
Pharmacogenomics of Drugs Used in β-Thalassemia and Sickle-Cell Disease: From Basic Research to Clinical Applications




Abstract
1. Introduction
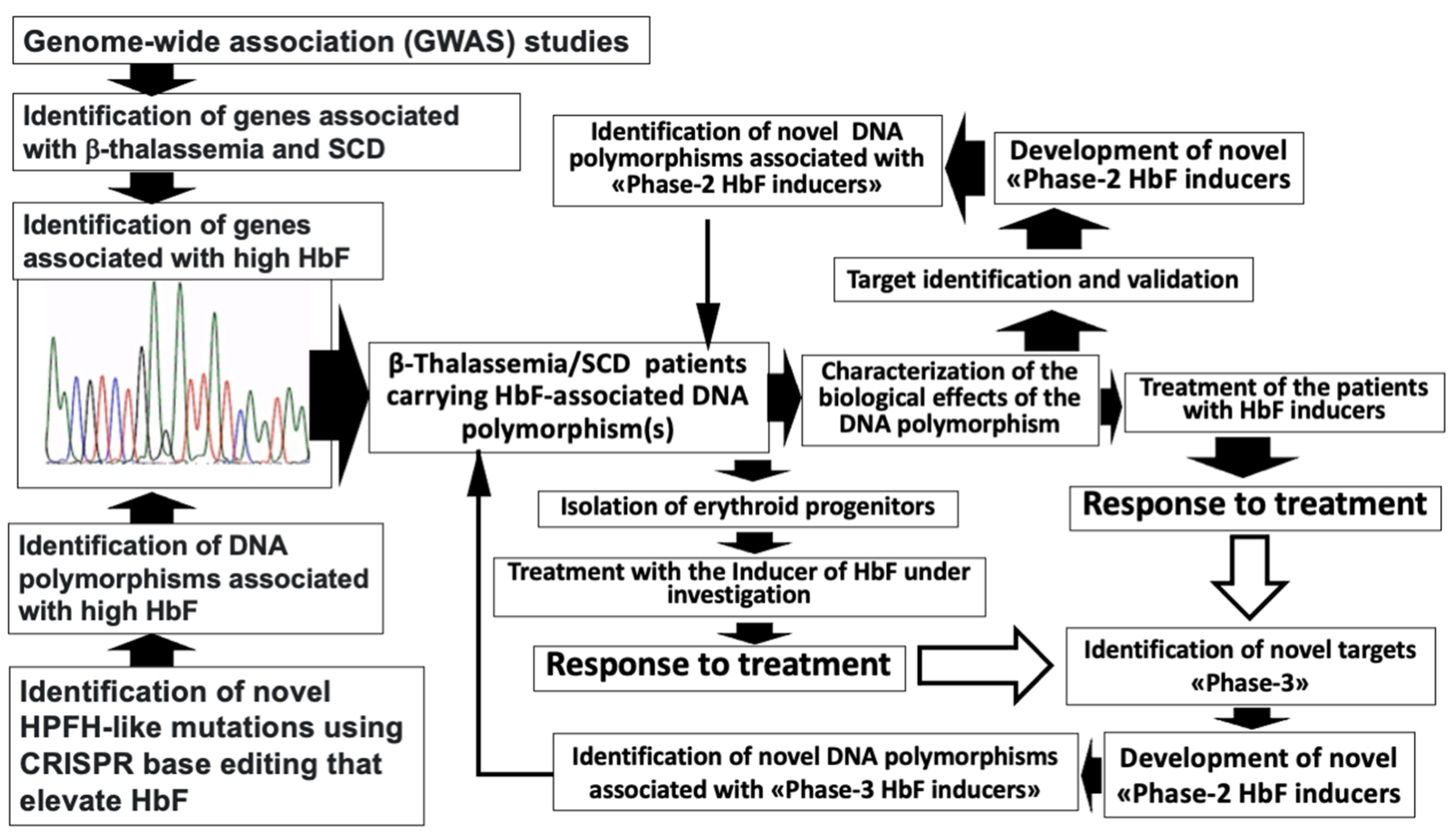
2. The Search of DNA Polymorphisms Associated with High Expression of γ-Globin Genes: GWAS Studies and CRISPR-Based Gene Editing Approaches
3. Major DNA Polymorphisms Associated with High Expression of γ-Globin Genes
3.1. The HBG2 XmnI rs7482144 Polymorphism
3.2. The LYAR rs368698783 (G>A) Polymorphism: Relevance and Structural Association with HBG2 XmnI
3.3. The BCL11A rs2071348 Polymorphisms
3.4. The KLF1 rs3817621, rs2072597, and rs2072596 Polymorphisms
3.5. Other Genetic Polymorphisms
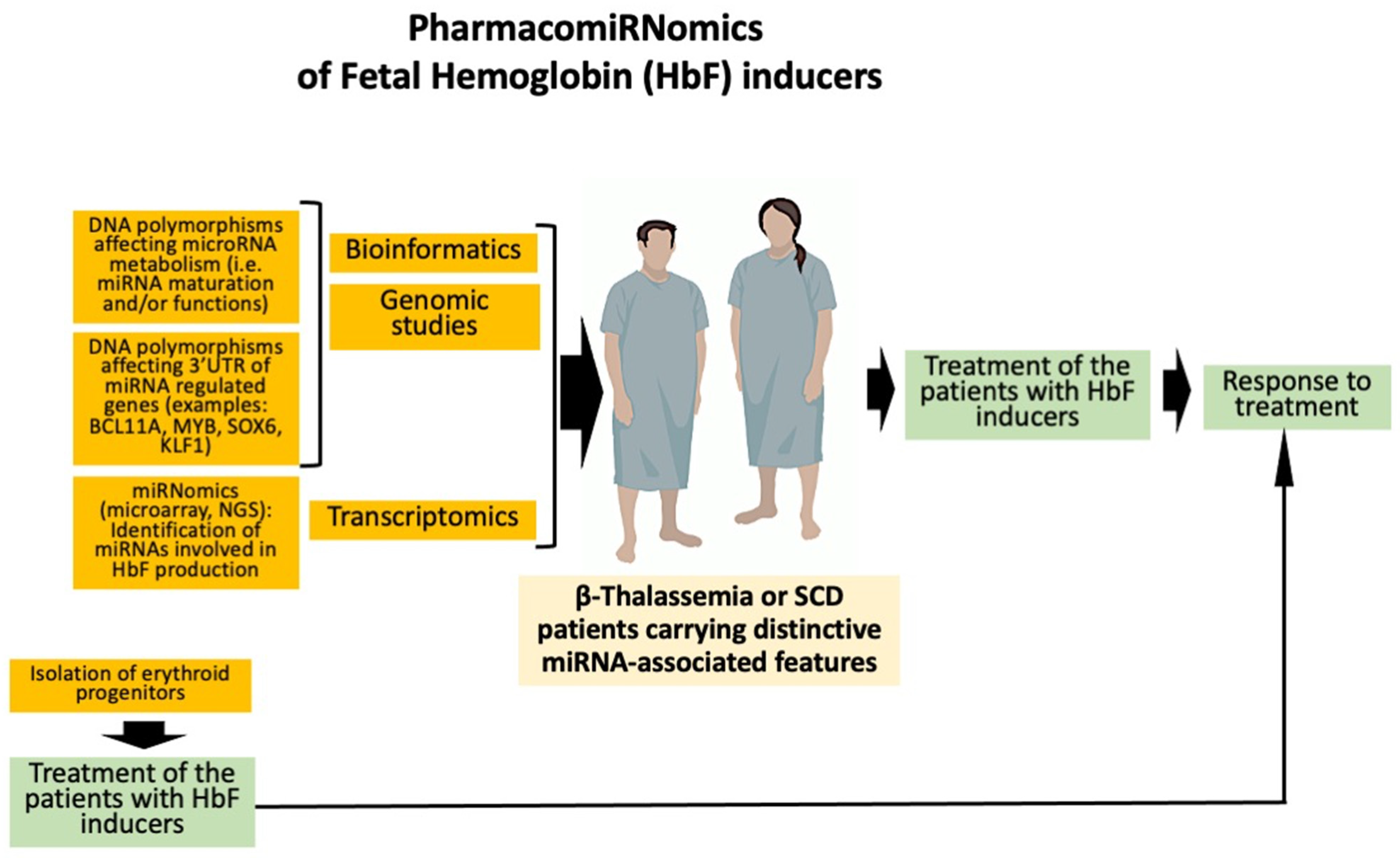
4. Expression of microRNAs Involved in the Regulation of γ-Globin Genes: Evidence Supporting PharmacomiRNomics
4.1. Non-Coding RNAs and HbF Production
4.2. MicroRNAs and HbF Production
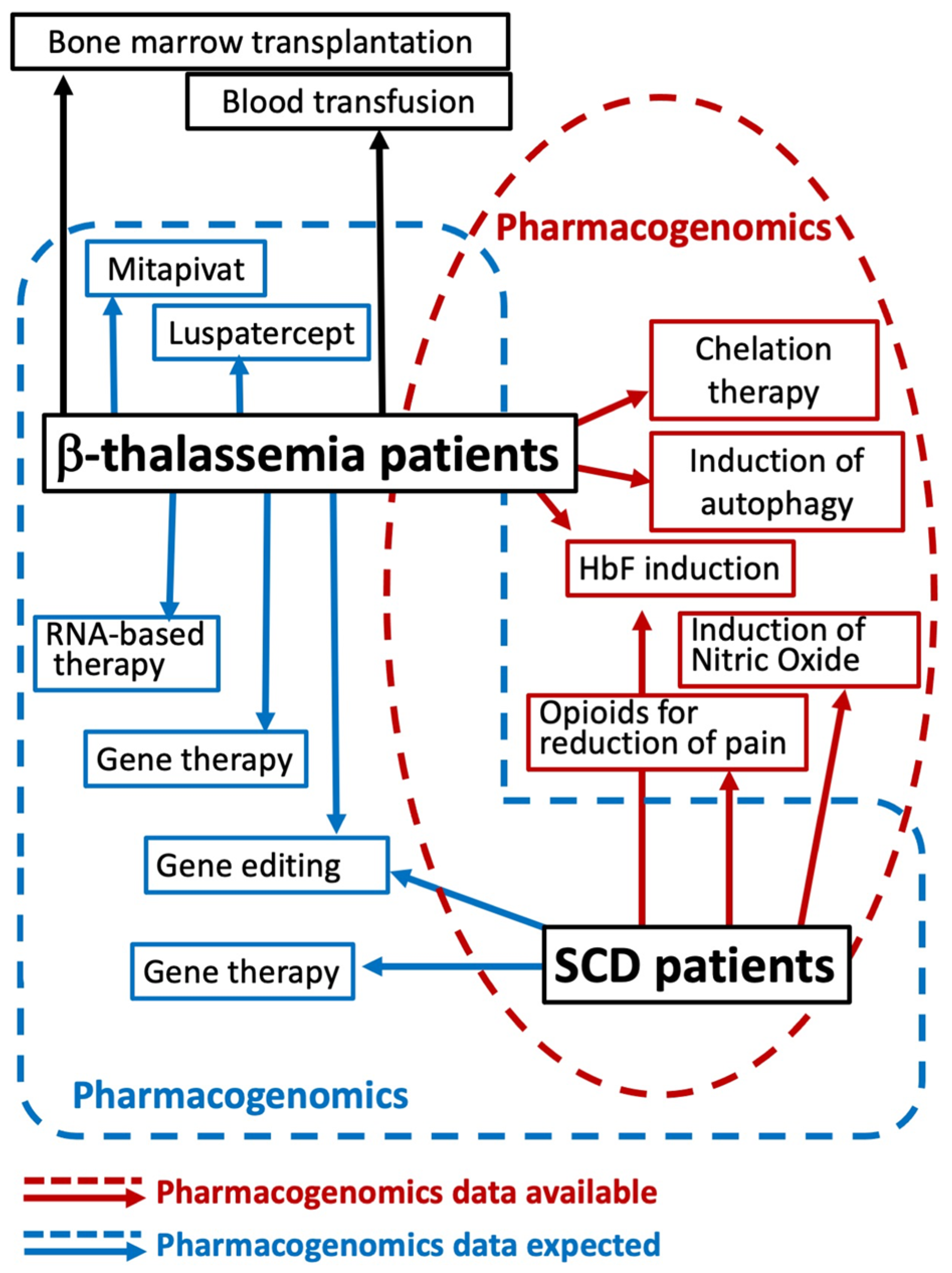
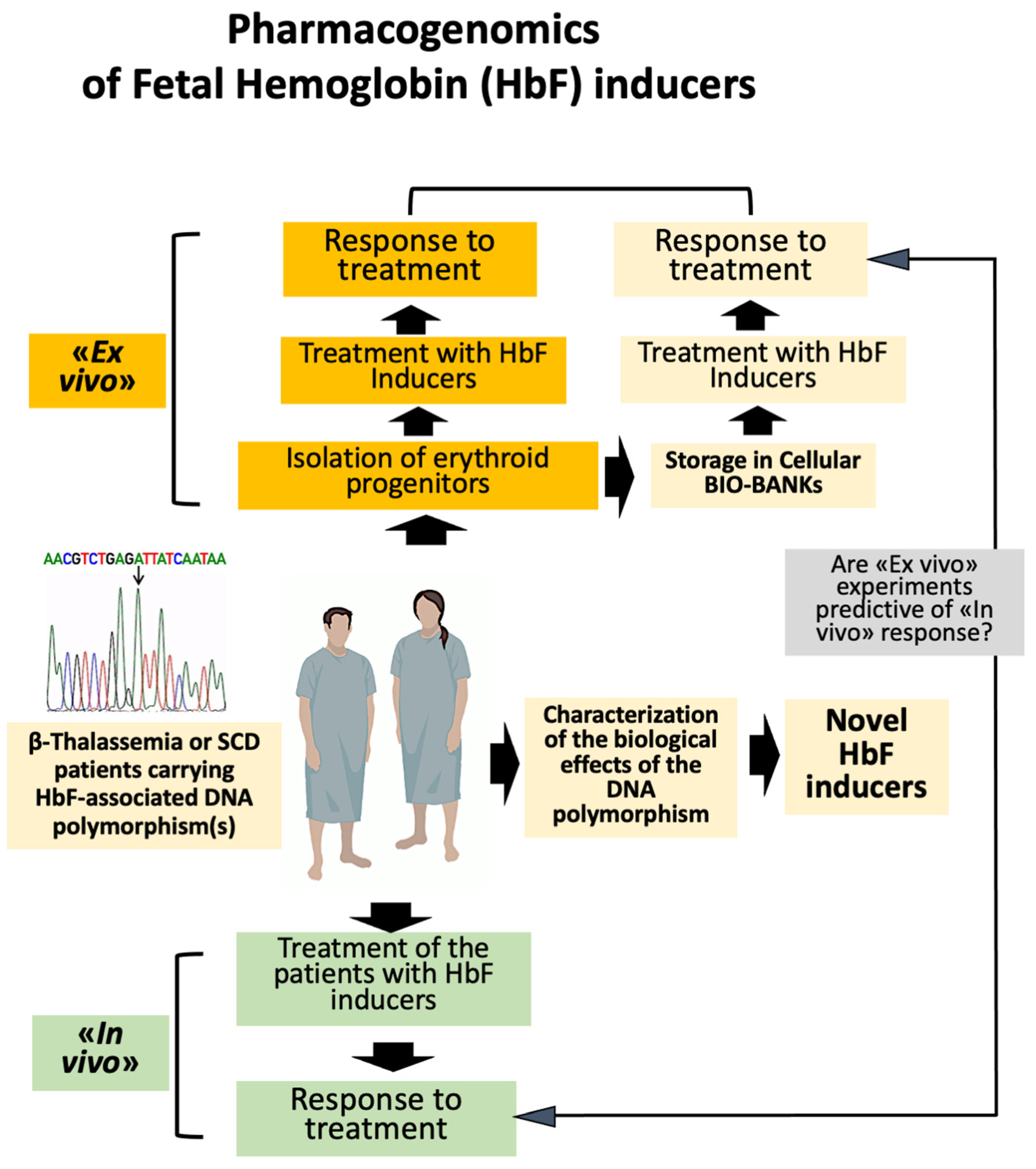
5. Pharmacogenomics of Fetal Hemoglobin Inducers in β-Thalassemia and Sickle-Cell Disease: Ex Vivo and In Vivo Studies
5.1. Hydroxyurea
5.2. Butyrate and Butyrate Analogues
5.3. Thalidomide
5.4. Rapamycin (Sirolimus)
6. Pharmacogenomics of Iron Chelators in β-Thalassemia and Other Hematopoietic Diseases Needing Blood Transfusions
6.1. Deferasirox
6.2. Deferiprone
7. Pharmacogenomics of Analgesics in Pain Management of SCD Patients
7.1. Pharmacogenomics of Opioids
7.2. Inducers of Nitric Oxide in SCD
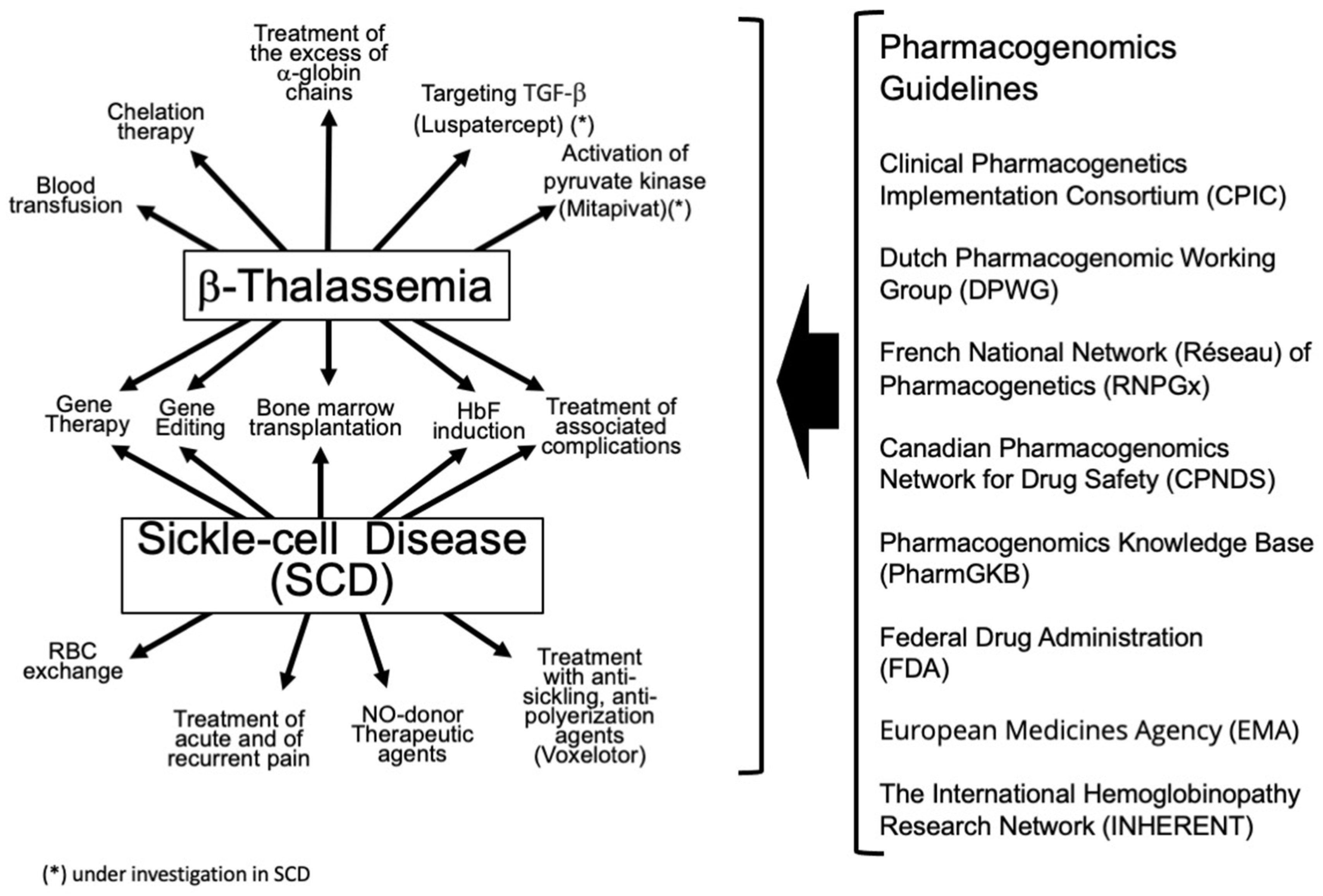
8. Guidelines and Clinical Trials Focusing on Pharmacogenomics/Pharmacogenetics in β-Thalassemia and SCD
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higgs, R.; Engel, J.D.; Stamatoyannopoulo, G. Thalassaemia. Lancet 2012, 37, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018, 70, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Kumar Chandraker, S.; Misha Singh, M.; Kumar, R. Global distribution of β-thalassemia mutations: An update. Gene 2024, 896, 148022. [Google Scholar] [CrossRef] [PubMed]
- Jaing, T.H.; Chang, T.Y.; Chen, S.H.; Lin, C.W.; Wen, Y.C.; Chiu, C.C. Molecular genetics of beta-thalassemia: A narrative review. Medicine 2021, 100, e27522. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, P.L.; Fasipe, T.A.; Wun, T. Sickle Cell Disease: A Review. JAMA 2022, 328, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bunn, H.F.; Noguchi, C.T.; Hofrichter, J.; Schechter, G.P.; Schechter, A.N.; Eaton, W.A. Molecular and cellular pathogenesis of hemoglobin SC disease. Proc. Natl. Acad. Sci. USA 1982, 79, 7527–7531. [Google Scholar] [CrossRef] [PubMed]
- May, A.; Choiseul, M. Sickle cell anaemia and thalassaemia: Symptoms, treatment and effects on lifestyle. Health Visit. 1988, 61, 212–215. [Google Scholar] [PubMed]
- Thein, S.L. Genetic Basis and Genetic Modifiers of β-Thalassemia and Sickle Cell Disease. Adv. Exp. Med. Biol. 2017, 1013, 27–57. [Google Scholar]
- Abdullah-Koolmees, H.; van Keulen, A.M.; Nijenhuis, M.; Deneer, V.H.M. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front. Pharmacol. 2021, 11, 595219. [Google Scholar] [CrossRef]
- Hulshof, E.C.; Deenen, M.J.; Nijenhuis, M.; Soree, B.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.J.F.; Risselada, A.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; et al. Dutch pharmacogenetics working group (DPWG) guideline for the gene-drug interaction between UGT1A1 and irinotecan. Eur. J. Hum. Genet. 2023, 31, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.C.; Caudle, K.E.; Whirl-Carrillo, M.; Gordon, R.J.; Hikino, K.; Prows, C.A.; Gaedigk, A.; Agundez, J.; Sadhasivam, S.; Klein, T.E.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 genotype and use of ondansetron and tropisetron. Clin. Pharmacol. Ther. 2017, 102, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A. Pharmacogenetics: Potential for individualized drug therapy through genetics. Trends Genet. 2003, 19, 660–666. [Google Scholar] [CrossRef]
- Pirmohamed, M. Pharmacogenomics: Current status and future perspectives. Nat. Rev. Genet. 2023, 24, 350–362. [Google Scholar] [CrossRef]
- Patrinos, G.P.; Grosveld, F.G. Pharmacogenomics and therapeutics of hemoglobinopathies. Hemoglobin 2008, 32, 229–236. [Google Scholar] [CrossRef]
- Peedicayil, J. Pharmacoepigenetics and Pharmacoepigenomics: An Overview. Curr. Drug Discov. Technol. 2019, 16, 392–399. [Google Scholar] [CrossRef]
- López-Camarillo, C.; Gallardo-Rincón, D.; Álvarez-Sánchez, M.E.; Marchat, L.A. Pharmaco-epigenomics: On the Road of Translation Medicine. Adv. Exp. Med. Biol. 2019, 1168, 31–42. [Google Scholar] [PubMed]
- Patrinos, G.P.; Innocenti, F. Pharmacogenomics: Paving the path to personalized medicine. Pharmacogenomics 2010, 11, 141–146. [Google Scholar] [CrossRef]
- Udagawa, C.; Zembutsu, H. Pharmacogenetics for severe adverse drug reactions induced by molecular-targeted therapy. Cancer Sci. 2020, 111, 3445–3457. [Google Scholar] [CrossRef]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and individualized drug therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef]
- Ahangari, N.; Doosti, M.; Ghayour Mobarhan, M.; Sahebkar, A.; Ferns, G.A.; Pasdar, A. Personalised medicine in hypercholesterolaemia: The role of pharmacogenetics in statin therapy. Ann. Med. 2020, 52, 462–470. [Google Scholar] [CrossRef]
- Wagner, J.B. Children Are Not Small Adults: Specific Findings in Statin Exposure and Response in a Growing Population. Clin. Pharmacol. Ther. 2019, 106, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Gummadi, A.C.; Guddati, A.K. Genetic Polymorphisms in Pharmaceuticals and Chemotherapy. World J. Oncol. 2021, 12, 149–154. [Google Scholar] [CrossRef]
- Del Re, M.; Cucchiara, F.; Rofi, E.; Fontanelli, L.; Petrini, I.; Gri, N.; Pasquini, G.; Rizzo, M.; Gabelloni, M.; Belluomini, L.; et al. A multiparametric approach to improve the prediction of response to immunotherapy in patients with metastatic NSCLC. Cancer Immunol. Immunother. 2021, 70, 1667–1678. [Google Scholar] [CrossRef]
- Patrinos, G.P.; Chui, D.H.K.; Hardison, R.C.; Steinberg, M.H. Strategies to improve pharmacogenomic-guided treatment options for patients with β-hemoglobinopathies. Expert Rev. Hematol. 2021, 14, 883–885. [Google Scholar] [CrossRef]
- Karamperis, K.; Tsoumpeli, M.T.; Kounelis, F.; Koromina, M.; Mitropoulou, C.; Moutinho, C.; Patrinos, G.P. Genome-based therapeutic interventions for β-type hemoglobinopathies. Hum. Genom. 2021, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Gravia, A.; Chondrou, V.; Sgourou, A.; Papantoni, I.; Borg, J.; Katsila, T.; Papachatzopoulou, A.; Patrinos, G.P. Individualizing fetal hemoglobin augmenting therapy for β-type hemoglobinopathies patients. Pharmacogenomics 2014, 15, 1355–1364. [Google Scholar] [CrossRef]
