irrigation on anthocyanin biosynthesis in Cabernet Sauvignon grape berries. Food Chem. 2020, 314, 126170. [Google Scholar] [CrossRef]
- Jiang, J.; Zhou, L.; Liu, S.; Zhou, L.; Tian, X. Species clarification of the medicinal wood-inhabiting fungus Phylloporia (Hymenochaetales, Basidiomycota) in China. Phytotaxa 2020, 446, 209–219. [Google Scholar] [CrossRef]
- Yang, L.; Gu, Y.; Zhou, J.; Yuan, P.; Jiang, N.; Wu, Z.; Tan, X. Whole-Genome Identification and Analysis of Multiple Gene Families Reveal Candidate Genes for Theasaponin Biosynthesis in Camellia oleifera. Int. J. Mol. Sci. 2022, 23, 6393. [Google Scholar] [CrossRef]
- Salvador, J.; Carvalho, J.; Neves, M.; Silvestre, S.; Leitão, A.; Silva, M.; Sá E Melo, M.L. Anticancer steroids: Linking natural and semi-synthetic compounds. Nat. Prod. Rep. 2012, 30, 324–374. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed]
- Huan, X.; Li, L.; Liu, Y.; Kong, Z.; Liu, Y.; Wang, Q.; Liu, J.; Zhang, P.; Guo, Y.; Qin, P. Integrating transcriptomics and metabolomics to analyze quinoa (Chenopodium quinoa Willd.) responses to drought stress and rewatering. Front. Plant Sci. 2022, 13, 988861. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Jeong, J.H.; Seo, J.W.; Shin, C.G.; Kim, Y.S.; In, J.G.; Yang, D.C.; Yi, J.S.; Choi, Y.E. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol. 2004, 45, 976–984. [Google Scholar] [CrossRef]
- Seo, J.W.; Jeong, J.H.; Shin, C.G.; Lo, S.C.; Han, S.S.; Yu, K.W.; Harada, E.; Han, J.Y.; Choi, Y.E. Overexpression of squalene synthase in Eleutherococcus senticosus increases phytosterol and triterpene accumulation. Phytochemistry 2005, 66, 869–877. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Wawrzak, Z.; Guengerich, F.P.; Lepesheva, G.I. Validation of Human Sterol 14α-Demethylase (CYP51) Druggability: Structure-Guided Design, Synthesis, and Evaluation of Stoichiometric, Functionally Irreversible Inhibitors. J. Med. Chem. 2019, 62, 10391–10401. [Google Scholar] [CrossRef]
- Liu, T.; Luo, T.; Guo, X.; Zou, X.; Zhou, D.; Afrin, S.; Li, G.; Zhang, Y.; Zhang, R.; Luo, Z. PgMYB2, a MeJA-Responsive Transcription Factor, Positively Regulates the Dammarenediol Synthase Gene Expression in Panax Ginseng. Int. J. Mol. Sci. 2019, 20, 2219. [Google Scholar] [CrossRef]
- Fröde, R.; Bröckelmann, M.; Steffan, B.; Steglich, W.; Marumoto, R. A novel type of triterpenoid quinone methide pigment from the toadstool Russula flavida (Agaricales). Tetrahedron 1995, 51, 2553–2560. [Google Scholar] [CrossRef]
- Payamnoor, V.; Kavosi, M.R.; Nazari, J. Polypore fungi of Caucasian alder as a source of antioxidant and antitumor agents. J. For. Res. 2019, 31, 1381–1390. [Google Scholar] [CrossRef]
- Hsu, B.Y.; Lu, T.J.; Chen, C.H.; Wang, S.J.; Hwang, L.S. Biotransformation of ginsenoside Rd in the ginseng extraction residue by fermentation with lingzhi (Ganoderma lucidum). Food Chem. 2013, 141, 4186–4193. [Google Scholar] [CrossRef]
