
A New Strategy to Investigate RNA:DNA Triplex Using Atomic Force Microscopy

Abstract
:1. Introduction
2. Results
2.1. Synthesis of the Target DNA and the RNA Probes
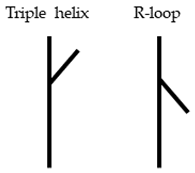
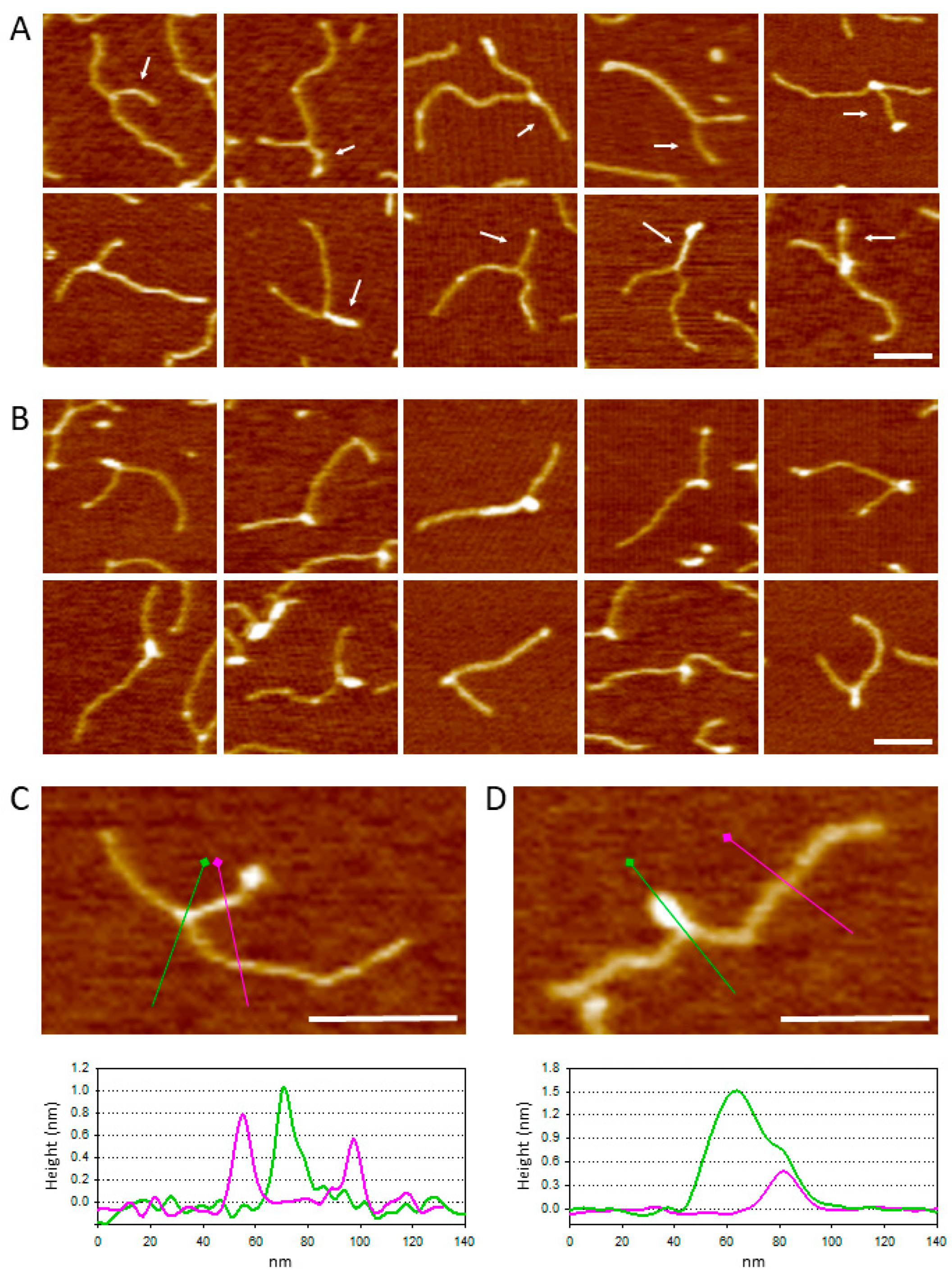
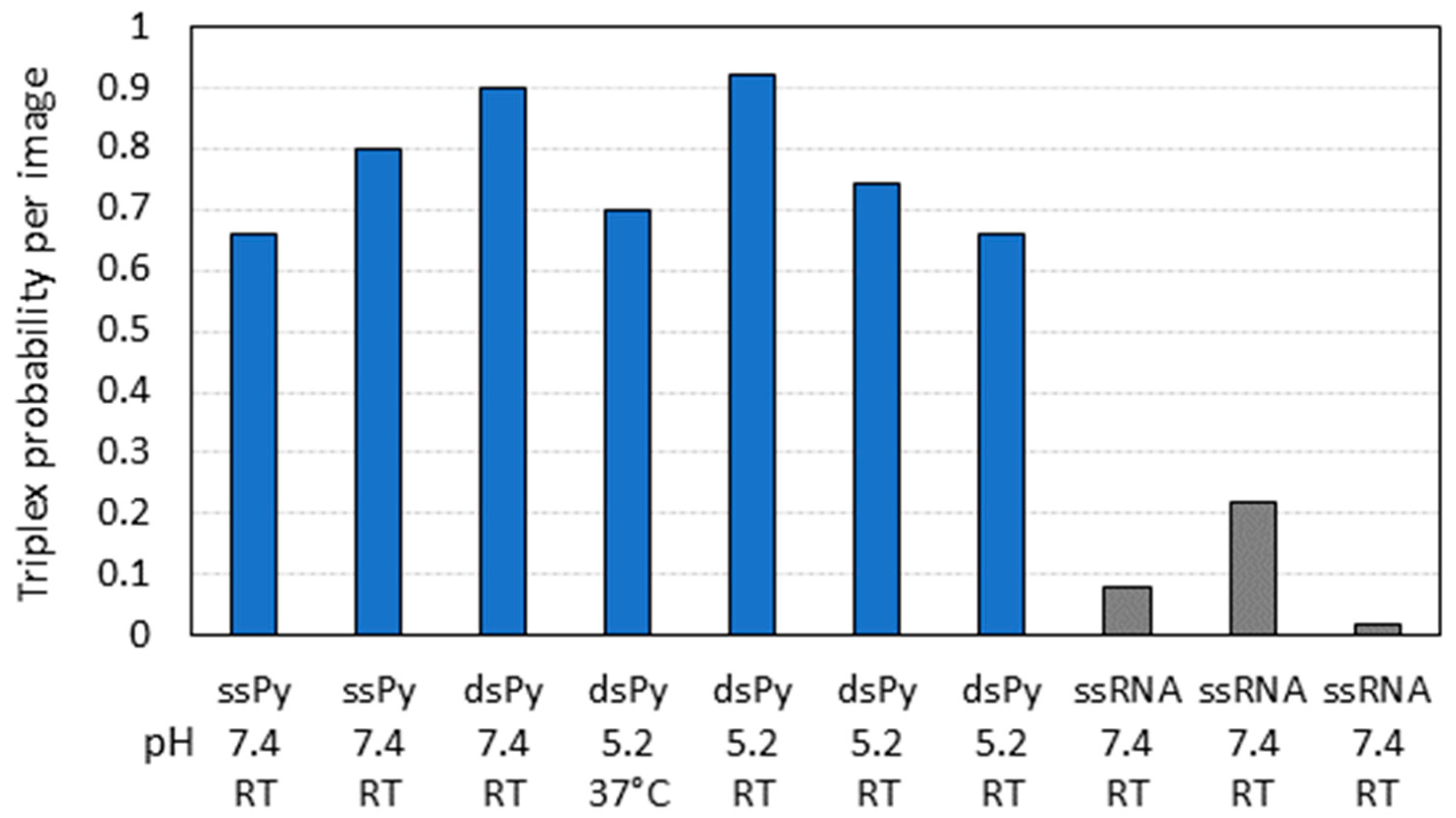
2.2. Triple-Helix Formation and AFM Imaging
3. Discussion
4. Materials and Methods
4.1. Cloning and Purification of the TFR2 DNA Template
4.2. Synthesis of the TFR2 RNA Probes
4.3. Triplex Formation and Atomic Force Microscopy
4.4. Image Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, J.; Wang, X.; Xiao, D.; Liu, S.; Tao, Y. The chromatin-associated RNAs in gene regulation and cancer. Mol. Cancer 2023, 22, 27. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long non-coding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.; Kanduri, C. Understanding Long Non-coding RNA and Chromatin Interactions: What We Know So Far. Non-coding RNA 2019, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Greifenstein, A.A.; Jo, S.; Bierhoff, H. RNA:DNA triple helices: From peculiar structures to pervasive chromatin regulators. Essays Biochem. 2021, 65, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Non-coding RNAs. Cell Chem. Biol. 2016, 23, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.; Langst, G. The chromatin—Triple-helix connection. Biol. Chem. 2023; ahead of print. [Google Scholar] [CrossRef]
- Xu, C.; Li, C.; Chen, J.; Xiong, Y.; Qiao, Z.; Fan, P.; Li, C.; Ma, S.; Liu, J.; Song, A.; et al. R-loop-dependent promoter-proximal termination ensures genome stability. Nature 2023, 621, 610–619. [Google Scholar] [CrossRef]