- Mnika, K.; Pule, G.D.; Dandara, C.; Wonkam, A. An Expert Review of Pharmacogenomics of Sickle Cell Disease Therapeutics: Not Yet Ready for Global Precision Medicine. OMICS 2016, 20, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Andolfo, I.; Russo, R. Red cells in post-genomic era: Impact of personalized medicine in the treatment of anemias. Haematologica 2015, 100, 3–6. [Google Scholar]
- Gallaway, K.A.; Sakon, C.; Ongeri, J.; Patel, K.S.; Oliver, J.; Patacca, H.; O’Brien, A.R.; Skaar, T.C.; Tillman, E.M. Opportunity for pharmacogenetics testing in patients with sickle cell anemia. Pharmacogenomics 2022, 23, 925–931. [Google Scholar] [CrossRef]
- Brewin, J.; Tewari, S.; Menzel, S.; Kirkham, F.; Inusa, B.; Renney, G.; Ward, M.; Rees, D.C. The effects of hydroxycarbamide on the plasma proteome of children with sickle cell anaemia. Br. J. Haematol. 2019, 186, 879–886. [Google Scholar] [CrossRef]
- Zohaib, M.; Ansari, S.H.; Shamsi, T.S.; Zubarev, R.A.; Zarina, S. Pharmacoproteomics Profiling of Plasma from β-Thalassemia Patients in Response to Hydroxyurea Treatment. J. Clin. Pharmacol. 2019, 59, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Lance, E.I.; Faulcon, L.M.; Fu, Z.; Yang, J.; Whyte-Stewart, D.; Strouse, J.J.; Barron-Casella, E.; Jones, K.; Van Eyk, J.E.; Casella, J.F.; et al. Proteomic discovery in sickle cell disease: Elevated neurogranin levels in children with sickle cell disease. Proteom. Clin. Appl. 2021, 15, e2100003. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Nag, A.; Ghosh, K.; Ray, R.; Roy, K.; Bandyopadhyay, A.; Bhattacharyya, M. Genetic determinants related to pharmacological induction of foetal haemoglobin in transfusion-dependent HbE-β thalassaemia. Ann. Hematol. 2019, 98, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Kargutkar, N.; Sawant-Mulay, M.; Hariharan, P.; Chandrakala, S.; Nadkarni, A. Role of microRNA in hydroxyurea mediated HbF induction in sickle cell anaemia patients. Sci. Rep. 2023, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Starlard-Davenport, A.; Gu, Q.; Pace, B.S. Targeting Genetic Modifiers of HBG Gene Expression in Sickle Cell Disease: The miRNA Option. Mol. Diagn. Ther. 2022, 26, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Okeke, C.; Silas, U.; Nnodu, O.; Clementina, O. HSC and miRNA Regulation with Implication for Foetal Haemoglobin Induction in Beta Haemoglobinopathies. Curr. Stem Cell Res. Ther. 2022, 17, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, S.A.; Ramakrishnan, V.; Kalra, I.S.; Li, W.; Pace, B.S. Histone deacetylase 9 activates gamma-globin gene expression in primary erythroid cells. J. Biol. Chem. 2011, 286, 2343–2353. [Google Scholar] [CrossRef]
- Zein, S.; Li, W.; Ramakrishnan, V.; Lou, T.F.; Sivanand, S.; Mackie, A.; Pace, B. Identification of fetal hemoglobin-inducing agents using the human leukemia KU812 cell line. Exp. Biol. Med. 2010, 235, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Boosalis, M.S.; Sangerman, J.I.; White, G.L.; Wolf, R.F.; Shen, L.; Dai, Y.; White, E.; Makala, L.H.; Li, B.; Pace, B.S.; et al. Novel Inducers of Fetal Globin Identified through High Throughput Screening (HTS) Are Active In Vivo in Anemic Baboons and Transgenic Mice. PLoS ONE 2015, 10, e0144660. [Google Scholar] [CrossRef]
- Perrine, S.P.; Mankidy, R.; Boosalis, M.S.; Bieker, J.J.; Faller, D.V. Erythroid Kruppel-like factor (EKLF) is recruited to the gamma-globin gene promoter as a co-activator and is required for gamma-globin gene induction by short-chain fatty acid derivatives. Eur. J. Haematol. 2009, 82, 466–476. [Google Scholar] [CrossRef]
- Zhou, G.; Zhang, H.; Lin, A.; Wu, Z.; Li, T.; Zhang, X.; Chen, H.; Lu, D. Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model. Int. J. Mol. Sci. 2022, 23, 2807. [Google Scholar] [CrossRef] [PubMed]
- Gotardo, É.M.F.; Brito, P.L.; Gushiken, L.F.S.; Chweih, H.; Leonardo, F.C.; Costa, F.F.; Conran, N. Molecular and cellular effects of in vivo chronic intravascular hemolysis and anti-inflammatory therapeutic approaches. Vasc. Pharmacol. 2023, 150, 107176. [Google Scholar] [CrossRef] [PubMed]
- Pradhan-Sundd, T.; Kato, G.J.; Novelli, E.M. Molecular mechanisms of hepatic dysfunction in sickle cell disease: Lessons from Townes mouse model. Am. J. Physiol. Cell Physiol. 2022, 323, C494–C504. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Friedman, A.; et al. Pooled Patient-Level Analysis of Inclisiran Trials in Patients with Familial Hypercholesterolemia or Atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T. Cardiovascular complications and risk of death in sickle-cell disease. Lancet 2016, 387, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, V.; Rosing, D.R.; Thein, S.L. Cardiovascular complications of sickle cell disease. Trends Cardiovasc. Med. 2021, 31, 187–193. [Google Scholar] [CrossRef]
- Sripichai, O.; Fucharoen, S. Fetal hemoglobin regulation in β-thalassemia: Heterogeneity, modifiers and therapeutic approaches. Expert Rev. Hematol. 2016, 9, 1129–1137. [Google Scholar] [CrossRef]
- Musallam, K.M.; Sankaran, V.G.; Cappellini, M.D.; Duca, L.; Nathan, D.G.; Taher, A.T. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood 2012, 119, 364–367. [Google Scholar] [CrossRef]
- Danjou, F.; Anni, F.; Perseu, L.; Satta, S.; Dessì, C.; Lai, M.E.; Fortina, P.; Devoto, M.; Galanello, R. Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica 2012, 97, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Mararenko, A.; Rozin, A.; Podrumar, A.; Gotlieb, V. Hereditary persistence of hemoglobin F is protective against red cell sickling. A case report and brief review. Hematol. Oncol. Stem Cell Ther. 2019, 12, 215–219. [Google Scholar] [PubMed]
- Adekile, A. The Genetic and Clinical Significance of Fetal Hemoglobin Expression in Sickle Cell Disease. Med. Princ. Pract. 2021, 30, 201–211. [Google Scholar] [CrossRef]
- Gambari, R.; Fibach, E. Medicinal chemistry of fetal hemoglobin inducers for treatment of β-thalassemia. Curr. Med. Chem. 2007, 14, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, D.; Engel, J.D.; Saunthararajah, Y. Fetal Hemoglobin Induction by Epigenetic Drugs. Semin. Hematol. 2018, 55, 60–67. [Google Scholar] [CrossRef]
