
Enhancing Clinical Applications by Evaluation of Sensitivity and Specificity in Whole Exome Sequencing

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
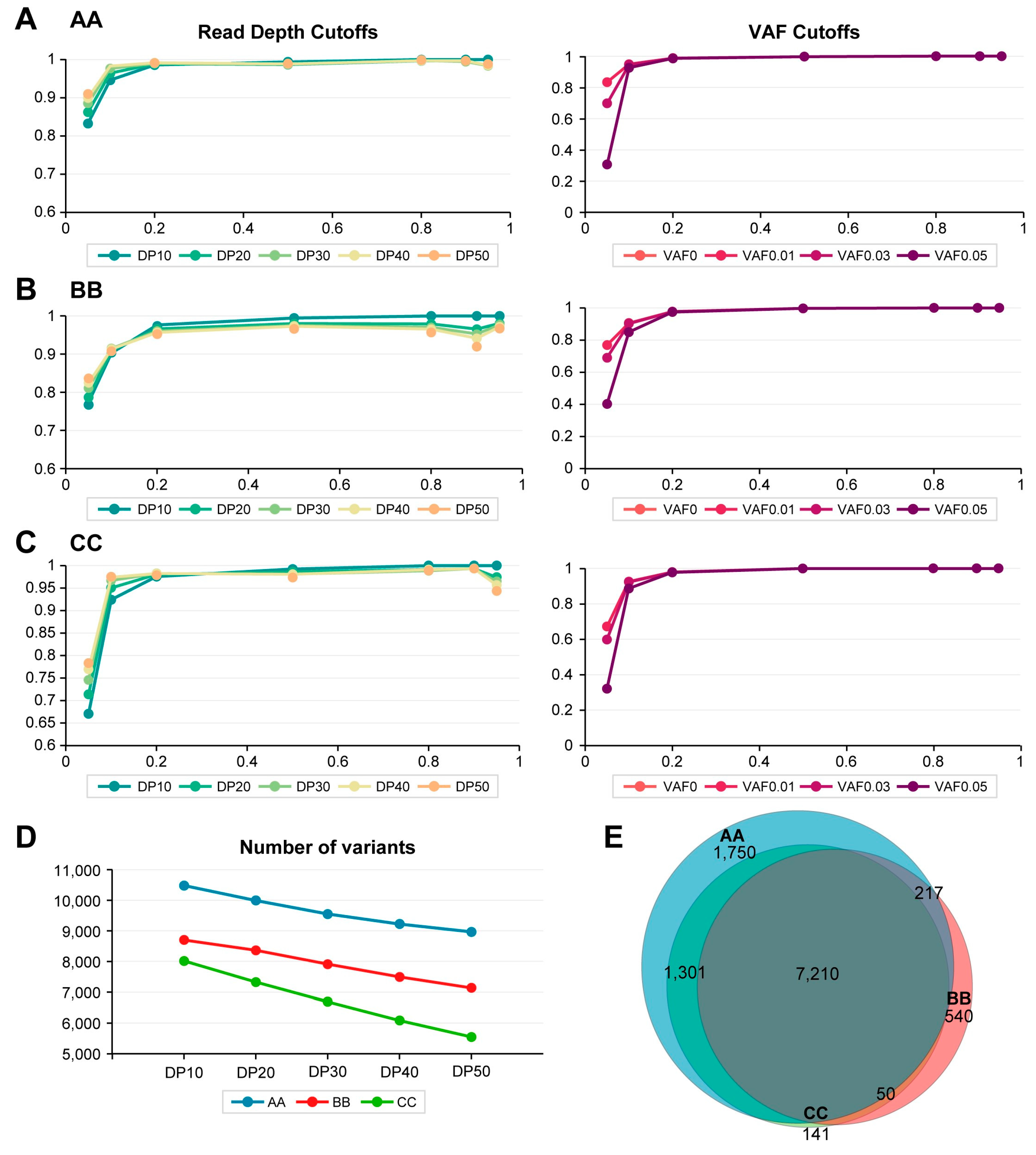
2.1. Evaluation of In-House WES Sensitivity
2.2. Differences in Sensitivity of In-House WES Results
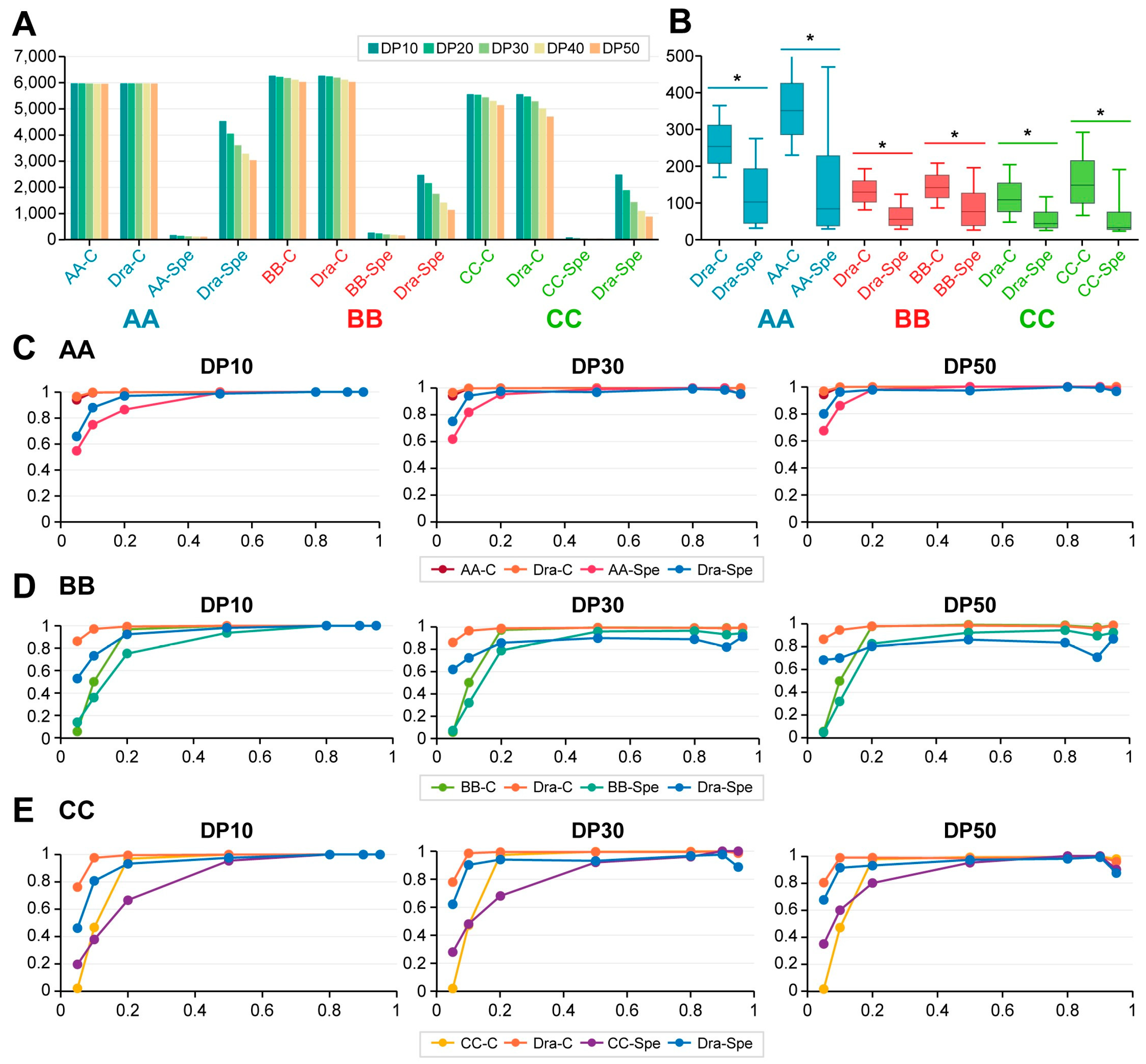
2.3. Evaluation of WES Sensitivity Using Dynamic Read Analysis for GENomics (DRAGEN) Bioinformatic Analysis
2.4. Sensitivity Comparisons Among Diverse Categories Based on DRAGEN and In-House Analyses
2.5. Superior WES Sensitivity and Greater Variant Call Rates Observed in Company AA
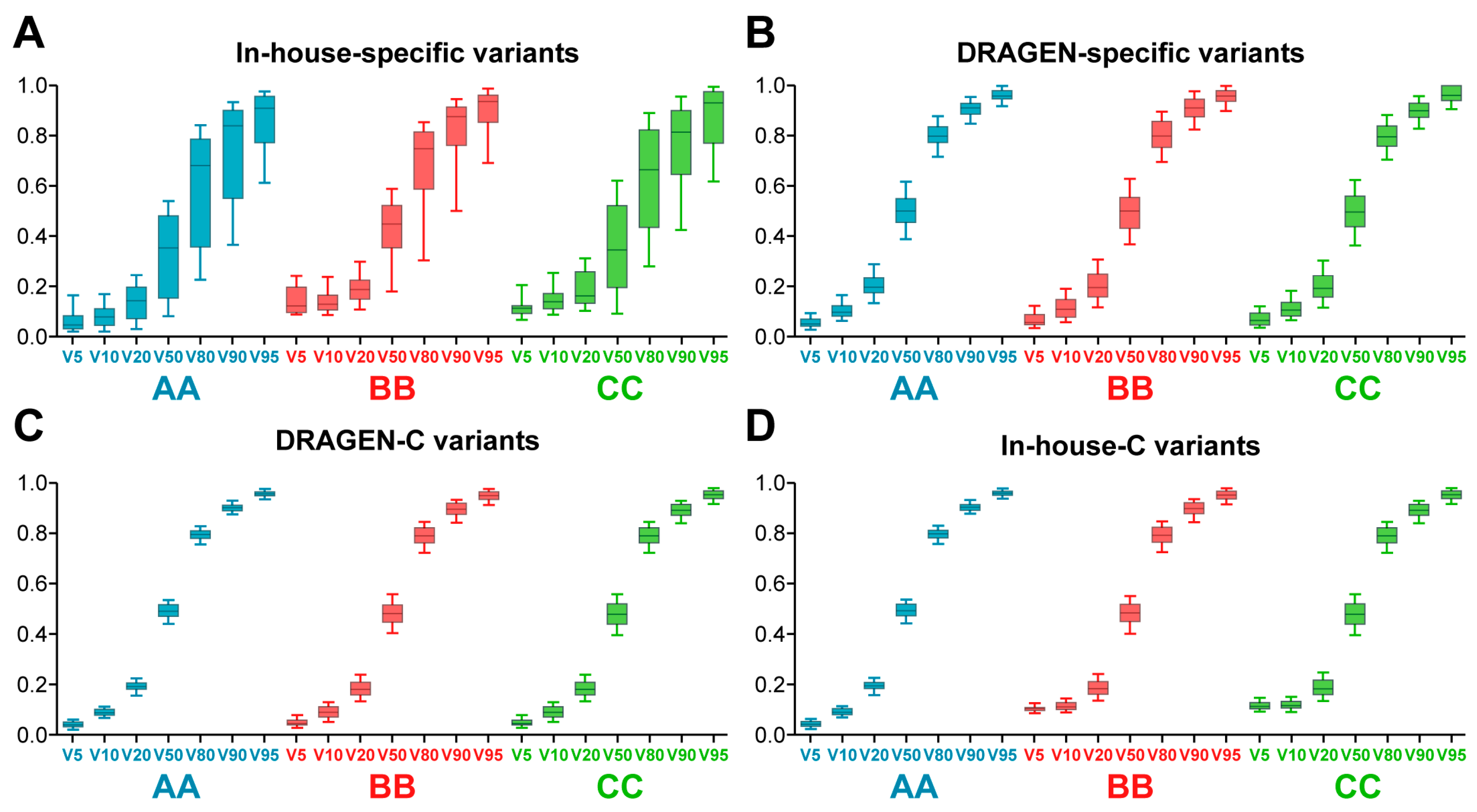
2.6. Analysis of False Positive Errors in WES Results
3. Discussion
4. Materials and Methods
4.1. Preparation of Reference-Standard DNAs
4.2. Whole Exome Sequencing (WES)
4.3. Variant Calling
4.4. Comparison of Variant Calls in Various Categories
4.5. False Positive (FP) Error Analysis
4.6. Statistical Analysis and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Luh, F.; Yen, Y. FDA guidance for next generation sequencing-based testing: Balancing regulation and innovation in precision medicine. NPJ Genom. Med. 2018, 3, 28. [Google Scholar] [CrossRef]
- Xiao, W.; Ren, L.; Chen, Z.; Fang, L.T.; Zhao, Y.; Lack, J.; Guan, M.; Zhu, B.; Jaeger, E.; Kerrigan, L.; et al. Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat. Biotechnol. 2021, 39, 1141–1150. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, L.T.; Shen, T.W.; Choudhari, S.; Talsania, K.; Chen, X.; Shetty, J.; Kriga, Y.; Tran, B.; Zhu, B.; et al. Whole genome and exome sequencing reference datasets from a multi-center and cross-platform benchmark study. Sci. Data 2021, 8, 296. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Lionel, A.C.; Costain, G.; Monfared, N.; Walker, S.; Reuter, M.S.; Hosseini, S.M.; Thiruvahindrapuram, B.; Merico, D.; Jobling, R.; Nalpathamkalam, T.; et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 2018, 20, 435–443. [Google Scholar] [CrossRef]
