
Pathogenic Variants Associated with Epigenetic Control and the NOTCH Pathway Are Frequent in Classic Hodgkin Lymphoma

 ,
,
Abstract
1. Introduction
2. Results
2.1. Characteristics of the Patients
2.2. Next-Generation Sequencing of Classic Hodgkin Lymphoma Reveals Intratumoral Heterogeneity
2.3. Mutational Landscape of cHL Patients Achieving a Complete Metabolic Response after Induction with Adriamycin, Bleomycin, Vinblastine and Dacarbazine (ABVD)
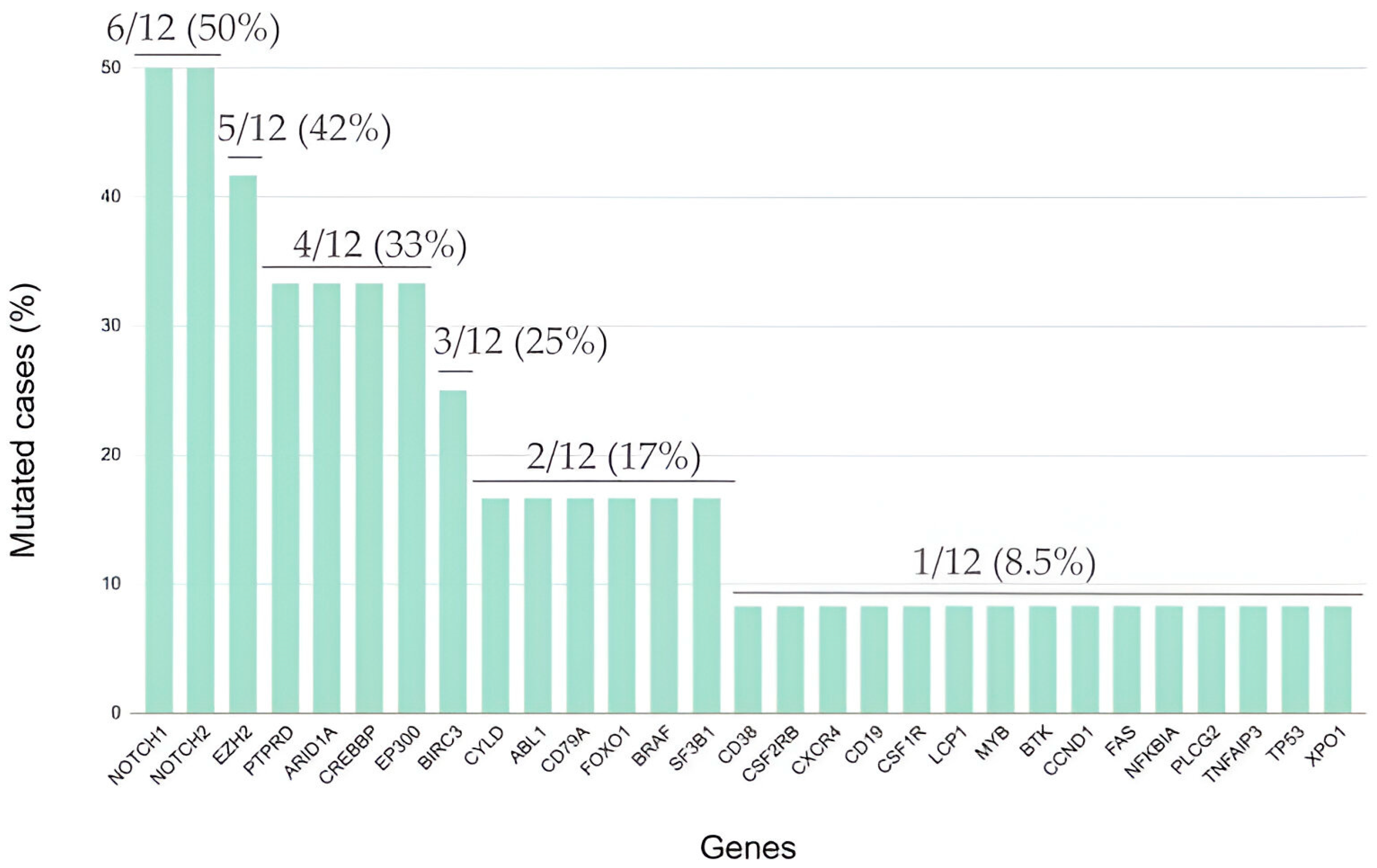
2.4. Somatic Mutations in Genes Associated with Epigenetic Control Are Frequent in Relapsed/Refractory Classic Hodgkin Lymphoma
2.5. Cell Tumor Burden and Variant Allele Frequency (VAF) at Diagnosis and Relapse
2.6. Temporal Dynamics and Clonal Selection in Relapsed/Refractory Classic Hodgkin Lymphoma
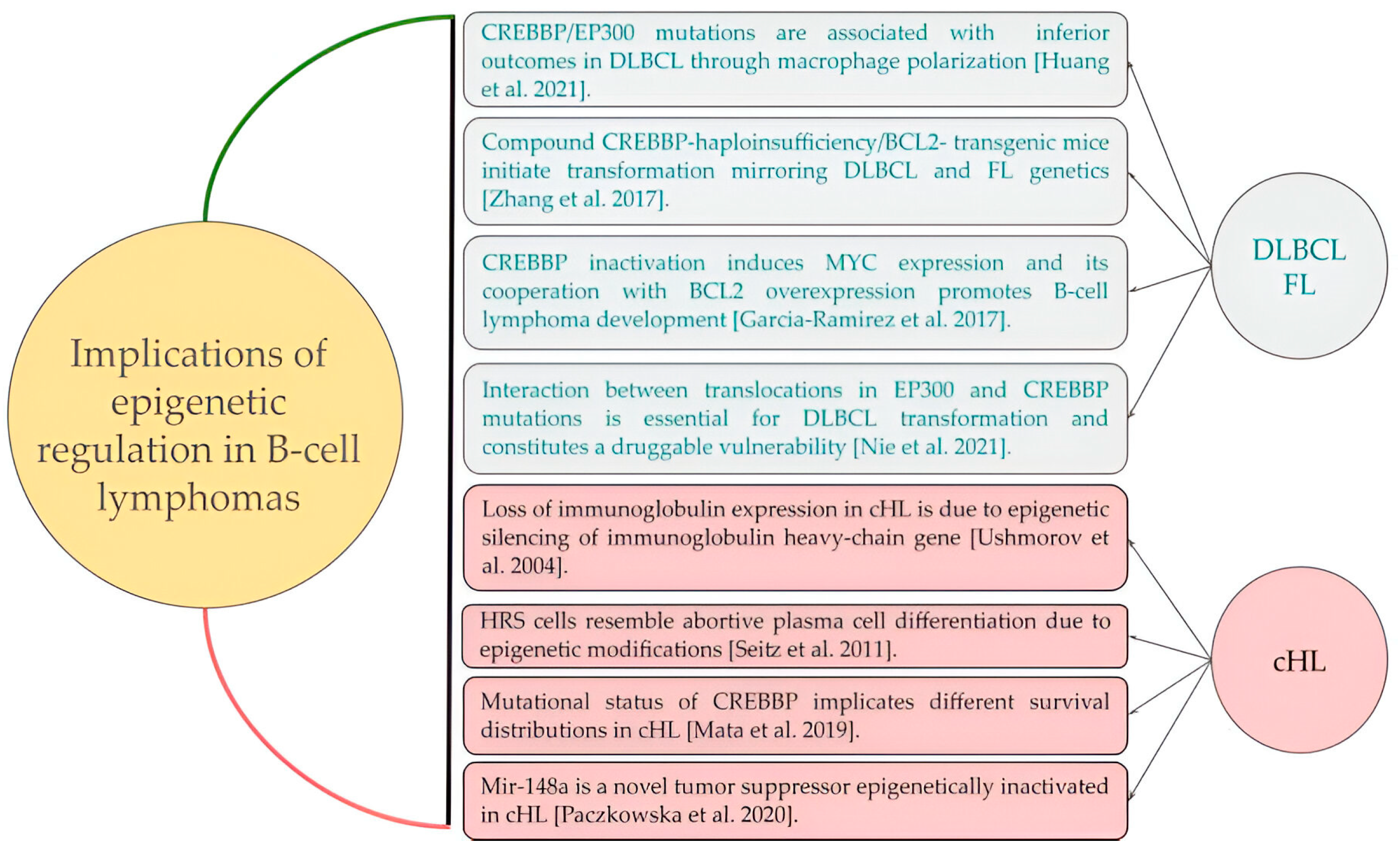
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. DNA Extraction and Construction of Libraries
4.3. Library Preparation and Templating
4.4. Next-Generation Sequencing, Variant Calling and Variant Categorization
4.5. Immunohistochemistry
4.6. Endpoints
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Küppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Küppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Eichenauer, D.A.; Aleman, B.M.P.; André, M.; Federico, M.; Hutchings, M.; Illidge, T.; Engert, A.; Ladetto, M.; ESMO Guidelines Committee. Hodgkin lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29(Suppl. 4), iv19–iv29. [Google Scholar] [CrossRef] [PubMed]
- Follows, G.A.; Barrington, S.F.; Bhuller, K.S.; Culligan, D.J.; Cutter, D.J.; Gallop-Evans, E.; Kassam, S.; Osborne, W.; Sadullah, S.; Townsend, W.; et al. Guideline for the first—Line management of Classical Hodgkin Lymphoma—A British Society for Haematology guideline. Br. J. Haematol. 2022, 197, 558–572. [Google Scholar] [CrossRef]
- Mata, E.; Díaz-López, A.; Martín-Moreno, A.M.; Sánchez-Beato, M.; Varela, I.; Mestre, M.J.; Santonja, C.; Burgos, F.; Menárguez, J.; Estévez, M.; et al. Analysis of the mutational landscape of classic Hodgkin lymphoma identifies disease heterogeneity and potential therapeutic targets. Oncotarget 2017, 8, 111386–111395. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Gomez, F.; Gomez, F.; Fisk, B.; Fisk, B.; McMichael, J.F.; McMichael, J.F.; Mosior, M.; Mosior, M.; Foltz, J.A.; Foltz, J.A.; et al. Ultra-Deep Sequencing Reveals the Mutational Landscape of Classical Hodgkin Lymphoma. Cancer Res. Commun. 2023, 3, 2312–2330. [Google Scholar] [CrossRef]
