
Integrating Dynamic 3D Chromatin Architecture and Gene Expression Alterations Reveal Heterosis in Brassica rapa

 ,
,  and
and 
Abstract
1. Introduction
2. Results
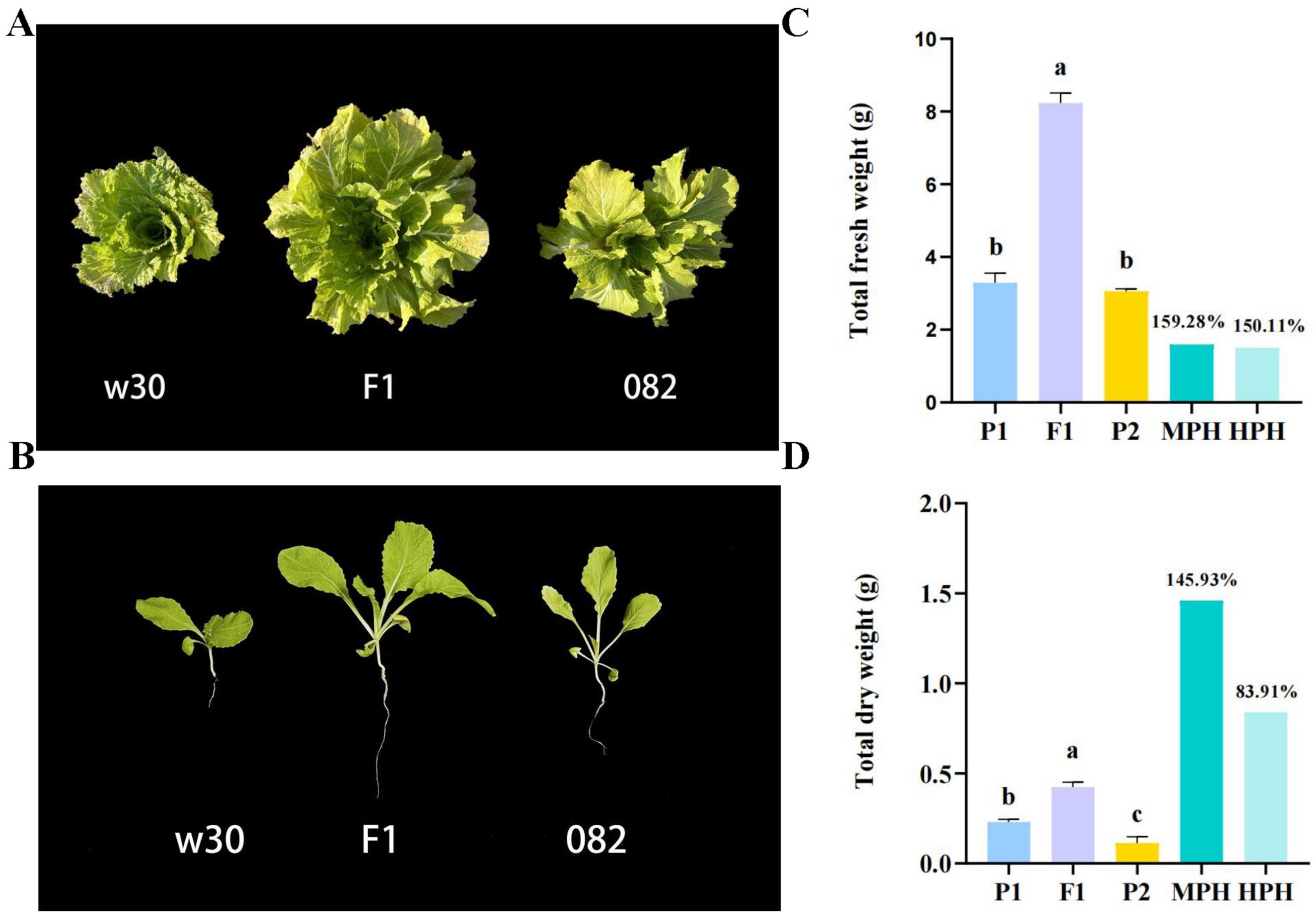
2.1. Heterosis Is Remarkable in the Hybrid F1
2.2. More Genes Were Actively Expressed in the Hybrid F1
2.3. Transcriptome Analysis of DEGs
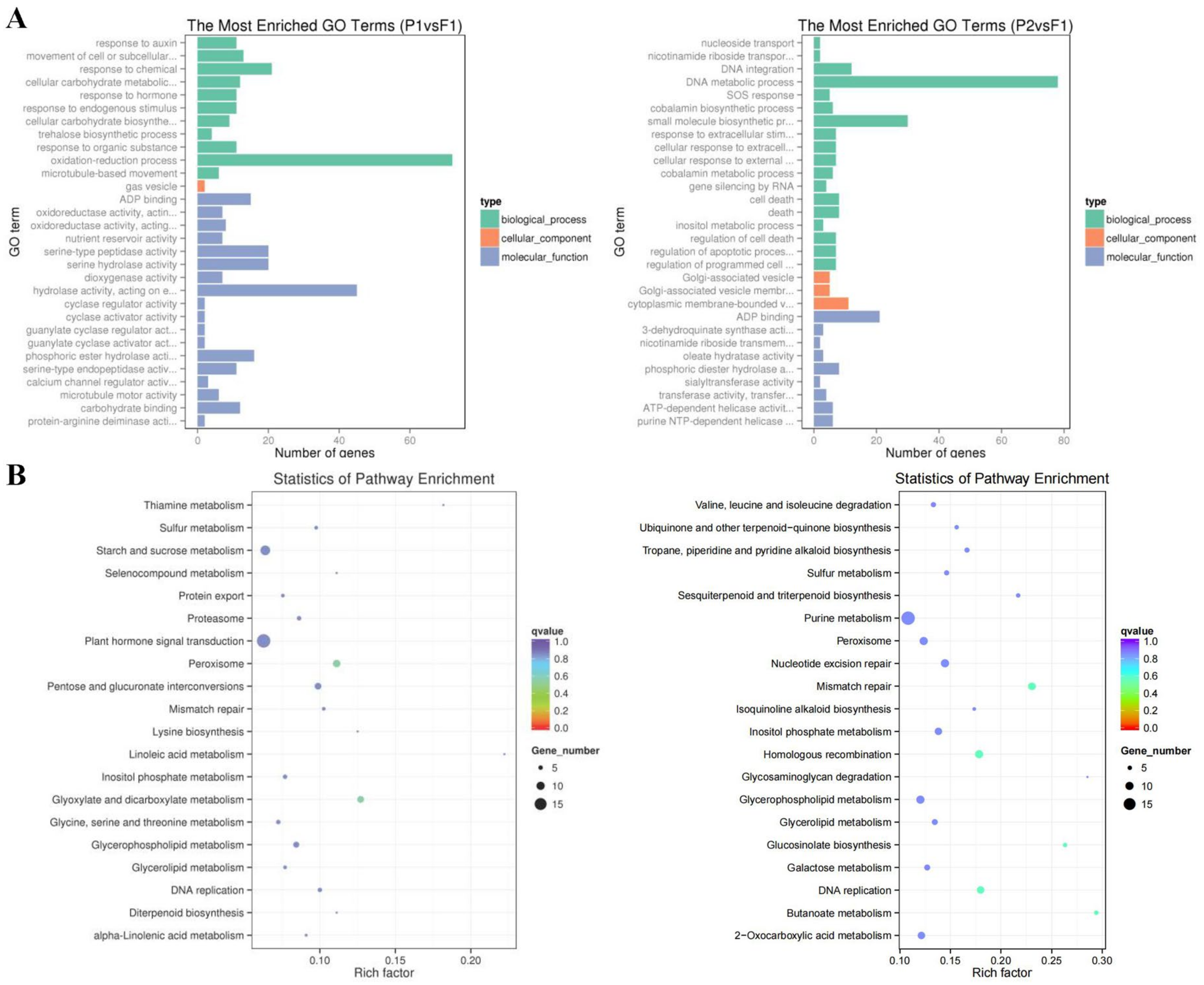
2.4. GO and KEGG Analysis of DEGs
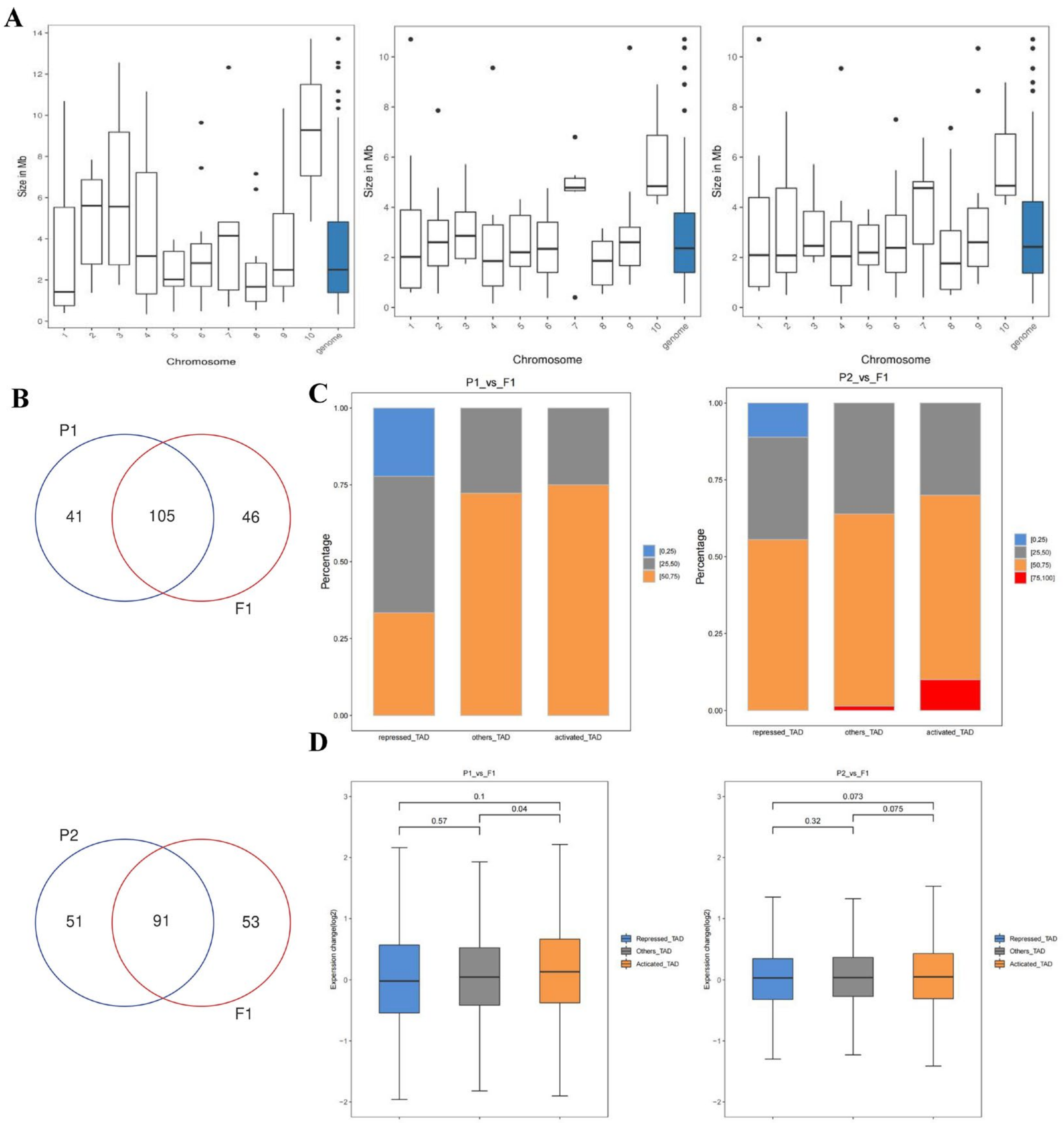
2.5. Genome-Wide Interaction Matrices of the Parents and F1
2.6. Identification of A/B Compartment Shifts
2.7. Changes in Compartments during Hybridization
2.8. Identification of Different Kinds of Topologically Associating Domains
2.9. Identification of Candidate Genes for Heterosis and Verification of qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Growth Conditions, and Sample Collection
4.2. Evaluation of Heterosis
4.3. RNA-Seq Experiment and Sequencing
4.4. Hi-C Libraries Construction and Sequencing
4.5. RNA-Seq Data Analysis
4.6. Hi-C Read Mapping
4.7. Quantitative Reverse-Transcription PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes; |
| FPKM | Fragments per kilobase of transcript sequence per million base pairs; |
| GO | Gene ontology; |
| MPV | Mid-parent value; |
| qRT-PCR | Quantitative real-time PCR; |
| QTL | Quantitative trait locus; |
| Hi-C | High throughput chromatin conformation capture; |
| RNA-seq | RNA-sequencing data. |
References
- Birchler, J.A.; Yao, H.; Chudalayandi, S.; Vaiman, D.; Veitia, R.A. Heterosis. Plant Cell 2010, 22, 2105–2112. [Google Scholar] [CrossRef]
- Hochholdinger, F.; Hoecker, N. Towards the molecular basis of heterosis. Trends Plant Sci. 2007, 12, 427–432. [Google Scholar] [CrossRef]
