
Molecular Simulation of SO2 Separation and Storage Using a Cryptophane-Based Porous Liquid

Abstract
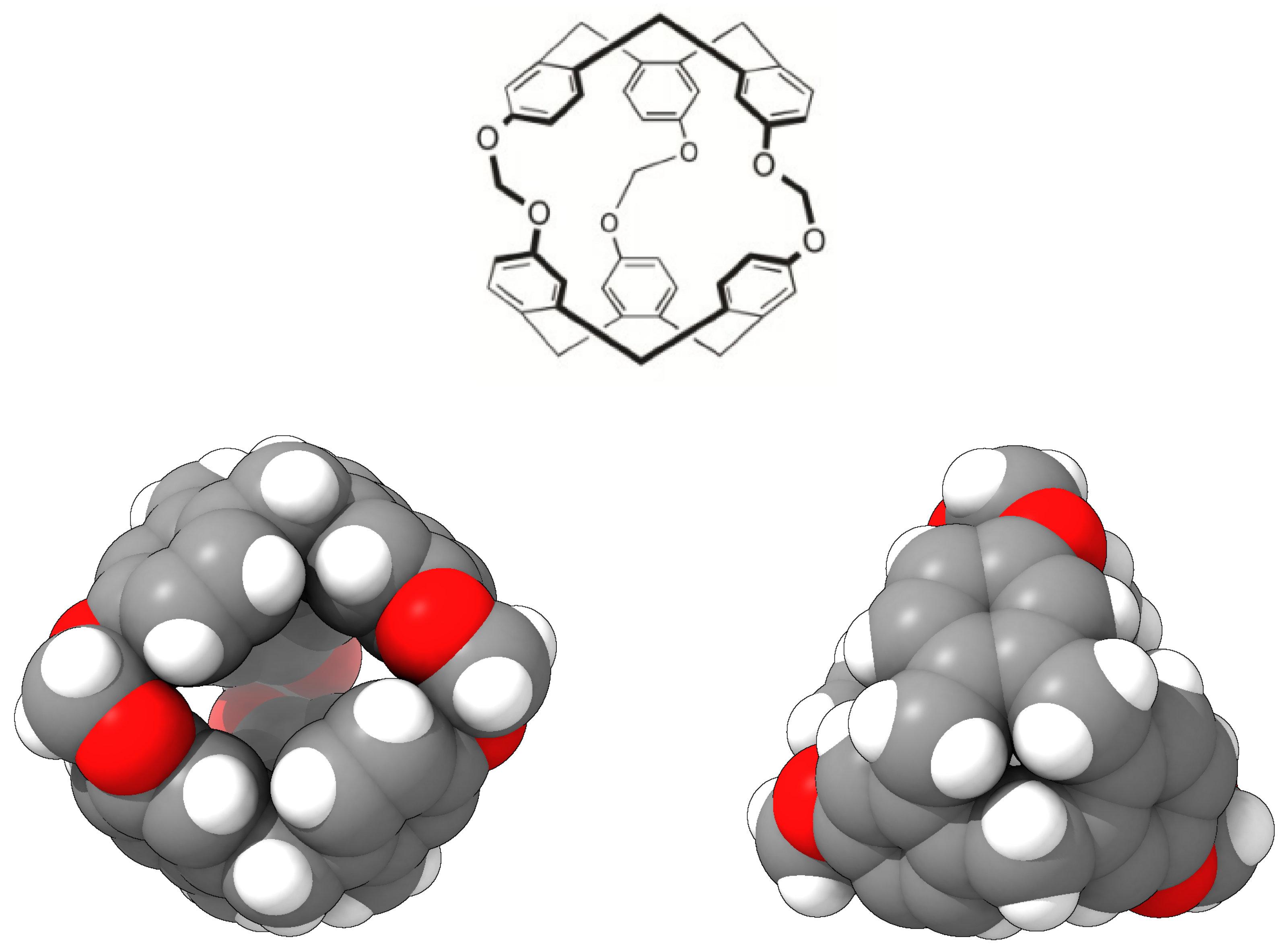
:1. Introduction
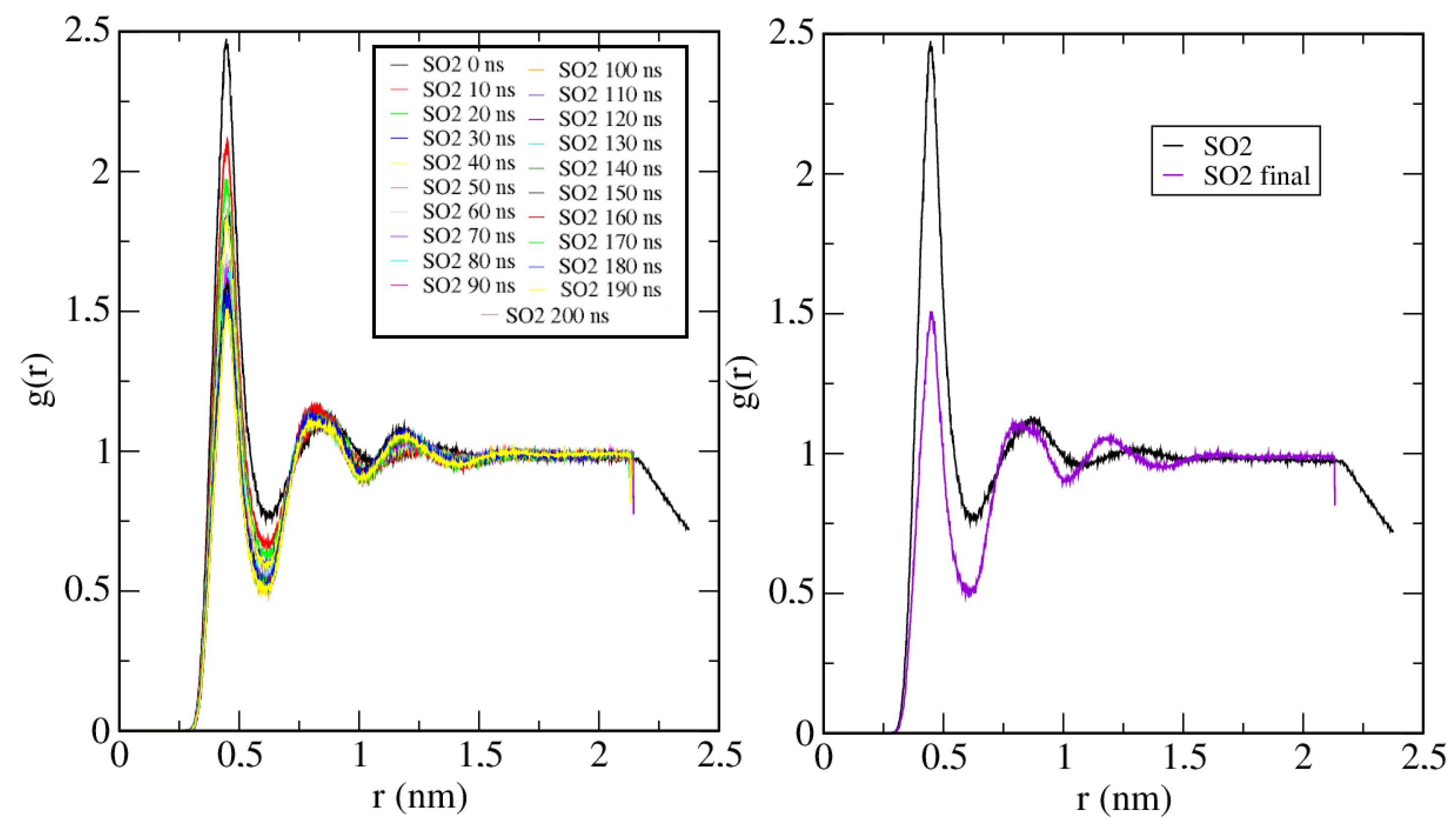
2. Results and Discussion
2.1. Case A
2.2. Case B
2.3. Case C
2.4. Case D
2.5. Case E
2.6. Future Approaches
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATB | Automatic Topology Builder |
| DCM | dichloromethane |
| C-111 | cryptophane-111 |
| MOF | metal organic framework |
| OR | occupancy ratio |
| PDB | Protein Data Bank |
| PL | porous liquid |
References
- O’Reilly, N.; Giri, N.; James, S.L. Porous liquids. Chem. Eur. J. 2007, 13, 3020–3025. [Google Scholar] [CrossRef]
- James, S.L. The dam bursts for porous liquids. Adv. Mater. 2016, 28, 5712–5716. [Google Scholar] [CrossRef]
- Bennett, T.D.; Coudert, F.X.; James, S.L.; Cooper, A.I. The changing state of porous materials. Nat. Mater. 2021, 20, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Jie, K.; Zhou, Y.; Ryan, H.P.; Dai, S.; Nitschke, J.R. Porous liquids: Engineering permanent porosity into liquids. Adv. Mater. 2021, 33, 2170136. [Google Scholar] [CrossRef]
- Melaugh, G.; Giri, N.; Davidson, C.E.; James, S.L.; Del Pópolo, M.G. Designing and understanding permanent microporosity in liquids. Phys. Chem. Chem. Phys. 2014, 16, 9422–9431. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, B.; Lin, L.C.; Chen, G.; Wu, Y.; Wang, J.; Gao, X.; Lv, Y.; Pan, Y.; Zhang, X.; et al. A hybrid absorption-adsorption method to efficiently capture carbon. Nat. Commun. 2014, 5, 5147. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Červinka, C.; Dugas, P.Y.; Pádua, A.A.H.; Costa Gomes, M. Porous ionic liquids: Structure, stability, and gas absorption mechanisms. Adv. Mater. Interfaces 2021, 8, 2001982. [Google Scholar] [CrossRef]
- Li, P.; Chen, H.; Schott, J.A.; Li, B.; Zheng, Y.; Mahurin, S.M.; Jiang, D.E.; Cui, G.; Hu, X.; Wang, Y.; et al. Porous liquid zeolites: Hydrogen bonding-stabilized H-ZSM-5 in branched ionic liquids. Nanoscale 2019, 11, 1515–1519. [Google Scholar] [CrossRef]
- Avila, J.; Lepre, L.F.; Santini, C.; Tiano, M.; Denis-Quanquin, S.; Szeto, K.C.; Padua, A.; Costa Gomes, M. High-performance porous ionic liquids for low pressure CO2 capture. Angew. Chem. 2021, 60, 12876–12882. [Google Scholar] [CrossRef]