- Duardo, R.C.; Guerra, F.; Pepe, S.; Capranico, G. Non-B DNA structures as a booster of genome instability. Biochimie 2023, 214, 176–192. [Google Scholar] [CrossRef]
- Zhou, Z.; Giles, K.E.; Felsenfeld, G. DNA.RNA triple-helix formation can function as a cis-acting regulatory mechanism at the human beta-globin locus. Proc. Natl. Acad. Sci. USA 2019, 116, 6130–6139. [Google Scholar] [CrossRef]
- Li, X.; Guo, G.; Lu, M.; Chai, W.; Li, Y.; Tong, X.; Li, J.; Jia, X.; Liu, W.; Qi, D.; et al. Long Non-coding RNA Lnc-MxA Inhibits Beta Interferon Transcription by Forming RNA-DNA Triplexes at Its Promoter. J. Virol. 2019, 93, e00786-19. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of non-coding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes. Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Senturk, N.; Song, C.; Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018, 32, 836–848. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, V.B.; Ovsepian, S.V.; Carrascosa, L.G.; Buske, F.A.; Radulovic, V.; Niyazi, M.; Moertl, S.; Trau, M.; Atkinson, M.J.; Anastasov, N. PARTICLE, a Triplex-Forming Long ncRNA, Regulates Locus-Specific Methylation in Response to Low-Dose Irradiation. Cell Rep. 2015, 11, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Yari, H.; Jin, L.; Teng, L.; Wang, Y.; Wu, Y.; Liu, G.Z.; Gao, W.; Liang, J.; Xi, Y.; Feng, Y.C.; et al. LncRNA REG1CP promotes tumorigenesis through an enhancer complex to recruit FANCJ helicase for REG3A transcription. Nat. Commun. 2019, 10, 5334. [Google Scholar] [CrossRef] [PubMed]