- Birchler, J.A.; Auger, D.L.; Riddle, N.C. In search of the molecular basis of heterosis. Plant Cell 2003, 15, 2236–2239. [Google Scholar] [CrossRef]
- Hochholdinger, F.; Baldauf, J.A. Heterosis in plants. Curr. Biol. 2018, 28, R1089–R1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, M.; Zhang, Q.; Wei, X.; Huang, X. Exploring the molecular basis of heterosis for plant breeding. J. Integr. Plant Biol. 2020, 62, 287–298. [Google Scholar] [CrossRef]
- Jones, D.F. Dominance of Linked Factors as a Means of Accounting for Heterosis. Proc. Natl. Acad. Sci. USA 1917, 3, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.B. The Mendelian theory of heredity and the augmentation of vigor. Science 1910, 32, 627–628. [Google Scholar] [CrossRef]
- Davenport, C.B. Degeneration, albinism, and inbreeding. Science 1908, 28, 454–455. [Google Scholar] [CrossRef]
- Crow, J.F. 90 years ago: The beginning of hybrid maize. Genetics 1998, 148, 923–928. [Google Scholar] [CrossRef] [PubMed]
- East, E.M. Heterosis. Genetics 1936, 21, 375–397. [Google Scholar] [CrossRef] [PubMed]
- Williams, W. Heterosis and the genetics of complex characters. Nature 1959, 184, 527–530. [Google Scholar] [CrossRef]
- Richey, F.D. Mock-dominance and hybrid vigor. Science 1942, 96, 280–281. [Google Scholar] [CrossRef]
- Fujimoto, R.; Uezone, K.; Ishikura, S.; Osabe, K.; Peacoc, W.J.; Dennis, E.S. Recent research on the mechanism of heterosis is important for crop and vegetable breeding systems. Breed. Sci. 2018, 68, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 2013, 14, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef]