- Tian, R.; Ge, T.; Kweon, H.; Rocha, D.B.; Lam, M.; Liu, J.Z.; Singh, K.; Biogen Biobank, T.; Levey, D.F.; Gelernter, J.; et al. Whole-exome sequencing in UK Biobank reveals rare genetic architecture for depression. Nat. Commun. 2024, 15, 1755. [Google Scholar] [CrossRef]
- Yang, L.; Ou, Y.N.; Wu, B.S.; Liu, W.S.; Deng, Y.T.; He, X.Y.; Chen, Y.L.; Kang, J.; Fei, C.J.; Zhu, Y.; et al. Large-scale whole-exome sequencing analyses identified protein-coding variants associated with immune-mediated diseases in 350,770 adults. Nat. Commun. 2024, 15, 5924. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef]
- Rennert, H.; Eng, K.; Zhang, T.; Tan, A.; Xiang, J.; Romanel, A.; Kim, R.; Tam, W.; Liu, Y.C.; Bhinder, B.; et al. Development and validation of a whole-exome sequencing test for simultaneous detection of point mutations, indels and copy-number alterations for precision cancer care. NPJ Genom. Med. 2016, 1, 16019. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]
- Ewans, L.J.; Minoche, A.E.; Schofield, D.; Shrestha, R.; Puttick, C.; Zhu, Y.; Drew, A.; Gayevskiy, V.; Elakis, G.; Walsh, C.; et al. Whole exome and genome sequencing in mendelian disorders: A diagnostic and health economic analysis. Eur. J. Hum. Genet. 2022, 30, 1121–1131. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef]
- Nurchis, M.C.; Radio, F.C.; Salmasi, L.; Heidar Alizadeh, A.; Raspolini, G.M.; Altamura, G.; Tartaglia, M.; Dallapiccola, B.; Pizzo, E.; Gianino, M.M.; et al. Cost-Effectiveness of Whole-Genome vs Whole-Exome Sequencing Among Children with Suspected Genetic Disorders. JAMA Netw. Open 2024, 7, e2353514. [Google Scholar] [CrossRef]