- Prosdocimi, M.; Zuccato, C.; Cosenza, L.C.; Borgatti, M.; Lampronti, I.; Finotti, A.; Gambari, R. A Rational Approach to Drug Repositioning in β-thalassemia: Induction of Fetal Hemoglobin by Established Drugs. Wellcome Open Res. 2022, 7, 150. [Google Scholar] [CrossRef]
- Pavan, A.R.; Lopes, J.R.; Dos Santos, J.L. The state of the art of fetal hemoglobin-inducing agents. Expert Opin. Drug Discov. 2022, 17, 1279–1293. [Google Scholar] [CrossRef]
- Lavelle, D.; Ibanez, V.; Vaitkus, K.; Zhang, X.; Ramasamy, J.; Rivers, A.E.; Saunthararajah, Y.; Molokie, R. Combinatorial targeting of epigenome-modifying enzymes with small molecule drugs synergistically increases HbF. Blood Adv. 2023, 7, 3891–3902. [Google Scholar]
- Kalantri, S.A.; Ray, R.; Chattopadhyay, A.; Bhattacharjee, S.; Biswas, A.; Bhattacharyya, M. Efficacy of decitabine as hemoglobin F inducer in HbE/β-thalassemia. Ann. Hematol. 2018, 97, 1689–1694. [Google Scholar] [CrossRef]
- Fucharoen, S.; Siritanaratkul, N.; Winichagoon, P.; Chowthaworn, J.; Siriboon, W.; Muangsup, W.; Chaicharoen, S.; Poolsup, N.; Chindavijak, B.; Pootrakul, P.; et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood 1996, 87, 887–892. [Google Scholar] [CrossRef]
- Bianchi, N.; Chiarabelli, C.; Zuccato, C.; Lampronti, I.; Borgatti, M.; Amari, G.; Delcanale, M.; Chiavilli, F.; Prus, E.; Fibach, E.; et al. Erythroid differentiation ability of butyric acid analogues: Identification of basal chemical structures of new inducers of foetal haemoglobin. Eur. J. Pharmacol. 2015, 752, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.E.; El Beshlawy, A.; Inati, A.; Kutlar, A.; Abboud, M.R.; Haynes, J., Jr.; Ward, R.; Sharon, B.; Taher, A.T.; Smith, W.; et al. A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am. J. Hematol. 2014, 89, 709–713. [Google Scholar] [CrossRef]
- Rönndahl, G.; Mönkemeyer, S.; Schulze, S.; Pekrun, A.; Eikel, D.; Nau, H.; Witt, O. Novel valproic acid derivatives with hemoglobin F inducing activity. Am. J. Hematol. 2006, 81, 374–376. [Google Scholar] [CrossRef]
- Aerbajinai, W.; Zhu, J.; Gao, Z.; Chin, K.; Rodgers, G.P. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood 2007, 110, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Moutouh-de Parseval, L.A.; Verhelle, D.; Glezer, E.; Jensen-Pergakes, K.; Ferguson, G.D.; Corral, L.G.; Morris, C.L.; Muller, G.; Brady, H.; Chan, K. Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells. J. Clin. Investig. 2008, 118, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Marianna, P.; Kollia, P.; Akel, S.; Papassotiriou, Y.; Stamoulakatou, A.; Loukopoulos, D. Valproic acid, trichostatin and their combination with hemin preferentially enhance gamma-globin gene expression in human erythroid liquid cultures. Haematologica 2001, 86, 700–705. [Google Scholar]
- Guo, L.; Chen, J.; Wang, Q.; Zhang, J.; Huang, W. Oridonin enhances γ-globin expression in erythroid precursors from patients with β-thalassemia via activation of p38 MAPK signaling. Mol. Med. Rep. 2020, 21, 909–917. [Google Scholar] [CrossRef]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Gasparello, J.; Papi, C.; D’Aversa, E.; Breveglieri, G.; Lampronti, I.; Finotti, A.; Borgatti, M.; et al. Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: Results from a pilot clinical trial (Sirthalaclin). Ther. Adv. Hematol. 2022, 13, 20406207221100648. [Google Scholar] [CrossRef]
- Zuccato, C.; Bianchi, N.; Borgatti, M.; Lampronti, I.; Massei, F.; Favre, C.; Gambari, R. Everolimus is a potent inducer of erythroid differentiation and gamma-globin gene expression in human erythroid cells. Acta Haematol. 2007, 117, 168–176. [Google Scholar] [CrossRef]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Prus, E.; Gambari, R. Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood 2003, 102, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Prus, E.; Bianchi, N.; Zuccato, C.; Breveglieri, G.; Salvatori, F.; Finotti, A.; Lipucci di Paola, M.; Brognara, E.; Lampronti, I.; et al. Resveratrol: Antioxidant activity and induction of fetal hemoglobin in erythroid cells from normal donors and β-thalassemia patients. Int. J. Mol. Med. 2012, 29, 974–982. [Google Scholar]
- Lampronti, I.; Bianchi, N.; Borgatti, M.; Fibach, E.; Prus, E.; Gambari, R. Accumulation of gamma-globin mRNA in human erythroid cells treated with angelicin. Eur. J. Haematol. 2003, 71, 189–195. [Google Scholar] [CrossRef]
- Iftikhar, F.; Ali, H.; Musharraf, S.G. Cinchona alkaloids as natural fetal hemoglobin inducing agents in human erythroleukemia cells. RSC Adv. 2019, 9, 17551–17559. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Lampronti, I.; Borgatti, M.; Scapoli, C.; Gambari, R.; Finotti, A. Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production. Int. J. Mol. Sci. 2021, 22, 13433. [Google Scholar] [CrossRef]
- Shi, L.; Cui, S.; Engel, J.D.; Tanabe, O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat. Med. 2013, 19, 291–294. [Google Scholar] [CrossRef]
- Mettananda, S.; Yasara, N.; Fisher, C.A.; Taylor, S.; Gibbons, R.; Higgs, D. Synergistic silencing of α-globin and induction of γ-globin by histone deacetylase inhibitor, vorinostat as a potential therapy for β-thalassaemia. Sci. Rep. 2019, 9, 11649. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, F.; Rahman, S.; Khan, M.B.N.; Khan, K.; Khan, M.N.; Uddin, R.; Musharraf, S.G. In Vitro and In Vivo Studies for the Investigation of γ-Globin Gene Induction by Adhatoda vasica: A Pre-Clinical Study of HbF Inducers for β-Thalassemia. Front. Pharmacol. 2022, 13, 797853. [Google Scholar] [CrossRef]
- Yasara, N.; Wickramarathne, N.; Mettananda, C.; Silva, I.; Hameed, N.; Attanayaka, K.; Rodrigo, R.; Wickramasinghe, N.; Perera, L.; Manamperi, A.; et al. A randomised double-blind placebo-controlled clinical trial of oral hydroxyurea for transfusion-dependent β-thalassaemia. Sci. Rep. 2022, 12, 2752. [Google Scholar] [CrossRef]
- Ansari, S.H.; Ansari, I.; Wasim, M.; Sattar, A.; Khawaja, S.; Zohaib, M.; Hussain, Z.; Adil, S.O.; Ansari, A.H.; Ansari, U.H.; et al. Evaluation of the combination therapy of hydroxyurea and thalidomide in β-thalassemia. Blood Adv. 2022, 6, 6162–6168. [Google Scholar] [CrossRef]
- Gamberini, M.R.; Prosdocimi, M.; Gambari, R. Sirolimus for treatment of β-thalassemia: From pre-clinical studies to the design of clinical trials. Health Educ. Public Health 2021, 4, 425–435. [Google Scholar]