- Probst, A.V.; Scheid, O.M. Stress-induced structural changes in plant chromatin. Curr. Opin. Plant Biol. 2015, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.; De Lucia, F.; Mylne, J.S.; Zhu, D.; Ohmido, N.; Pendle, A.; Kato, N.; Shaw, P.; Dean, C. Physical clustering of FLC alleles during Polycomb-mediated epigenetic silencing in vernalization. Genes Dev. 2013, 27, 1845–1850. [Google Scholar] [CrossRef]
- Ouyang, W.; Xiong, D.; Li, G.; Li, X. Unraveling the 3D Genome Architecture in Plants: Present and Future. Mol. Plant 2020, 13, 1676–1693. [Google Scholar] [CrossRef]
- Hu, Y.; Xiong, J.; Shalby, N.; Zhuo, C.; Jia, Y.; Yang, Q.-Y.; Tu, J. Comparison of dynamic 3D chromatin architecture uncovers heterosis for leaf size in Brassica napus. J. Adv. Res. 2022, 42, 289–301. [Google Scholar] [CrossRef]
- Sexton, T.; Cavalli, G. The Role of Chromosome Domains in Shaping the Functional Genome. Cell 2015, 160, 1049–1059. [Google Scholar] [CrossRef]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Hom, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2022, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Song, G.-S.; Zhai, H.-L.; Peng, Y.-G.; Zhang, L.; Wei, G.; Chen, X.-Y.; Xiao, Y.-G.; Wang, L.; Chen, Y.-J.; Wu, B.; et al. Comparative Transcriptional Profiling and Preliminary Study on Heterosis Mechanism of Super-Hybrid Rice. Mol. Plant 2010, 3, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, T.; Steinfath, M.; Andorf, S.; Lisec, J.; Meyer, R.C.; Altmann, T.; Willmitzer, L.; Selbig, J. Improved Heterosis Prediction by Combining Information on DNA- and Metabolic Markers. PLoS ONE 2009, 4, e5220. [Google Scholar] [CrossRef]