- Meynert, A.M.; Ansari, M.; FitzPatrick, D.R.; Taylor, M.S. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinform. 2014, 15, 247. [Google Scholar] [CrossRef]
- Moon, Y.; Kim, Y.-H.; Kim, J.-K.; Hong, C.H.; Kang, E.-K.; Choi, H.W.; Lee, D.-E.; Kim, T.-M.; Heo, S.G.; Han, N.; et al. Evaluation of false positive and false negative errors in targeted next generation sequencing. Genome Biol. 2024; submitted. [Google Scholar]
- Carey, L.; Nash, B.M.; Wright, D.C. Molecular genetic studies of complete hydatidiform moles. Transl. Pediatr. 2015, 4, 181–188. [Google Scholar]
- Nguyen, N.M.P.; Khawajkie, Y.; Mechtouf, N.; Rezaei, M.; Breguet, M.; Kurvinen, E.; Jagadeesh, S.; Solmaz, A.E.; Aguinaga, M.; Hemida, R.; et al. The genetics of recurrent hydatidiform moles: New insights and lessons from a comprehensive analysis of 113 patients. Mod. Pathol. 2018, 31, 1116–1130. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, M.; Li, X.; Wang, Z.; Pan, D.; Shi, Y. Performance comparison of four types of target enrichment baits for exome DNA sequencing. Hereditas 2021, 158, 10. [Google Scholar] [CrossRef]
- Kim, H.; Yun, J.W.; Lee, S.T.; Kim, H.J.; Kim, S.H.; Kim, J.W.; Korean Society for Genetic Diagnostics Clinical Guidelines Committee. Korean Society for Genetic Diagnostics Guidelines for Validation of Next-Generation Sequencing-Based Somatic Variant Detection in Hematologic Malignancies. Ann. Lab. Med. 2019, 39, 515–523. [Google Scholar] [CrossRef]
- Menzel, M.; Ossowski, S.; Kral, S.; Metzger, P.; Horak, P.; Marienfeld, R.; Boerries, M.; Wolter, S.; Ball, M.; Neumann, O.; et al. Multicentric pilot study to standardize clinical whole exome sequencing (WES) for cancer patients. NPJ Precis. Oncol. 2023, 7, 106. [Google Scholar] [CrossRef]
- Kim, Y.H.; Song, Y.; Kim, J.K.; Kim, T.M.; Sim, H.W.; Kim, H.L.; Jang, H.; Kim, Y.W.; Hong, K.M. False-negative errors in next-generation sequencing contribute substantially to inconsistency of mutation databases. PLoS ONE 2019, 14, e0222535. [Google Scholar] [CrossRef]