- Mata, E.; Fernández, S.; Astudillo, A.; Fernández, R.; García-Cosío, M.; Sánchez-Beato, M.; Provencio, M.; Estévez, M.; Montalbán, C.; Piris, M.A.; et al. Genomic analyses of microdissected Hodgkin and Reed-Sternberg cells: Mutations in epigenetic regulators and p53 are frequent in refractory classic Hodgkin lymphoma. Blood Cancer J. 2019, 9, 34. [Google Scholar] [CrossRef]
- Sujobert, P.; Le Bris, Y.; de Leval, L.; Gros, A.; Merlio, J.P.; Pastoret, C.; Huet, S.; Sarkozy, C.; Davi, F.; Callanan, M.; et al. The Need for a Consensus Next—Generation Sequencing Panel for Mature Lymphoid Malignancies. HemaSphere 2019, 3, e169. [Google Scholar] [CrossRef]
- Sánchez-Beato, M.; Méndez, M.; Guirado, M.; Pedrosa, L.; Sequero, S.; Yanguas-Casás, N.; de la Cruz-Merino, L.; Gálvez, L.; Llanos, M.; García, J.F.; et al. A genetic profiling guideline to support diagnosis and clinical management of lymphomas. Clin. Transl. Oncol. 2023, in press. [CrossRef]
- Santisteban-Espejo, A.; Bernal-Florindo, I.; Perez-Requena, J.; Atienza-Cuevas, L.; Moran-Sanchez, J.; Fernandez-Valle, M.d.C.; Romero-Garcia, R.; Garcia-Rojo, M. The Need for Standardization in Next-Generation Sequencing Studies for Classic Hodgkin Lymphoma: A Systematic Review. Diagnostics 2022, 12, 963. [Google Scholar] [CrossRef]
- Weniger, M.A.; Küppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef]
- Schwarzer, R.; Jundt, F. Notch and NF-?B signaling pathways in the biology of classical Hodgkin lymphoma. Curr. Mol. Med. 2011, 11, 236–245. [Google Scholar] [CrossRef]
- Jundt, F.; Acikgöz, O.; Kwon, S.-H.; Schwarzer, R.; Anagnostopoulos, I.; Wiesner, B.; Mathas, S.; Hummel, M.; Stein, H.; Reichardt, H.M.; et al. Aberrant expression of Notch1 interferes with the B-lymphoid phenotype of neoplastic B cells in classical Hodgkin lymphoma. Leukemia 2008, 22, 1587–1594. [Google Scholar] [CrossRef]