- Smith, E.C.; Orkin, S.H. Hemoglobin genetics: Recent contributions of GWAS and gene editing. Hum. Mol. Genet. 2016, 25, R99–R105. [Google Scholar] [CrossRef] [PubMed]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Nuinoon, M.; Makarasara, W.; Mushiroda, T.; Setianingsih, I.; Wahidiyat, P.A.; Sripichai, O.; Kumasaka, N.; Takahashi, A.; Svasti, S.; Munkongdee, T.; et al. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum. Genet. 2010, 127, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Sherva, R.; Sripichai, O.; Abel, K.; Ma, Q.; Whitacre, J.; Angkachatchai, V.; Makarasara, W.; Winichagoon, P.; Svasti, S.; Fucharoen, S.; et al. Genetic modifiers of Hb E/beta0 thalassemia identified by a two-stage genome-wide association study. BMC Med. Genet. 2010, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Habara, A.; Steinberg, M.H. Minireview: Genetic basis of heterogeneity and severity in sickle cell disease. Exp. Biol. Med. 2016, 241, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Solovieff, N.; Milton, J.N.; Hartley, S.W.; Sherva, R.; Sebastiani, P.; Dworkis, D.A.; Klings, E.S.; Farrer, L.A.; Garrett, M.E.; Ashley-Koch, A.; et al. Fetal hemoglobin in sickle cell anemia: Genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood 2010, 115, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.S.; Wienert, B.; Wyman, S.K.; Bell, H.W.; George, A.; Mahalingam, G.; Vu, J.T.; Prasad, K.; Bandlamudi, B.P.; Devaraju, N.; et al. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. Elife 2022, 11, e65421. [Google Scholar] [CrossRef]
- Venkatesan, V.; Christopher, A.C.; Rhiel, M.; Azhagiri, M.K.K.; Babu, P.; Walavalkar, K.; Saravanan, B.; Andrieux, G.; Rangaraj, S.; Srinivasan, S.; et al. Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of β-hemoglobinopathies. Mol. Ther. Nucleic Acids 2023, 32, 671–688. [Google Scholar] [CrossRef]
- Antoniou, P.; Hardouin, G.; Martinucci, P.; Frati, G.; Felix, T.; Chalumeau, A.; Fontana, L.; Martin, J.; Masson, C.; Brusson, M.; et al. Base-editing-mediated dissection of a γ-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nat. Commun. 2022, 13, 6618. [Google Scholar] [CrossRef]
- Quagliano, A.; Acevedo, D.; Hardigan, P.; Prasad, S. Using Clustered Regularly Interspaced Short Palindromic Repeats gene editing to induce permanent expression of fetal hemoglobin in β-thalassemia and sickle cell disease: A comparative meta-analysis. Front. Med. 2022, 9, 943631. [Google Scholar] [CrossRef] [PubMed]
- Manco, L.; Santos, R.; Rocha, C.; Relvas, L.; Bento, C.; Maia, T.; Gomes, V.; Amorim, A.; Prata, M.J. Hb F Levels in β-Thalassemia Carriers and Normal Individuals: Known and Unknown Quantitative Trait Loci in the β-Globin Gene Cluster. Hemoglobin 2022, 46, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Mahmood, S.; Mohsin, S.; Tabassum, I.; Ghafoor, M.; Sajjad, O. Modulatory effect of single nucleotide polymorphism in Xmn1, BCL11A and HBS1L-MYB loci on foetal haemoglobin levels in β-thalassemia major and Intermedia patients. J. Pak. Med. Assoc. 2021, 71, 1394–1398. [Google Scholar] [PubMed]
- Roy, P.; Bhattacharya, G.; Mandal, A.; Dasgupta, U.B.; Banerjee, D.; Chandra, S.; Das, M. Influence of BCL11A, HBS1L-MYB, HBBP1 single nucleotide polymorphisms and the HBG2 XmnI polymorphism on Hb F levels. Hemoglobin 2012, 36, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Pace, B.; Zein, S. Understanding mechanisms of gamma-globin gene regulation to develop strategies for pharmacological fetal hemoglobin induction. Dev. Dyn. 2006, 235, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Aktuna, S.; Thongjuea, S.; Aghajanirefah, A.; Pourfarzad, F.; van Ijcken, W.; Lenhard, B.; Rooks, H.; Best, S.; Menzel, S.; et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Investig. 2014, 124, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Tepakhan, W.; Kanjanaopas, S.; Srewaradachpisal, K. Association Between Genetic Polymorphisms and Hb F Levels in Heterozygous β-Thalassemia 3.5 kb Deletions. Hemoglobin 2020, 44, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.K.; Joly, P.; Bardel, C.; Moulsma, M.; Bonello-Palot, N.; Francina, A. The XmnI (G)gamma polymorphism influences hemoglobin F synthesis contrary to BCL11A and HBS1L-MYB SNPs in a cohort of 57 beta-thalassemia intermedia patients. Blood Cells Mol. Dis. 2010, 45, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Nuinoon, M.; Rattanaporn, P.; Benjchareonwong, T.; Choowet, A.; Suwanno, K.; Saekoo, N.; Lekpetch, K.; Thipthara, O.; Svasti, S.; Fucharoen, S. Genetic predictions of life expectancy in southern Thai patients with β0-thalassemia/Hb E. Biomed. Rep. 2022, 16, 52. [Google Scholar] [CrossRef]
- Bhagat, S.; Patra, P.K.; Thakur, A.S. Association between XmnI Polymorphism and HbF Level in Sickle Cell Disease Patients from Chhattisgarh. Int. J. Biomed. Sci. 2012, 8, 36–39. [Google Scholar] [CrossRef]
- Menzel, S.; Thein, S.L. Genetic Modifiers of Fetal Haemoglobin in Sickle Cell Disease. Mol. Diagn. Ther. 2019, 23, 235–244. [Google Scholar] [CrossRef]
- Ju, J.; Wang, Y.; Liu, R.; Zhang, Y.; Xu, Z.; Wang, Y.; Wu, Y.; Liu, M.; Cerruti, L.; Zou, F.; et al. Human fetal globin gene expression is regulated by LYAR. Nucleic Acids Res. 2014, 42, 9740–9752. [Google Scholar] [CrossRef]
- Chen, D.; Zuo, Y.; Zhang, X.; Ye, Y.; Bao, X.; Huang, H.; Tepakhan, W.; Wang, L.; Ju, J.; Chen, G.; et al. A Genetic Variant Ameliorates β-Thalassemia Severity by Epigenetic-Mediated Elevation of Human Fetal Hemoglobin Expression. Am. J. Hum. Genet. 2017, 101, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, N.; Cosenza, L.C.; Lampronti, I.; Finotti, A.; Breveglieri, G.; Zuccato, C.; Fabbri, E.; Marzaro, G.; Chilin, A.; De Angelis, G.; et al. Structural and Functional Insights on an Uncharacterized Aγ-Globin-Gene Polymorphism Present in Four β0-Thalassemia Families with High Fetal Hemoglobin Levels. Mol. Diagn. Ther. 2016, 20, 161–173. [Google Scholar] [CrossRef]
- Breveglieri, G.; Bianchi, N.; Cosenza, L.C.; Gamberini, M.R.; Chiavilli, F.; Zuccato, C.; Montagner, G.; Borgatti, M.; Lampronti, I.; Finotti, A.; et al. An Aγ-globin G->A gene polymorphism associated with β039 thalassemia globin gene and high fetal hemoglobin production. BMC Med. Genet. 2017, 18, 93. [Google Scholar] [CrossRef]
- Gemmo, C.; Breveglieri, G.; Marzaro, G.; Lampronti, I.; Cosenza, L.C.; Gasparello, J.; Zuccato, C.; Fabbri, E.; Borgatti, M.; Chilin, A.; et al. Surface plasmon resonance based analysis of the binding of LYAR protein to the rs368698783 (G>A) polymorphic Aγ-globin gene sequences mutated in β-thalassemia. Anal. Bioanal. Chem. 2019, 411, 7699–7707. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Breveglieri, G.; Bianchi, N.; Lampronti, I.; Gasparello, J.; Scapoli, C.; Borgatti, M.; Finotti, A.; et al. The rs368698783 (G>A) Polymorphism Affecting LYAR Binding to the Aγ-Globin Gene Is Associated with High Fetal Hemoglobin (HbF) in β-Thalassemia Erythroid Precursor Cells Treated with HbF Inducers. Int. J. Mol. Sci. 2023, 24, 776. [Google Scholar] [CrossRef]