- Cheng, C.; Fei, Z.; Xiao, P. Methods to improve the accuracy of next-generation sequencing. Front. Bioeng. Biotechnol. 2023, 11, 982111. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Grober, C.; Blank, M.; Handler, K.; Beyer, M.; Schultze, J.L.; Mayer, G. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep. 2018, 8, 10950. [Google Scholar] [CrossRef]
- Zhang, T.H.; Wu, N.C.; Sun, R. A benchmark study on error-correction by read-pairing and tag-clustering in amplicon-based deep sequencing. BMC Genom. 2016, 17, 108. [Google Scholar] [CrossRef]
- Hudson, A.M.; Yates, T.; Li, Y.; Trotter, E.W.; Fawdar, S.; Chapman, P.; Lorigan, P.; Biankin, A.; Miller, C.J.; Brognard, J. Discrepancies in cancer genomic sequencing highlight opportunities for driver mutation discovery. Cancer Res. 2014, 74, 6390–6396. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Ushakov, M.O.; Lazareva, T.E.; Nasykhova, Y.A.; Glotov, A.S.; Predeus, A.V. Bioinformatics of germline variant discovery for rare disease diagnostics: Current approaches and remaining challenges. Brief. Bioinform. 2024, 25, bbad508. [Google Scholar] [CrossRef]
- Riess, O.; Sturm, M.; Menden, B.; Liebmann, A.; Demidov, G.; Witt, D.; Casadei, N.; Admard, J.; Schutz, L.; Ossowski, S.; et al. Genomes in clinical care. NPJ Genom. Med. 2024, 9, 20. [Google Scholar] [CrossRef]
- Garcia-Garcia, G.; Baux, D.; Faugere, V.; Moclyn, M.; Koenig, M.; Claustres, M.; Roux, A.F. Assessment of the latest NGS enrichment capture methods in clinical context. Sci. Rep. 2016, 6, 20948. [Google Scholar] [CrossRef]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef]
- Hedges, D.J.; Guettouche, T.; Yang, S.; Bademci, G.; Diaz, A.; Andersen, A.; Hulme, W.F.; Linker, S.; Mehta, A.; Edwards, Y.J.; et al. Comparison of three targeted enrichment strategies on the SOLiD sequencing platform. PLoS ONE 2011, 6, e18595. [Google Scholar] [CrossRef]