- Li, P.; Wang, D.; Zhang, L.; Liu, C.; Wu, F.; Wang, Y.; Wang, Z.; Zhao, Z.; Wu, W.; Liang, Y.; et al. An in situ coupling strategy toward porous carbon liquid with permanent porosity. Small 2021, 17, e2006687. [Google Scholar] [CrossRef]
- Ahmad, M.Z.; Fuoco, A. Porous liquids—Future for CO2 Capture and Separation? Curr. Res. Green Sust. Chem. 2021, 4, 100070. [Google Scholar] [CrossRef]
- Brand, M.C.; Rankin, N.; Cooper, A.I.; Greenaway, R.L. Photoresponsive Type III Porous Liquids. Chem. Eur. J. 2023, 29, e202202848. [Google Scholar] [CrossRef]
- Christian, M.S.; Hurlock, M.J.; Nenoff, T.M.; Rimsza, J.M. CO2 adsorption mechanisms at the ZIF-8 interface in a Type 3 porous liquid. J. Mol. Liq. 2024, 395, 123913. [Google Scholar] [CrossRef]
- Zou, Y.H.; Huang, Y.B.; Si, D.H.; Yin, Q.; Wu, Q.J.; Weng, Z.; Cao, R. Porous Metal-Organic Framework Liquids for Enhanced CO2 Adsorption and Catalytic Conversion. Angew. Chem. Int. Ed. 2021, 60, 20915–20920. [Google Scholar] [CrossRef]
- Oltean, M. Theoretical Study of Chemical Reactivity: From Nano-Objects to Follow the Entropy via Metadynamics Simulations. Ph.D. Thesis, Université Joseph-Fourier, Grenoble, France, 2010. Available online: https://theses.hal.science/tel-00561804 (accessed on 21 February 2024).
- Mahdavi, H.; Smith, S.J.D.; Mulet, X.; Hill, M.R. Practical considerations in the design and use of porous liquids. Mater. Horiz. 2022, 9, 1577–1601. [Google Scholar] [CrossRef] [PubMed]
- Collado, P.; Piñeiro, M.M.; Pérez-Rodríguez, M. Molecular Simulation of CO2 and H2 Encapsulation in a Nanoscale Porous Liquid. Nanomaterials 2023, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.A.; Berthault, P.; Brotin, T.; Huber, G.; Desvaux, H.; Dutasta, J.P. A cryptophane core optimized for xenon encapsulation. J. Am. Chem. Soc. 2007, 129, 10332–10333. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.I.; Lapidus, S.H.; Kane, C.M.; Holman, K.T. Extreme confinement of xenon by cryptophane-111 in the solid state. Angew. Chem. Weinheim Bergstr. Ger. 2015, 127, 1491–1495. [Google Scholar] [CrossRef]
- Mecozzi, S.; Rebek, J., Jr. The 55% solution: A formula for molecular recognition in the liquid state. Chemistry 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- El-Ayle, G.; Holman, K.T. Cryptophanes; Elsevier: Amsterdam, The Netherlands, 2017; pp. 199–249. [Google Scholar]