- Tiacci, E.; Döring, C.; Brune, V.; van Noesel, C.J.M.; Klapper, W.; Mechtersheimer, G.; Falini, B.; Küppers, R.; Hansmann, M.-L. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood 2012, 120, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Dörken, B.; Jundt, F. Notch is an essential upstream regulator of NF-κB and is relevant for survival of Hodgkin and Reed–Sternberg cells. Leukemia 2011, 26, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Gulley, M.L.; Glaser, S.L.; Craig, F.E.; Borowitz, M.; Mann, R.B.; Shema, S.J.; Ambinder, R.F. Guidelines for Interpreting EBER In Situ Hybridization and LMP1 Immunohistochemical Tests for Detecting Epstein-Barr Virus in Hodgkin Lymphoma. Am. J. Clin. Pathol. 2002, 117, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Ushmorov, A.; Leithauser, F.; Sakk, O.; Weinhausel, A.; Popov, S.W.; Moller, P.; Wirth, T. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood 2006, 107, 2493–2500. [Google Scholar] [CrossRef]
- Ushmorov, A.; Ritz, O.; Hummel, M.; Leithauser, F.; Moller, P.; Stein, H.; Wirth, T. Epigenetic silencing of the immunoglobulin heavy-chain gene in classical Hodgkin lymphoma-derived cell lines contributes to the loss of immunoglobulin expression. Blood 2004, 104, 3326–3334. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Cai, K.; Xu, P.-P.; Wang, L.; Huang, C.-X.; Fang, Y.; Cheng, S.; Sun, X.-J.; Liu, F.; Huang, J.-Y.; et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-NOTCH-CCL2/CSF1 axis. Signal Transduct. Target. Ther. 2021, 6, 10. [Google Scholar] [CrossRef]
- Nie, M.; Du, L.; Ren, W.; Joung, J.; Ye, X.; Shi, X.; Ciftci, S.; Liu, D.; Wu, K.; Zhang, F.; et al. Genome-wide CRISPR screens reveal synthetic lethal interaction between CREBBP and EP300 in diffuse large B-cell lymphoma. Cell Death Dis. 2021, 12, 419. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Krysiak, K.; Ramirez, C.; Mosior, M.; Watkins, M.P.; Gomez, F.; Skidmore, Z.L.; Trani, L.; Gao, F.; Geyer, S.; et al. Mutations associated with progression in follicular lymphoma predict inferior outcomes at diagnosis: Alliance A151303. Blood Adv. 2023, 7, 5524–5539. [Google Scholar] [CrossRef]
- Green, M.R. Chromatin modifying gene mutations in follicular lymphoma. Blood 2018, 131, 595–604. [Google Scholar] [CrossRef]
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017, 7, 322–337. [Google Scholar] [CrossRef] [PubMed]