- Kallenborn, F.; Cascitti, J.; Schmidt, B. CARE 2.0: Reducing false-positive sequencing error corrections using machine learning. BMC Bioinform. 2022, 23, 227. [Google Scholar] [CrossRef]
- Dai, P.; Wu, L.R.; Chen, S.X.; Wang, M.X.; Cheng, L.Y.; Zhang, J.X.; Hao, P.; Yao, W.; Zarka, J.; Issa, G.C.; et al. Calibration-free NGS quantitation of mutations below 0.01% VAF. Nat. Commun. 2021, 12, 6123. [Google Scholar] [CrossRef]
- Eren, K.K.; Cinar, E.; Karakurt, H.U.; Ozgur, A. Improving the filtering of false positive single nucleotide variations by combining genomic features with quality metrics. Bioinformatics 2023, 39, btad694. [Google Scholar] [CrossRef]
- Karimnezhad, A.; Palidwor, G.A.; Thavorn, K.; Stewart, D.J.; Campbell, P.A.; Lo, B.; Perkins, T.J. Accuracy and reproducibility of somatic point mutation calling in clinical-type targeted sequencing data. BMC Med. Genom. 2020, 13, 156. [Google Scholar] [CrossRef]
- Larson, N.B.; Oberg, A.L.; Adjei, A.A.; Wang, L. A Clinician’s Guide to Bioinformatics for Next-Generation Sequencing. J. Thorac. Oncol. 2023, 18, 143–157. [Google Scholar] [CrossRef]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef]
- Shin, H.T.; Choi, Y.L.; Yun, J.W.; Kim, N.K.D.; Kim, S.Y.; Jeon, H.J.; Nam, J.Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun. 2017, 8, 1377. [Google Scholar] [CrossRef]
- Yadav, D.; Patil-Takbhate, B.; Khandagale, A.; Bhawalkar, J.; Tripathy, S.; Khopkar-Kale, P. Next-Generation sequencing transforming clinical practice and precision medicine. Clin. Chim. Acta 2023, 551, 117568. [Google Scholar] [CrossRef]