- Giannopoulou, E.; Bartsakoulia, M.; Tafrali, C.; Kourakli, A.; Poulas, K.; Stavrou, E.F.; Papachatzopoulou, A.; Georgitsi, M.; Patrinos, G.P. A single nucleotide polymorphism in the HBBP1 gene in the human β-globin locus is associated with a mild β-thalassemia disease phenotype. Hemoglobin 2012, 36, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Xu, X.; Higgs, D.R.; Patrinos, G.P.; Arnaud, L.; Bieker, J.J.; Philipsen, S.; KLF1 Consensus Workgroup. Krüppeling erythropoiesis: An unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 2016, 127, 1856–1862. [Google Scholar] [CrossRef]
- Kumar, R.; Yadav, R.; Mishra, S.; Singh, M.P.S.S.; Gwal, A.; Bharti, P.K.; Rajasubramaniam, S. Krüppel-like factor 1 (KLF1) gene single nucleotide polymorphisms in sickle cell disease and its association with disease-related morbidities. Ann. Hematol. 2021, 100, 365–373. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, K.; Sun, C.W.; Pawlik, K.M.; Townes, T.M. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef] [PubMed]
- Khamphikham, P.; Sripichai, O.; Munkongdee, T.; Fucharoen, S.; Tongsima, S.; Smith, D.R. Genetic variation of Krüppel-like factor 1 (KLF1) and fetal hemoglobin (HbF) levels in β0-thalassemia/HbE disease. Int. J. Hematol. 2018, 107, 297–310. [Google Scholar] [CrossRef]
- Pereira, C.; Relvas, L.; Bento, C.; Abade, A.; Ribeiro, M.L.; Manco, L. Polymorphic variations influencing fetal hemoglobin levels: Association study in beta-thalassemia carriers and in normal individuals of Portuguese origin. Blood Cells Mol. Dis. 2015, 54, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Zaker-Kandjani, B.; Namdar-Aligoodarzi, P.; Azarkeivan, A.; Najmabadi, H.; Banan, M. Mutation screening of the Krüppel-like factor 1 gene using single-strand conformational polymorphism in a cohort of Iranian β-thalassemia patients. Hemoglobin 2015, 39, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Borgio, J.F.; AbdulAzeez, S.; Al-Muslami, A.M.; Naserullah, Z.A.; Al-Jarrash, S.; Al-Suliman, A.M.; Al-Madan, M.S.; Al-Ali, A.K. KLF1 gene and borderline hemoglobin A2 in Saudi population. Arch. Med. Sci. 2018, 14, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Kolliopoulou, A.; Siamoglou, S.; John, A.; Sgourou, A.; Kourakli, A.; Symeonidis, A.; Vlachaki, E.; Chalkia, P.; Theodoridou, S.; Ali, B.R.; et al. Role of Genomic Biomarkers in Increasing Fetal Hemoglobin Levels Upon Hydroxyurea Therapy and in β-Thalassemia Intermedia: A Validation Cohort Study. Hemoglobin 2019, 43, 27–33. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Jia, S.; Chen, P.; He, Y. Construction and Analysis of a Long Non-Coding RNA (lncRNA)-Associated ceRNA Network in β-Thalassemia and Hereditary Persistence of Fetal Hemoglobin. Med. Sci. Monit. 2019, 25, 7079–7086. [Google Scholar] [CrossRef]
- Yang, F.; Ruan, H.; Li, S.; Hou, W.; Qiu, Y.; Deng, L.; Su, S.; Chen, P.; Pang, L.; Lai, K. Analysis of circRNAs and circRNA-associated competing endogenous RNA networks in β-thalassemia. Sci. Rep. 2022, 12, 8071. [Google Scholar] [CrossRef]
- Fakhr-Eldeen, A.; Toraih, E.A.; Fawzy, M.S. Long non-coding RNAs MALAT1, MIAT and ANRIL gene expression profiles in beta-thalassemia patients: A cross-sectional analysis. Hematology 2019, 24, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Finotti, A.; Fabbri, E.; Lampronti, I.; Gasparello, J.; Borgatti, M.; Gambari, R. MicroRNAs and Long Non-coding RNAs in Genetic Diseases. Mol. Diagn. Ther. 2019, 23, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Wonkam, A. The future of sickle cell disease therapeutics rests in genomics. Dis. Model. Mech. 2023, 16, dmm049765. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, E.J.; Carthew, R.W. Silence from within: Endogenous siRNAs and miRNAs. Cell 2005, 122, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kowdley, K.V. MicroRNAs in common human diseases. Genom. Proteom. Bioinform. 2012, 10, 246–253. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef]
- Das, J.; Podder, S.; Ghosh, T.C. Insights into the miRNA regulations in human disease genes. BMC Genom. 2014, 15, 1010. [Google Scholar] [CrossRef][Green Version]
- Paunovska, K.; Loughrey, K.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Buck, J.; Grossen, P.; Cullis, P.R.; Huwyler, J.; Witzigmann, D. Lipid-based DNA therapeutics: Hallmarks of non-viral gene delivery. ACS Nano 2019, 13, 3754–3782. [Google Scholar] [CrossRef] [PubMed]
- Sristi Almalki, W.H.; Karwasra, R.; Gupta, G.; Singh, S.; Sharma, A.; Sahebkar, A.; Kesharwani, P. Advances in the polymeric nanoparticulate delivery systems for RNA therapeutics. Prog. Mol. Biol. Transl. Sci. 2024, 204, 219–248. [Google Scholar]
- Roberts, T.C.; Langer, R.; Wood MJ, A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Mukai, Y.; Wada, F.; Terada, C.; Kayaba, Y.; Oh, K.; Yamayoshi, A.; Obika, S.; Harada-Shiba, M. Highly Potent GalNAc-Conjugated Tiny LNA Anti-miRNA-122 Antisense Oligonucleotides. Pharmaceutics 2021, 13, 817. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.M.; Li, B.; Pace, B.S. Original Research: Stable expression of miR-34a mediates fetal hemoglobin induction in K562 cells. Exp. Biol. Med. 2016, 241, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ling, L.; Yu, D. MicroRNAs in β-thalassemia. Am. J. Med. Sci. 2021, 362, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Saki, N.; Abroun, S.; Soleimani, M.; Kavianpour, M.; Shahjahani, M.; Mohammadi-Asl, J.; Hajizamani, S. MicroRNA Expression in β-Thalassemia and Sickle Cell Disease: A Role in the Induction of Fetal Hemoglobin. Cell J. 2016, 17, 583–592. [Google Scholar]
- Starlard-Davenport, A.; Fitzgerald, A.; Pace, B.S. Exploring epigenetic and microRNA approaches for γ-globin gene regulation. Exp. Biol. Med. 2021, 246, 2347–2357. [Google Scholar] [CrossRef]
- Das, S.S.; Das, S.; Byram, P.K.; Rahaman, M.; Dolai, T.K.; Chatterjee, A.; Chakravorty, N. MicroRNA expression patterns in HbE/β-thalassemia patients: The passwords to unlock fetal hemoglobin expression in β-hemoglobinopathies. Blood Cells Mol. Dis. 2021, 87, 102523. [Google Scholar] [CrossRef] [PubMed]