- García-Ramírez, I.; Tadros, S.; González-Herrero, I.; Martín-Lorenzo, A.; Rodríguez-Hernández, G.; Moore, D.; Ruiz-Roca, L.; Blanco, O.; Alonso-López, D.; Rivas, J.D.L.; et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood 2017, 129, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Seitz, V.; Thomas, P.E.; Zimmermann, K.; Paul, U.; Ehlers, A.; Joosten, M.; Dimitrova, L.; Lenze, D.; Sommerfeld, A.; Oker, E.; et al. Classical Hodgkin’s lymphoma shows epigenetic features of abortive plasma cell differentiation. Haematologica 2011, 96, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Paczkowska, J.; Janiszewska, J.; Bein, J.; Schneider, M.; Bednarek, K.; Ustaszewski, A.; Hartmann, S.; Hansmann, M.-L.; Giefing, M. The Tumor Suppressive mir-148a Is Epigenetically Inactivated in Classical Hodgkin Lymphoma. Cells 2020, 9, 2292. [Google Scholar] [CrossRef] [PubMed]
- Santisteban-Espejo, A.; Bernal-Florindo, I.; Perez-Requena, J.; Atienza-Cuevas, L.; Maira-Gonzalez, N.; Garcia-Rojo, M. Whole-slide image analysis identifies a high content of Hodgkin Reed-Sternberg cells and a low content of T lymphocytes in tumor microenvironment as predictors of adverse outcome in patients with classic Hodgkin lymphoma treated with ABVD. Front. Oncol. 2022, 12, 1000762. [Google Scholar] [CrossRef]
- Santisteban-Espejo, A.; Bernal-Florindo, I.; Perez-Requena, J.; Atienza-Cuevas, L.; Catalina-Fernandez, I.; Fernandez-Valle, M.d.C.; Romero-Garcia, R.; Garcia-Rojo, M. Identification of prognostic factors in classic Hodgkin lymphoma by integrating whole slide imaging and next generation sequencing. Mol. Omics 2022, 18, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Canellos, G.P.; Rosenberg, S.A.; Friedberg, J.W.; Lister, T.A.; DeVita, V.T. Treatment of Hodgkin Lymphoma: A 50-Year Perspective. J. Clin. Oncol. 2014, 32, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Alcoceba, M.; García-Álvarez, M.; Chillón, M.C.; Jiménez, C.; Medina, A.; Antón, A.; Blanco, O.; Díaz, L.G.; Tamayo, P.; González-Calle, V.; et al. Liquid biopsy: A non—Invasive approach for Hodgkin lymphoma genotyping. Br. J. Haematol. 2021, 195, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Sadaf, H.; Ambroziak, M.; Binkowski, R.; Kluebsoongnoen, J.; Paszkiewicz-Kozik, E.; Steciuk, J.; Markowicz, S.; Walewski, J.; Sarnowska, E.; Sarnowski, T.J.; et al. New molecular targets in Hodgkin and Reed-Sternberg cells. Front. Immunol. 2023, 14, 1155468. [Google Scholar] [CrossRef] [PubMed]