- Carter, T.C.; He, M.M. Challenges of Identifying Clinically Actionable Genetic Variants for Precision Medicine. J. Healthc. Eng. 2016, 2016, 3617572. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, L.; Roberts, R.; Tong, W. Toward Clinical Implementation of Next-Generation Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends Genet. 2019, 35, 852–867. [Google Scholar] [CrossRef]
- Sheng, X.; Xia, L.; Cahoon, J.L.; Conti, D.V.; Haiman, C.A.; Kachuri, L.; Chiang, C.W.K. Inverted genomic regions between reference genome builds in humans impact imputation accuracy and decrease the power of association testing. HGG Adv. 2023, 4, 100159. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, Y.; Hong, C.H.; Kim, Y.-H.; Kim, J.-K.; Ye, S.-H.; Kang, E.-K.; Choi, H.W.; Cho, H.; Choi, H.; Lee, D.-e.; et al. Enhancing Clinical Applications by Evaluation of Sensitivity and Specificity in Whole Exome Sequencing. Int. J. Mol. Sci. 2024, 25, 13250. https://doi.org/10.3390/ijms252413250
Moon Y, Hong CH, Kim Y-H, Kim J-K, Ye S-H, Kang E-K, Choi HW, Cho H, Choi H, Lee D-e, et al. Enhancing Clinical Applications by Evaluation of Sensitivity and Specificity in Whole Exome Sequencing. International Journal of Molecular Sciences. 2024; 25(24):13250. https://doi.org/10.3390/ijms252413250
Chicago/Turabian StyleMoon, Youngbeen, Chung Hwan Hong, Young-Ho Kim, Jong-Kwang Kim, Seo-Hyeon Ye, Eun-Kyung Kang, Hye Won Choi, Hyeri Cho, Hana Choi, Dong-eun Lee, and et al. 2024. "Enhancing Clinical Applications by Evaluation of Sensitivity and Specificity in Whole Exome Sequencing" International Journal of Molecular Sciences 25, no. 24: 13250. https://doi.org/10.3390/ijms252413250
APA StyleMoon, Y., Hong, C. H., Kim, Y.-H., Kim, J.-K., Ye, S.-H., Kang, E.-K., Choi, H. W., Cho, H., Choi, H., Lee, D.-e., Choi, Y., Kim, T.-M., Heo, S. G., Han, N., & Hong, K.-M. (2024). Enhancing Clinical Applications by Evaluation of Sensitivity and Specificity in Whole Exome Sequencing. International Journal of Molecular Sciences, 25(24), 13250. https://doi.org/10.3390/ijms252413250