- Postepska-Igielska, A.; Giwojna, A.; Gasri-Plotnitsky, L.; Schmitt, N.; Dold, A.; Ginsberg, D.; Grummt, I. LncRNA Khps1 Regulates Expression of the Proto-oncogene SPHK1 via Triplex-Mediated Changes in Chromatin Structure. Mol. Cell 2015, 60, 626–636. [Google Scholar] [CrossRef]
- Blank-Giwojna, A.; Postepska-Igielska, A.; Grummt, I. lncRNA KHPS1 Activates a Poised Enhancer by Triplex-Dependent Recruitment of Epigenomic Regulators. Cell Rep. 2019, 26, 2904–2915.e4. [Google Scholar] [CrossRef]
- Bustamante, C.; Rivetti, C. Visualizing protein-nucleic acid interactions on a large scale with the scanning force microscope. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 395–429. [Google Scholar] [CrossRef]
- Main, K.H.S.; Provan, J.I.; Haynes, P.J.; Wells, G.; Hartley, J.A.; Pyne, A.L.B. Atomic force microscopy-A tool for structural and translational DNA research. APL Bioeng. 2021, 5, 031504. [Google Scholar] [CrossRef]
- Cherny, D.I.; Fourcade, A.; Svinarchuk, F.; Nielsen, P.E.; Malvy, C.; Delain, E. Analysis of various sequence-specific triplexes by electron and atomic force microscopies. Biophys. J. 1998, 74, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Klinov, D.; Dwir, B.; Kapon, E.; Borovok, N.; Molotsky, T.; Kotlyar, A. High-resolution atomic force microscopy of duplex and triplex DNA molecules. Nanotechnology 2007, 18, 225102. [Google Scholar] [CrossRef]
- Tiner, W.J., Sr.; Potaman, V.N.; Sinden, R.R.; Lyubchenko, Y.L. The structure of intramolecular triplex DNA: Atomic force microscopy study. J. Mol. Biol. 2001, 314, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Rivetti, C.; Codeluppi, S. Accurate length determination of DNA molecules visualized by atomic force microscopy: Evidence for a partial B- to A-form transition on mica. Ultramicroscopy 2001, 87, 55–66. [Google Scholar] [CrossRef]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple-helix: 50 years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef]