- van Bladel, D.A.G.; Brand, M.v.D.; Rijntjes, J.; Naga, S.P.; Haacke, D.L.C.M.; Luijks, J.A.C.W.; Hebeda, K.M.; van Krieken, J.H.J.M.; Groenen, P.J.T.A.; Scheijen, B. Clonality assessment and detection of clonal diversity in classic Hodgkin lymphoma by next-generation sequencing of immunoglobulin gene rearrangements. Mod. Pathol. 2021, 35, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Venanzi, A.; Marra, A.; Schiavoni, G.; Milner, S.G.; Limongello, R.; Santi, A.; Pettirossi, V.; Ultimo, S.; Tasselli, L.; Pucciarini, A.; et al. Dissecting Clonal Hematopoiesis in Tissues of Patients with Classical Hodgkin Lymphoma Patients. Blood Cancer Discov. 2021, 2, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Buedts, L.; Wlodarska, I.; Finalet-Ferreiro, J.; Gheysens, O.; Dehaspe, L.; Tousseyn, T.; Fornecker, L.-M.; Lazarovici, J.; Casasnovas, R.-O.; Gac, A.-C.; et al. The landscape of copy number variations in classical Hodgkin lymphoma: A joint KU Leuven and LYSA study on cell-free DNA. Blood Adv. 2021, 5, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Hasenclever, D.; Diehl, V.; Armitage, J.O.; Assouline, D.; Björkholm, M.; Brusamolino, E.; Canellos, G.P.; Carde, P.; Crowther, D.; Cunningham, D.; et al. A Prognostic Score for Advanced Hodgkin’s Disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N. Engl. J. Med. 1998, 339, 1506–1514. [Google Scholar] [CrossRef]
- Engert, A.; Plütschow, A.; Eich, H.T.; Lohri, A.; Dörken, B.; Borchmann, P.; Berger, B.; Greil, R.; Willborn, K.C.; Wilhelm, M.; et al. Reduced treatment intensity in patients with early-stage Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 363, 640–652. [Google Scholar] [CrossRef]
- Rodday, A.M.; Parsons, S.K.; Upshaw, J.N.; Friedberg, J.W.; Gallamini, A.; Hawkes, E.; Hodgson, D.; Johnson, P.; Link, B.K.; Mou, E.; et al. The Advanced-Stage Hodgkin Lymphoma International Prognostic Index: Development and Validation of a Clinical Prediction Model From the HoLISTIC Consortium. J. Clin. Oncol. 