- Gasparello, J.; Fabbri, E.; Bianchi, N.; Breveglieri, G.; Zuccato, C.; Borgatti, M.; Gambari, R.; Finotti, A. BCL11A mRNA Targeting by miR-210, A Possible Network Regulating γ-Globin Gene Expression. Int. J. Mol. Sci. 2017, 18, 2530. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lin, R.; Li, H.; Ou, R.; Wang, K.; Lin, J.; Li, C. MicroRNA-92a-3p-mediated inhibition of BCL11A upregulates γ-globin expression and inhibits oxidative stress and apoptosis in erythroid precursor cells. Hematology 2022, 27, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Gholampour, M.A.; Asadi, M.; Naderi, M.; Azarkeivan, A.; Soleimani, M.; Atashi, A. miR-30a regulates γ-globin expression in erythoid precursors of intermedia thalassemia through targeting, BCL11A. Mol. Biol. Rep. 2020, 47, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Shang, X.; Chen, D.; Pang, D.; Zhao, C.; Xu, X. MicroRNA-2355-5p regulates γ-globin expression in human erythroid cells by inhibiting KLF6. Br. J. Haematol. 2021, 193, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Srinoun, K.; Sathirapongsasuti, N.; Paiboonsukwong, K.; Sretrirutchai, S.; Wongchanchailert, M.; Fucharoen, S. miR-144 regulates oxidative stress tolerance of thalassemic erythroid cell via targeting NRF2. Ann. Hematol. 2019, 98, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, D.; Zhang, X.; Li, Z.; Ye, Y.; Liu, Q.; Shen, J.; Chen, Z.; Huang, H.; Liang, Y.; et al. miR-326 regulates HbF synthesis by targeting EKLF in human erythroid cells. Exp. Hematol. 2018, 63, 33–40.e2. [Google Scholar] [CrossRef]
- Srinoun, K.; Nopparatana, C.; Wongchanchailert, M.; Fucharoen, S. MiR-155 enhances phagocytic activity of β-thalassemia/HbE monocytes via targeting of BACH1. Int. J. Hematol. 2017, 106, 638–647. [Google Scholar] [CrossRef]
- Pule, G.D.; Mowla, S.; Novitzky, N.; Wonkam, A. Hydroxyurea down-regulates BCL11A, KLF-1 and MYB through miRNA-mediated actions to induce γ-globin expression: Implications for new therapeutic approaches of sickle cell disease. Clin. Transl. Med. 2016, 5, 15. [Google Scholar] [CrossRef]
- Starlard-Davenport, A.; Smith, A.; Vu, L.; Li, B.; Pace, B.S. MIR29B mediates epigenetic mechanisms of HBG gene activation. Br. J. Haematol. 2019, 186, 91–100. [Google Scholar] [CrossRef]
- Lulli, V.; Romania, P.; Morsilli, O.; Cianciulli, P.; Gabbianelli, M.; Testa, U.; Giuliani, A.; Marziali, G. MicroRNA-486-3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS ONE 2013, 8, e60436. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Menne, T.F.; Šćepanović, D.; Vergilio, J.A.; Ji, P.; Kim, J.; Thiru, P.; Orkin, S.H.; Lander, E.S.; Lodish, H.F. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc. Natl. Acad. Sci. USA 2011, 108, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, X.; Wang, H.; Zhang, M.; Chen, L.; Chen, H.; Pan, Y.; Zhang, Y.; Xu, L.; Huang, H. The clinical value of hsa-miR-190b-5p in peripheral blood of pediatric beta-thalassemia and its regulation on BCL11A expression. PLoS ONE 2023, 18, e0292031. [Google Scholar]
- Chen, M.; Lv, A.; Zhang, S.; Zheng, J.; Lin, N.; Xu, L.; Huang, H. Peripheral blood circular RNA circ-0008102 may serve as a novel clinical biomarker in beta-thalassemia patients. Eur. J. Pediatr. 2024, 183, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Han, Y.; Zhang, W.; Wu, J.; An, B.; Huang, S.; Sun, F. Long Non-Coding RNA H19 Leads to Upregulation of gamma-Globin Gene Expression during Erythroid Differentiation. Hemoglobin 2024, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.P.; Gao, X.X.; Zhou, G.Q.; Zhang, H.K.; Yang, J.M.; Wang, W.J.; Song, X.M.; Chen, H.Y.; Lu, D.R. Reactivation of gamma-globin expression using a minicircle DNA system to treat beta-thalassemia. Gene 2022, 820, 146289. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.A.; Wilcox, I.; Luo, H.Y.; Farrell, J.J.; Kurita, R.; Nakamura, Y.; Murphy, G.J.; Cui, S.; Steinberg, M.H.; Chui, D.H.K. A long noncoding RNA from the HBS1L-MYB intergenic region on chr6q23 regulates human fetal hemoglobin expression. Blood Cells Mol. Dis. 2018, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Papasavva, P.L.; Papaioannou, N.Y.; Patsali, P.; Kurita, R.; Nakamura, Y.; Sitarou, M.; Christou, S.; Kleanthous, M.; Lederer, C.W. Distinct miRNA Signatures and Networks Discern Fetal from Adult Erythroid Differentiation and Primary from Immortalized Erythroid Cells. Int. J. Mol. Sci. 2021, 22, 3626. [Google Scholar] [CrossRef]
- Rahaman, M.; Mukherjee, M.; Bhattacharya, S.; Mukherjee, B.; Shukla, P.C.; Dolai, T.K.; Chakravorty, N. Exploring the crosstalk between long non-coding RNAs and microRNAs to unravel potential prognostic and therapeutic biomarkers in β-thalassemia. Mol. Biol. Rep. 2022, 49, 7057–7068. [Google Scholar] [CrossRef]
- Kalaigar, S.S.; Rajashekar, R.B.; Nataraj, S.M.; Vishwanath, P.; Prashant, A. Bioinformatic Tools for the Identification of MicroRNAs Regulating the Transcription Factors in Patients with β-Thalassemia. Bioinform. Biol. Insights 2022, 16, 11779322221115536. [Google Scholar] [CrossRef]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional, microRNA. Biochim. Biophys. Acta 2009, 1792, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Mitra, A.; Chakravorty, N. Diseases and their clinical heterogeneity—Are we ignoring the SNiPers and micRomaNAgers? An illustration using Beta-thalassemia clinical spectrum and fetal hemoglobin levels. Genomics 2019, 111, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, M.T.; Azarkeivan, A.; Pourfathollah, A.A.; Amirizadeh, N. Comparison of MicroRNAs Mediated in Reactivation of the γ-Globin in β-Thalassemia Patients, Responders and Non-Responders to Hydroxyurea. Hemoglobin 2017, 41, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Sales, R.R.; Nogueira, B.L.; Luizon, M.R. Pharmacogenomics of hydroxyurea therapy and fetal hemoglobin levels in sickle cell anemia. Pharmacogenomics 2022, 23, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Sales, R.R.; Nogueira, B.L.; Tosatti, J.A.G.; Gomes, K.B.; Luizon, M.R. Do Genetic Polymorphisms Affect Fetal Hemoglobin (HbF) Levels in Patients with Sickle Cell Anemia Treated with Hydroxyurea? A Systematic Review and Pathway Analysis. Front. Pharmacol. 2022, 12, 779497. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Ayyub, M.; Khan, S.A.; Ahmed, S.; Abbas, K.; Malik, H.S.; Tashfeen, S. Frequency of Ggamma-globin promoter −158 (C>T) XmnI polymorphism in patients with homozygous/compound heterozygous beta thalassaemia. Hematol. Oncol. Stem Cell Ther. 2015, 8, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Banan, M.; Bayat, H.; Azarkeivan, A.; Mohammadparast, S.; Kamali, K.; Farashi, S.; Bayat, N.; Khani, M.H.; Neishabury, M.; Najmabadi, H. The XmnI and BCL11A single nucleotide polymorphisms may help predict hydroxyurea response in Iranian beta-thalassemia patients. Hemoglobin 2012, 36, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Levin, C.; Dgany, O.; Kransnov, T.; Elhasid, R.; Zalman, L.; Palmor, H.; Tamary, H. Response to hydroxyurea therapy in beta-thalassemia. Am. J. Hematol. 2008, 83, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Panja, A.; Saha, D.; Banerjee, U.; Datta, A.K.; Basu, A. Drug Repurposing: Hydroxyurea Therapy Improves the Transfusion-Free Interval in HbE/Beta-Thalassemia-Major Patients with the XmnI Polymorphism. Genet. Test. Mol. Biomark. 2021, 25, 563–570. [Google Scholar] [CrossRef]