- Lisec, J.; Steinfath, M.; Meyer, R.C.; Selbig, J.; Melchinger, A.E.; Willmitzer, L.; Altmann, T. Identification of heterotic metabolite QTL in Arabidopsis thaliana RIL and IL populations. Plant J. 2009, 59, 777–788. [Google Scholar] [CrossRef]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695–7701. [Google Scholar] [CrossRef]
- Hu, X.; Wang, H.; Diao, X.; Liu, Z.; Li, K.; Wu, Y.; Liang, Q.; Wang, H.; Huang, C. Transcriptome profiling and comparison of maize ear heterosis during the spikelet and floret differentiation stages. BMC Genom. 2016, 17, 959. [Google Scholar] [CrossRef]
- Cai, C.; Wang, X.; Liu, B.; Wu, J.; Liang, J.; Cui, Y.; Cheng, F.; Wang, X. Brassica rapa Genome 2.0: A Reference Upgrade through Sequence Re-assembly and Gene Re-annotation. Mol. Plant 2017, 10, 649–651. [Google Scholar] [CrossRef]
- Tanabe, H.; Muller, S.; Neusser, M.; von Hase, J.; Calcagno, E.; Cremer, M.; Solovei, I.; Cremer, C.; Cremer, T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl. Acad. Sci. USA 2002, 99, 4424–4429. [Google Scholar] [CrossRef] [PubMed]
- Ghavi-Helm, Y.; Jankowski, A.; Meiers, S.; Viales, R.R.; Korbel, J.O.; Furlong, E.E.M. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 2019, 51, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Grob, S.; Schmid, M.W.; Grossniklaus, U. Hi-C Analysis in Arabidopsis Identifies the KNOT, a Structure with Similarities to the flamenco Locus of Drosophila. Mol. Cell 2014, 55, 678–693. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Du, Z.; Wei, L.; Fang, H.; Dong, Q.; Niu, J.; Li, Y.; Gao, J.; Zhang, M.Q.; et al. The loss of heterochromatin is associated with multiscale three-dimensional genome reorganization and aberrant transcription during cellular senescence. Genome Res. 2021, 31, 1121–1135. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Dekker, J. The Hierarchy of the 3D Genome. Mol. Cell 2013, 49, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, F.-G.; Zhang, H.-Y.; Wang, X.-T.; Hu, J.-H.; Wu, X.-M. Biased gene retention during diploidization in Brassica linked to three-dimensional genome organization. Nat. Plants 2019, 5, 822–832. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Lin, M.; Ye, Z.; Li, G.; Tu, L.; Shen, C.; Li, J.; Yang, Q.; Zhang, X. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat. Plants 2018, 4, 90–97. [Google Scholar] [CrossRef]
- Dong, Q.; Li, N.; Li, X.; Yuan, Z.; Xie, D.; Wang, X.; Li, J.; Yu, Y.; Wang, J.; Ding, B.; et al. Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice. Plant J. 2018, 94, 1141–1156. [Google Scholar] [CrossRef]
- Kong, S.; Zhang, Y. Deciphering Hi-C: From 3D genome to function. Cell Biol. Toxicol. 2019, 35, 15–32. [Google Scholar] [CrossRef]
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010, 39, e1869. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wingett, S.; Ewels, P.; Furlan-Magaril, M.; Nagano, T.; Schoenfelder, S.; Fraser, P.; Andrews, S. HiCUP: Pipeline for mapping and processing Hi-C data. F1000Research 2015, 4, 1310. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Szalaj, P.; Tang, Z.; Michalski, P.; Pietal, M.J.; Luo, O.J.; Sadowski, M.; Li, X.; Radew, K.; Ruan, Y.; Plewczynski, D. An integrated 3-Dimensional Genome Modeling Engine for data-driven simulation of spatial genome organization. Genome Res. 2016, 26, 1697–1709. [Google Scholar] [CrossRef]