2023, 41, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Horgan, D.; Hamdi, Y.; Lal, J.A.; Nyawira, T.; Meyer, S.; Kondji, D.; Francisco, N.M.; De Guzman, R.; Paul, A.; Bernard, B.; et al. Framework for Adoption of Next-Generation Sequencing (NGS) Globally in the Oncology Area. Healthcare 2023, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Krueger, K.E. Neo-Darwinian Principles Exemplified in Cancer Genomics. Mol. Cancer Res. 2023, 21, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.A.; Oli, M.W.; Oli, M.K. Eco—Oncology: Applying ecological principles to understand and manage cancer. Ecol. Evol. 2020, 10, 8538–8553. [Google Scholar] [CrossRef]
- Metz, C.W.; Bridges, C.B. Incompatibility of Mutant Races in Drosophila. Proc. Natl. Acad. Sci. USA 1917, 3, 673–678. [Google Scholar] [CrossRef]
- Mohr, O.L. Character Changes Caused by Mutation of an Entire Region of a Chromosome in Drosophila. Genetics 1919, 4, 275–282. [Google Scholar] [CrossRef]
- Reynolds, T.C.; Smith, S.D.; Sklar, J. Analysis of DNA surrounding the breakpoints of chromosomal translocations involving the β T cell receptor gene in human lymphoblastic neoplasms. Cell 1987, 50, 107–117. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Jundt, F.; Anagnostopoulos, I.; Forster, R.; Mathas, S.; Stein, H.; Dorken, B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood 2002, 99, 3398–3403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Z.; Li, Y.; Peng, H.; Liu, J.; Zhang, J.; Xiao, X. The Role of CREBBP/EP300 and Its Therapeutic Implications in Hematological Malignancies. Cancers 2023, 15, 1219. [Google Scholar] [CrossRef]
- Horton, S.J.; Giotopoulos, G.; Yun, H.; Vohra, S.; Sheppard, O.; Bashford-Rogers, R.; Rashid, M.; Clipson, A.; Chan, W.-I.; Sasca, D.; et al. Early loss of Crebbp confers malignant stem cell properties on lymphoid progenitors. Nat. Cell Biol. 2017, 19, 1093–1104. [Google Scholar] [CrossRef]
- van Bladel, D.A.G.; Stevens, W.B.C.; Kroeze, L.I.; de Groen, R.A.L.; de Groot, F.A.; van der Last-Kempkes, J.L.M.; Berendsen, M.R.; Rijntjes, J.; Luijks, J.A.C.W.; Bonzheim, I.; et al. A significant proportion of classic Hodgkin lymphoma recurrences represents clonally unrelated second primary lymphoma. Blood Adv. 2023, 7, 5911–5924. [Google Scholar] [CrossRef]