- Perrine, S.P.; Miller, B.A.; Faller, D.V.; Cohen, R.A.; Vichinsky, E.P.; Hurst, D.; Lubin, B.H.; Papayannopoulou, T. Sodium butyrate enhances fetal globin gene expression in erythroid progenitors of patients with Hb SS and beta thalassemia. Blood 1989, 74, 454–459. [Google Scholar] [CrossRef]
- Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.; Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N. Engl. J. Med. 1993, 328, 81–86. [Google Scholar] [CrossRef]
- Faller, D.V.; Perrine, S.P. Butyrate in the treatment of sickle cell disease and beta-thalassemia. Curr. Opin. Hematol. 1995, 2, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Panja, A.; Basu, A. Pharmacogenomics of the Drugs used for the Treatment of Thalassemia. J. Cytol. Histol. 2015, 6, 5. [Google Scholar]
- Testa, U. Fetal hemoglobin chemical inducers for treatment of hemoglobinopathies. Ann. Hematol. 2009, 88, 505–528. [Google Scholar] [CrossRef][Green Version]
- Tayebi, B.; Abrishami, F.; Alizadeh, S.; Minayi, N.; Mohammadian, M.; Soleimani, M.; Dehghanifard, A.; Atwan, H.; Ajami, M.; Ajami, M. Modulation of microRNAs expression in hematopoietic stem cells treated with sodium butyrate in inducing fetal hemoglobin expression. Artif. Cells Nanomed. Biotechnol. 2016, 45, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, Z.; Yang, G.; Lai, Y.; Liu, R. Investigating the Efficacy and Safety of Thalidomide for Treating Patients with ß-Thalassemia: A Meta-Analysis. Front. Pharmacol. 2022, 12, 814302. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Y.; Ma, Y.; Xiao, J.; Zhou, Y.; Yin, X. The association of HBG2, BCL11A, and HBS1L-MYB polymorphisms to thalidomide response in Chinese β-thalassemia patients. Blood Cells Mol. Dis. 2020, 84, 102442. [Google Scholar] [CrossRef]
- Taher, A.; Cappellini, M.D. Update on the use of deferasirox in the management of iron overload. Ther. Clin. Risk Manag. 2009, 5, 857–868. [Google Scholar] [CrossRef]
- Lee, J.W.; Kang, H.J.; Choi, J.Y.; Kim, N.H.; Jang, M.K.; Yeo, C.W.; Lee, S.S.; Kim, H.; Park, J.D.; Park, K.D.; et al. Pharmacogenetic study of deferasirox, an iron chelating agent. PLoS ONE 2013, 8, e64114. [Google Scholar] [CrossRef]
- Ricchi, P.; Marsella, M. Profile of deferasirox for the treatment of patients with non-transfusion-dependent thalassemia syndromes. Drug Des. Dev. Ther. 2015, 9, 6475–6482. [Google Scholar] [CrossRef][Green Version]
- Cusato, J.; Allegra, S.; De Francia, S.; Massano, D.; Piga, A.; D’Avolio, A. Role of pharmacogenetics on deferasirox AUC and efficacy. Pharmacogenomics 2016, 17, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Cusato, J.; Allegra, S.; Massano, D.; De Francia, S.; Piga, A.; D’Avolio, A. Influence of single-nucleotide polymorphisms on deferasirox C trough levels and effectiveness. Pharmacogenomics J. 2015, 15, 263–271. [Google Scholar] [CrossRef]
- Biancamano, B.M.; Connelly, J.; Villeneuve, L.; Caron, P.; Guillemette, C. Deferiprone Glucuronidation by Human Tissues and Recombinant UDP Glucuronosyltransferase1A6, An In Vitro Investigation of Genetic and Splice Variants. Am. Soc. Pharmacol. Exp. Ther. 1997, 37, 322–329. [Google Scholar]
- Dadheech, S.; Rao, A.V.; Shaheen, U.; Hussien, M.D.; Jain, S.; Jyothy, A.; Munshi, A. Three most common nonsynonymous UGT1A6*2 polymorphisms (Thr181Ala, Arg184Ser and Ser7Ala) and therapeutic response to deferiprone in β-thalassemia major patients. Gene 2013, 531, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Huang, S.Q.; Su, H.H.; Zhou, S.F. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr. Drug Metab. 2009, 10, 781–834. [Google Scholar] [CrossRef]
- Zobdeh, F.; Eremenko, I.I.; Akan, M.A.; Tarasov, V.V.; Chubarev, V.N.; Schiöth, H.B.; Mwinyi, J. Pharmacogenetics and PainTreatment with a Focus on NonSteroidal Anti-Inflammatory Drugs (NSAIDs) and Antidepressants: A Systematic Review. Pharmaceutics 2022, 14, 1190. [Google Scholar] [CrossRef] [PubMed]
- Agúndez, J.A.; García-Martín, E.; Martínez, C. Genetically based impairment in CYP2C8- and CYP2C9-dependent NSAID metabolism as a risk factor for gastrointestinal bleeding: Is a combination of pharmacogenomics and metabolomics required to improve personalized medicine? Expert Opin. Drug Metab. Toxicol. 2009, 5, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Hartman, A.D.; Desai, P. Pharmacogenomics of sickle cell disease: Steps toward personalized medicine. Pharmgenomics Pers. Med. 2017, 10, 261–265. [Google Scholar] [CrossRef]
- Joly, P.; Gagnieu, M.C.; Bardel, C.; Francina, A.; Pondarre, C.; Martin, C. Genotypic screening of the main opiate-related polymorphisms in a cohort of 139 sickle cell disease patients. Am. J. Hematol. 2012, 87, 534–536. [Google Scholar] [CrossRef]
- Jhun, E.H.; Yao, Y.; He, Y.; Mack, A.K.; Wilkie, D.J.; Molokie, R.E.; Wang, Z.J. Prevalence of pain-related single nucleotide polymorphisms in patients of African origin with sickle cell disease. Pharmacogenomics 2015, 16, 1795–1806. [Google Scholar] [CrossRef]
- Ofoegbu, A.; BEttienne, E. Pharmacogenomics and Morphine. J. Clin. Pharmacol. 2021, 61, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Somogyi, A.A.; Rubio, J.; Philip, J. The Role of Pharmacogenomics in Opioid Prescribing. Curr. Treat. Options Oncol. 2022, 23, 1353–1369. [Google Scholar] [CrossRef]