- Ye, L.; Zhou, C.; Zhou, W.; Zhou, P.; Chen, D.; Liu, X.; Shi, X.; Feng, M. Biotransformation of ginsenoside Rb1 to ginsenoside Rd by highly substrate-tolerant Paecilomyces bainier 229-7. Bioresour. Technol. 2010, 101, 7872–7876. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hu, T.; Gao, L.; Su, P.; Zhang, Y.; Zhao, Y.; Chen, S.; Tu, L.; Song, Y.; Wang, X.; et al. Friedelane-type triterpene cyclase in celastrol biosynthesis from Tripterygium wilfordii and its application for triterpenes biosynthesis in yeast. New Phytol. 2019, 223, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Nakai, A.; Ando, E.; Fujimoto, J.; Leach, S.; Arimori, T.; Higo, D.; van Eerden, F.J.; Tulyeu, J.; Liu, Y.C.; et al. Celastrol suppresses humoral immune responses and autoimmunity by targeting the COMMD3/8 complex. Sci. Immunol. 2023, 8, eadc9324. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.; Alvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Yi, Z.; Zhang, J.; Lu, B.; Sung, B.; Qu, W.; Aggarwal, B.B.; Liu, M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010, 70, 1951–1959. [Google Scholar] [CrossRef]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, T.; Liu, Y.; Tu, L.; Song, Y.; Lu, Y.; Zhang, Y.; Tong, Y.; Zhao, Y.; Su, P.; et al. Cytochrome P450 catalyses the 29-carboxyl group formation of celastrol. Phytochemistry 2021, 190, 112868. [Google Scholar] [CrossRef]
- Sułkowska-Ziaja, K.; Galanty, A.; Szewczyk, A.; Paśko, P.; Kała, K.; Apola, A.; Podolak, I.; Muszyńska, B. Effect of Methyl Jasmonate Elicitation on Triterpene Production and Evaluation of Cytotoxic Activity of Mycelial Culture Extracts of Ganoderma applanatum (Pers.). Plants 2023, 12, 294. [Google Scholar] [CrossRef]
- Bai, M.; Zhou, L.; Tong, Y.; Yin, F. Risk assessment and warning system for strategic biological resources in China. Innov. Life 2023, 1, 100004. [Google Scholar] [CrossRef]
- Wei, F. Toward post-2020 global biodiversity conservation: Footprint and direction in China. Innovation 2021, 2, 100175. [Google Scholar] [CrossRef]
- Liu, S.-L.; Liu, D.-M.; Zhou, L.-W. The future is now: How to conserve fungi. Biological Diversity 2024, 1, 6–8. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2017, 7, 1. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, D.; Wang, Z. Effects of diclofenac on the pharmacokinetics of celastrol in rats and its transport. Pharmaceutical Biology 2018, 56, 269–274. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, J.-H.; Li, Q.-Z.; Luo, X.; Yu, J.; Zhou, L.-W. Transcriptome and Metabolome Reveal Accumulation of Key Metabolites with Medicinal Properties of Phylloporia pulla. Int. J. Mol. Sci. 2024, 25, 11070. https://doi.org/10.3390/ijms252011070
Jiang J-H, Li Q-Z, Luo X, Yu J, Zhou L-W. Transcriptome and Metabolome Reveal Accumulation of Key Metabolites with Medicinal Properties of Phylloporia pulla. International Journal of Molecular Sciences. 2024; 25(20):11070. https://doi.org/10.3390/ijms252011070
Chicago/Turabian StyleJiang, Ji-Hang, Qian-Zhu Li, Xing Luo, Jia Yu, and Li-Wei Zhou. 2024. "Transcriptome and Metabolome Reveal Accumulation of Key Metabolites with Medicinal Properties of Phylloporia pulla" International Journal of Molecular Sciences 25, no. 20: 11070. https://doi.org/10.3390/ijms252011070
APA StyleJiang, J.-H., Li, Q.-Z., Luo, X., Yu, J., & Zhou, L.-W. (2024). Transcriptome and Metabolome Reveal Accumulation of Key Metabolites with Medicinal Properties of Phylloporia pulla. International Journal of Molecular Sciences, 25(20), 11070. https://doi.org/10.3390/ijms252011070