- Traoré, T.; Delacour, L.; Garcia-Argote, S.; Berthault, P.; Cintrat, J.C.; Rousseau, B. Scalable Synthesis of Cryptophane-1.1.1 and its Functionalization. Org. Lett. 2010, 12, 960–962. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Buffeteau, T.; Pitrat, D.; Daugey, N.; Calin, N.; Jean, M.; Vanthuyne, N.; Ducasse, L.; Wien, F.; Brotin, T. Chiroptical properties of cryptophane-111. Phys. Chem. Chem. Phys. 2017, 19, 18303–18310. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, M.; Otero-Fernández, J.; Comesaña, A.; Fernández-Fernández, A.M.; Piñeiro, M.M. Simulation of Capture and Release Processes of Hydrogen by β-Hydroquinone Clathrate. ACS Omega 2018, 3, 18771–18782. [Google Scholar] [CrossRef] [PubMed]
- Torré, J.P.; Gornitzka, H.; Coupan, R.; Dicharry, C.; Pérez-Rodríguez, M.; Comesaña, A.; Piñeiro, M.M. Insights into the Crystal Structure and Clathration Selectivity of Organic Clathrates Formed with Hydroquinone and (CO2 + CH4) Gas Mixtures. J. Phys. Chem. C 2019, 123, 14582–14590. [Google Scholar] [CrossRef]
- Fernández-Fernández, A.M.; Conde, M.M.; Pérez-Sánchez, G.; Pérez-Rodríguez, M.; Piñeiro, M.M. Molecular simulation of methane hydrate growth confined into a silica pore. J. Mol. Liq. 2022, 362, 119698. [Google Scholar] [CrossRef]
- Méndez-Morales, T.; Montes-Campos, H.; Pérez-Rodríguez, M.; Piñeiro, M.M. Evaluation of hydrogen storage ability of hydroquinone clathrates using molecular simulations. J. Mol. Liq. 2022, 360, 119487. [Google Scholar] [CrossRef]
- Rodríguez-García, B.; Piñeiro, M.M.; Pérez-Rodríguez, M. Diffusion and dynamics of noble gases in hydroquinone clathrate channels. J. Chem. Phys. 2023, 158, 044503. [Google Scholar] [CrossRef]
- Rodríguez-García, B.; Piñeiro, M.M.; Pérez-Rodríguez, M. Influence of Lennard-Jones Parameters in the Temperature Dependence of Real Gases Diffusion through Nanochannels. Nanomaterials 2023, 13, 1534. [Google Scholar] [CrossRef]