- Alig, S.K.; Esfahani, M.S.; Garofalo, A.; Li, M.Y.; Rossi, C.; Flerlage, T.; Flerlage, J.E.; Adams, R.; Binkley, M.S.; Shukla, N.; et al. Distinct Hodgkin lymphoma subtypes defined by noninvasive genomic profiling. Nature 2023, 625, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef]
- Cheson, B.D.; Pfistner, B.; Juweid, M.E.; Gascoyne, R.D.; Specht, L.; Horning, S.J.; Coiffier, B.; Fisher, R.I.; Hagenbeek, A.; Zucca, E.; et al. Revised Response Criteria for Malignant Lymphoma. J. Clin. Oncol. 2007, 25, 579–586. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Result |
|---|---|
| Female sex, no. (%) | 13 (59.1) |
| Age, years, median (range) | 33 (19–69) |
| Ann Arbor stage, no. (%) | |
| Initial stages (I–II) | 10 (45.5) |
| Advanced stages (III–IV) | 12 (54.5) |
| Histology, no. (%) | |
| Nodular sclerosis | 17 (77.3) |
| Lymphocyte-rich | 2 (9.1) |
| Not otherwise specified (NOS) | 3 (13.6) |
| Bulky disease, no. (%) | 19 (86.4) |
| B symptoms, no. (%) | 14 (63.6) |
| Epstein–Barr virus latent membrane protein-1, no. (%) | 6 (27.3) |
| Treatment response, no. (%) | |
| Complete metabolic response | 12 (54.5) |
| Relapse/refractory | 10 (45.5) |
| Refractory Cases | Genes Mutated at Diagnosis (N1–N10) | Genes Mutated at Relapse (N11–N20) |
|---|---|---|
| Patient 1 (Samples N1 and N11) | NOTCH2 * (W2436Ter) CREBBP * (P1943S) | NOTCH2 * (C745Y) CREBBP * (S2338F) |
| Patient 2 (Samples N2 and N12) | NOTCH1 (S2341F) ARID1A (S740F) CREBBP (S2361F) EZH2 (R690C) NOTCH2 *(F1017S) SF3B1 (S541F) CSF1R (C278Y) MYB (S245F) TNFAIP3 (C627Y) PTPRD (S874F) CSF2RB (S322F) EP300 (C1163Y) | NOTCH2 * (S2437L) |
| Patient 3 (Samples N3 and N13) | NOTCH1 (C1363Y) PLCG2 (Y293C) EP300 (S1642F) | PTPRD (G61R) |
| Patient 4 (Samples N4 and N14) | NOTCH2 (S2437L) ARID1A (G151R) KLF2 (R145C) | NOTCH1 (C398Y) |
| Patient 5 (Samples N5 and N15) | CREBBP (S1917L) SF3B1 (S57L) | ARID1A (S1570F) TNFAIP3 (C612Y) EP300 (C1177Y) |
| Patient 6 (Samples N6 and N16) | PTPRD (M282I) CYLD (S544L) EP300 (L1788F) BTK (E488K) | NOTCH1 (C1521Y) |
| Patient 7 (Samples N7 and N17) | NOTCH2 * (S2437L) CREBBP (A884T) ARID1A (G151E) CARD11 (D446N) | NOTCH2 * (R568C) BTK (S371F) SF3B1 (R568C) PTPRD (S623F) BCL10 (R25C) |