- Dheda, K.; Huggett, J.F.; Bustin, S.A.; Johnson, M.A.; Rook, G.; Zumla, A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004, 37, 112–119. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C-T method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid Set Samples | Up | Down | B2P | Total |
|---|---|---|---|---|
| 082 vs. w30 | 888 (41.19%) | 1268 (58.81%) | 2156 | |
| F1 vs. 082 | 885 (80.38%) | 216 (19.62%) | 1101 | |
| F1 vs. w30 | 1115 (63.24%) | 648 (36.76%) | 1763 | |
| F1 vs. (082 and w30) | 173 (58.45%) | 32 (10.81%) | 91 (30.74%) | 296 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
E, L.; Lyu, S.; Wang, Y.; Xiao, D.; Liu, T.; Hou, X.; Li, Y.; Zhang, C. Integrating Dynamic 3D Chromatin Architecture and Gene Expression Alterations Reveal Heterosis in Brassica rapa. Int. J. Mol. Sci. 2024, 25, 2568. https://doi.org/10.3390/ijms25052568
E L, Lyu S, Wang Y, Xiao D, Liu T, Hou X, Li Y, Zhang C. Integrating Dynamic 3D Chromatin Architecture and Gene Expression Alterations Reveal Heterosis in Brassica rapa. International Journal of Molecular Sciences. 2024; 25(5):2568. https://doi.org/10.3390/ijms25052568
Chicago/Turabian StyleE, Liu, Shanwu Lyu, Yaolong Wang, Dong Xiao, Tongkun Liu, Xilin Hou, Ying Li, and Changwei Zhang. 2024. "Integrating Dynamic 3D Chromatin Architecture and Gene Expression Alterations Reveal Heterosis in Brassica rapa" International Journal of Molecular Sciences 25, no. 5: 2568. https://doi.org/10.3390/ijms25052568
APA StyleE, L., Lyu, S., Wang, Y., Xiao, D., Liu, T., Hou, X., Li, Y., & Zhang, C. (2024). Integrating Dynamic 3D Chromatin Architecture and Gene Expression Alterations Reveal Heterosis in Brassica rapa. International Journal of Molecular Sciences, 25(5), 2568. https://doi.org/10.3390/ijms25052568