- Egleston, B.D.; Mroz, A.; Jelfs, K.E.; Greenaway, R.L. Porous liquids—The future is looking emptier. Chem. Sci. 2022, 13, 5042–5054. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated force field Topology Builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated topology builder version 3.0: Prediction of solvation free enthalpies in water and hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M. Gromacs: A parallel computer for molecular dynamics simulations. In Physics Computing 92; DeGroot, R., Nadrchal, J., Eds.; World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Berendsen, H.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale: International Conference on Exascale Applications and Software; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Gullingsrud, J.; Grayson, P.; Schulten, K. A System for Interactive Molecular Dynamics Simulation. In Proceedings of the 2001 ACM Symposium on Interactive 3D Graphics, Chapel Hill, NC, USA, 26–29 March 2001; Hughes, J.F., Séquin, C.H., Eds.; ACM: New York, NY, USA, 2001; pp. 191–194. [Google Scholar]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 300 K | |
| Simulations | Occupation (Out of 7) |
| REP1 | 6 |
| REP2 | 5 |
| REP3 | 6 |
| REP4 | 6 |
| REP5 | 7 |
| REP6 | 7 |
| REP7 | 5 |
| REP8 | 4 |
| REP9 | 7 |
| Average | |
| 283 K | |
| Simulations | Occupation (Out of 7) |
| REP1 | 5 |
| REP2 | 5 |
| REP3 | 6 |
| REP4 | 7 |
| REP5 | 6 |
| REP6 | 6 |
| REP7 | 7 |
| REP8 | 7 |
| REP9 | 6 |
| Average | |
| 300 K | |
| Simulations | Occupation (Out of 14) |
| REP1 | 11 |
| REP2 | 10 |
| REP3 | 10 |
| Average | 10.3 |
| 283 K | |
| Simulations | Occupation (Out of 14) |
| REP1 | 11 |
| REP2 | 11 |
| REP3 | 10 |
| Average | 10.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collado, P.; Piñeiro, M.M.; Pérez-Rodríguez, M. Molecular Simulation of SO2 Separation and Storage Using a Cryptophane-Based Porous Liquid. Int. J. Mol. Sci. 2024, 25, 2718. https://doi.org/10.3390/ijms25052718
Collado P, Piñeiro MM, Pérez-Rodríguez M. Molecular Simulation of SO2 Separation and Storage Using a Cryptophane-Based Porous Liquid. International Journal of Molecular Sciences. 2024; 25(5):2718. https://doi.org/10.3390/ijms25052718
Chicago/Turabian StyleCollado, Pablo, Manuel M. Piñeiro, and Martín Pérez-Rodríguez. 2024. "Molecular Simulation of SO2 Separation and Storage Using a Cryptophane-Based Porous Liquid" International Journal of Molecular Sciences 25, no. 5: 2718. https://doi.org/10.3390/ijms25052718