| Patient 8 (Samples N8 and N18) | SF3B1 (C1123Y) PTPRD (R1181C) XPO1 * (R553C) CD38 (C99Y) EZH2 (S114F) CYLD (S384F) PLCG2 (S152F) CSF2RB (C289Y) | XPO1 *(P552L) CREBBP (P1476L) |
| Patient 9 (Samples N9 and N19) | NOTCH1 * (C440Y) NOTCH2 (S2078F) PTPRD (C1465Y) XPO1 (C595Y) CXCR4 (S330F) SF3B1 (S637F) TNFAIP3 (R562C) BRAF (C748Y) CASP8 (C371Y) CARD11 (R35C) FOXO1 (S363F) CREBBP (C398Y) PLCG2 (S1179F) TCF3 (S157F) BTK (R641C) | NOTCH1 * (S2530F) |
| Patient 10 (Samples N10 and N20) | SF3B1 (R568C) PTPRD * (R174C) CREBBP * (S566F) | NOTCH1 (S1856F) NOTCH2 (S2421F) SF3B1 (S611F) CXCR4 (S319F) CSF1R (S840F) ABL1 (S438F) PTPRD * (R1181C) CREBBP * (S1432F) EP300 (C1177Y) ARID1A (S1604F) BRAF (S125F) PLCG2 (S1179F) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santisteban-Espejo, A.; Bernal-Florindo, I.; Montero-Pavon, P.; Perez-Requena, J.; Atienza-Cuevas, L.; Fernandez-Valle, M.d.C.; Villalba-Fernandez, A.; Garcia-Rojo, M. Pathogenic Variants Associated with Epigenetic Control and the NOTCH Pathway Are Frequent in Classic Hodgkin Lymphoma. Int. J. Mol. Sci. 2024, 25, 2457. https://doi.org/10.3390/ijms25052457
Santisteban-Espejo A, Bernal-Florindo I, Montero-Pavon P, Perez-Requena J, Atienza-Cuevas L, Fernandez-Valle MdC, Villalba-Fernandez A, Garcia-Rojo M. Pathogenic Variants Associated with Epigenetic Control and the NOTCH Pathway Are Frequent in Classic Hodgkin Lymphoma. International Journal of Molecular Sciences. 2024; 25(5):2457. https://doi.org/10.3390/ijms25052457
Chicago/Turabian StyleSantisteban-Espejo, Antonio, Irene Bernal-Florindo, Pedro Montero-Pavon, Jose Perez-Requena, Lidia Atienza-Cuevas, Maria del Carmen Fernandez-Valle, Ana Villalba-Fernandez, and Marcial Garcia-Rojo. 2024. "Pathogenic Variants Associated with Epigenetic Control and the NOTCH Pathway Are Frequent in Classic Hodgkin Lymphoma" International Journal of Molecular Sciences 25, no. 5: 2457. https://doi.org/10.3390/ijms25052457
APA StyleSantisteban-Espejo, A., Bernal-Florindo, I., Montero-Pavon, P., Perez-Requena, J., Atienza-Cuevas, L., Fernandez-Valle, M. d. C., Villalba-Fernandez, A., & Garcia-Rojo, M. (2024). Pathogenic Variants Associated with Epigenetic Control and the NOTCH Pathway Are Frequent in Classic Hodgkin Lymphoma. International Journal of Molecular Sciences, 25(5), 2457. https://doi.org/10.3390/ijms25052457