- Ferre, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein-lncRNA interaction. Brief. Bioinform. 2016, 17, 106–116. [Google Scholar] [CrossRef]
- Badmalia, M.D.; Sette Pereira, H.; Siddiqui, M.Q.; Patel, T.R. A comprehensive review of methods to study lncRNA-protein interactions in solution. Biochem. Soc. Trans. 2022, 50, 1415–1426. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| FA01 TH01 | ATATGAATTCAATGACAAGAATGAGGG |
| FA02 TH02 | ATATGAATTCCTCCGAGAAACAGGAAC |
| FA07 TH07 | GCCCCAGGAGGAAAAAAAAAGGGGGGATTAGCAGCCGGATCTCAGTGG |
| FA03 TH03 | GAAATTAATACGACTCACTATAGGGGAATTGTG |
| FA08 TH08 | GAAATTAATACGACTCACTATAGGGTTAGCAGCCGGATCTCAGTGG |
| FA06 TH06 | GGGAATTGTGAGCGGATAACAATTCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merici, G.; Amidani, D.; Dieci, G.; Rivetti, C. A New Strategy to Investigate RNA:DNA Triplex Using Atomic Force Microscopy. Int. J. Mol. Sci. 2024, 25, 3035. https://doi.org/10.3390/ijms25053035
Merici G, Amidani D, Dieci G, Rivetti C. A New Strategy to Investigate RNA:DNA Triplex Using Atomic Force Microscopy. International Journal of Molecular Sciences. 2024; 25(5):3035. https://doi.org/10.3390/ijms25053035
Chicago/Turabian StyleMerici, Giovanni, Davide Amidani, Giorgio Dieci, and Claudio Rivetti. 2024. "A New Strategy to Investigate RNA:DNA Triplex Using Atomic Force Microscopy" International Journal of Molecular Sciences 25, no. 5: 3035. https://doi.org/10.3390/ijms25053035