- Gammal, R.S.; Crews, K.R.; Haidar, C.E.; Hoffman, J.M.; Baker, D.K.; Barker, P.J.; Estepp, J.H.; Pei, D.; Broeckel, U.; Wang, W.; et al. Pharmacogenetics for Safe Codeine Use in Sickle Cell Disease. Pediatrics 2016, 138, e20153479. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Kumar, R. Nitric oxide: A potential etiological agent for vaso-occlusive crises in sickle cell disease. Nitric Oxide 2024, 144, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.L.; Hibberd, P.L.; Betit, P.; Cooper, A.B.; Botelho, C.A.; Brugnara, C. Preliminary assessment of inhaled nitric oxide for acute vaso-occlusive crisis in pediatric patients with sickle cell disease. JAMA 2003, 289, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, T.; Yokoyama, K.; Arai, T.; Takemoto, F.; Hara, S.; Yamada, A.; Kawaguchi, Y.; Hosoya, T.; Igari, J. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochem. Biophys. Res. Commun. 1998, 245, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Yousry, S.M.; Ellithy, H.N.; Shahin, G.H. Endothelial nitric oxide synthase gene polymorphisms and the risk of vasculopathy in sickle cell disease. Hematology 2016, 21, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Nishank, S.S.; Singh, M.P.; Yadav, R.; Gupta, R.B.; Gadge, V.S.; Gwal, A. Endothelial nitric oxide synthase gene polymorphism is associated with sickle cell disease patients in India. J. Hum. Genet. 2013, 58, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Vercellotti, G.M. Multiple inducers of endothelial NOS (eNOS) dysfunction in sickle cell disease. Am. J. Hematol. 2021, 96, 1505–1517. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Schechter, A.N. Nitric oxide therapy in sickle cell disease. Semin. Hematol. 2001, 38, 333–342. [Google Scholar] [CrossRef]
- King, S.B. Nitric oxide production from hydroxyurea. Free Radic. Biol. Med. 2004, 37, 737–744. [Google Scholar] [CrossRef]
- Caudle, K.E.; Klein, T.E.; Hoffman, J.M.; Muller, D.J.; Whirl-Carrillo, M.; Gong, L.; McDonagh, E.M.; Sangkuhl, K.; Thorn, C.F.; Schwab, M.; et al. Incorporation of pharmacogenomics into routine clinical practice: The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 2014, 15, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.; Anie, K.A.; Buckton, J.; Kiilu, P.; Layton, M.; Alexander, L.; Hemmaway, C.; Sutton, D.; Amos, C.; Doré, C.J.; et al. SWIM (sickle with ibuprofen and morphine) randomised controlled trial fails to recruit: Lessons learnt. BMJ Open 2016, 6, e011276. [Google Scholar] [CrossRef]
- Adam, S.S.; Hoppe, C. Potential role for statins in sickle cell disease. Pediatr. Blood Cancer 2013, 60, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Selby, R.; Nisbet-Brown, E.; Basran, R.K.; Chang, L.; Olivieri, N.F. Valproic acid and augmentation of fetal hemoglobin in individuals with and without sickle cell disease. Blood 1997, 90, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Lampronti, I.; Bianchi, N.; Zuccato, C.; Medici, A.; Bergamini, P.; Gambari, R. Effects on erythroid differentiation of platinum(II) complexes of synthetic bile acid derivatives. Bioorg. Med. Chem. 2006, 14, 5204–5210. [Google Scholar] [CrossRef]
- Kolliopoulou, A.; Stratopoulos, A.; Siamoglou, S.; Sgourou, A.; Ali, B.R.; Papachatzopoulou, A.; Katsila, T.; Patrinos, G.P. Key Pharmacogenomic Considerations for Sickle Cell Disease Patients. OMICS 2017, 21, 314–322. [Google Scholar] [CrossRef]
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef]
- Gambari, R. The Role of OMICS Research in Understanding Phenotype Variation in Thalassaemia: The THALAMOSS Project. Thalass. Rep. 2014, 4, 4877. [Google Scholar] [CrossRef]
- Mañú Pereira, M.D.M.; Colombatti, R.; Alvarez, F.; Bartolucci, P.; Bento, C.; Brunetta, A.L.; Cela, E.; Christou, S.; Collado, A.; de Montalembert, M.; et al. Sickle cell disease landscape and challenges in the EU: The ERN-EuroBloodNet perspective. Lancet Haematol. 2023, 10, e687–e694. [Google Scholar] [CrossRef]
- Halim-Fikri, B.H.; Lederer, C.W.; Baig, A.A.; Mat-Ghani, S.N.A.; Syed-Hassan, S.R.; Yusof, W.; Abdul Rashid, D.; Azman, N.F.; Fucharoen, S.; Panigoro, R.; et al. Global Globin Network Consensus Paper: Classification and Stratified Roadmaps for Improved Thalassaemia Care and Prevention in 32 Countries. J. Pers. Med. 2022, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. ClinGen Hemoglobinopathy Variant Curation Expert Panel. Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum. Mutat. 2022, 43, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Stephanou, C.; Archer, N.; Bonifazi, F.; Giannuzzi, V.; Kuo, K.H.M.; Maggio, A.; Makani, J.; Mañú-Pereira, M.D.M.; Michailidou, K.; et al. The International Hemoglobinopathy Research Network (INHERENT): An international initiative to study the role of genetic modifiers in hemoglobinopathies. Am. J. Hematol. 2021, 96, E416–E420. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Nong, C.; Lai, F.; Tang, Y.; Wang, T. Research Progress of Cell-Free Fetal DNA in Non-Invasive Prenatal Diagnosis of Thalassemia. Hemoglobin 2023, 47, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ye, Y.; Fan, D.; Lin, S.; Li, M.; Hou, H.; Zhang, J.; Yang, X. Non-invasive prenatal diagnosis of thalassemia through multiplex PCR, target capture and next-generation sequencing. Mol. Med. Rep. 2020, 22, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Maskoen, A.M.; Rahayu, N.S.; Laksono, B.; Fibriani, A.; Soewondo, W.; Mose, J.C.; Sahiratmadja, E.; Panigoro, R. Cell-free fetal DNA as a non-invasive method using pyrosequencing in detecting beta-globin gene mutation: A pilot study from area with limited facilities in Indonesia. Front. Pediatr. 2022, 10, 902879. [Google Scholar] [CrossRef]
- Sawakwongpra, K.; Tangmansakulchai, K.; Ngonsawan, W.; Promwan, S.; Chanchamroen, S.; Quangkananurug, W.; Sriswasdi, S.; Jantarasaengaram, S.; Ponnikorn, S. Droplet-based digital PCR for non-invasive prenatal genetic diagnosis of α and β-thalassemia. Biomed. Rep. 2021, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- D’Aversa, E.; Breveglieri, G.; Boutou, E.; Balassopoulou, A.; Voskaridou, E.; Pellegatti, P.; Guerra, G.; Scapoli, C.; Gambari, R.; Borgatti, M. Droplet Digital PCR for Non-Invasive Prenatal Detection of Fetal Single-Gene Point Mutations in Maternal Plasma. Int. J. Mol. Sci. 2022, 23, 2819. [Google Scholar] [CrossRef]
- Constantinou, C.G.; Karitzi, E.; Byrou, S.; Stephanou, C.; Michailidou, K.; Makariou, C.; Hadjilambi, G.; Christofides, A.; Kleanthous, M.; Papasavva, T. Optimized Droplet Digital PCR Assay on Cell-Free DNA Samples for Non-Invasive Prenatal Diagnosis: Application to Beta-Thalassemia. Clin. Chem. 2022, 68, 1053–1063. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Comments | References |
|---|---|---|
| 5-Azacytidine | DNA hypomethylation | Kalantri et al., 2018 [60] |
| Hydroxyurea | DNA hypomethylation | Fucharoen et al., 1996 [61] |
| Butyrate | HDAC activity inhibition | Bianchi et al., 2015 [62] |
| 2,2-dimethylbutyrate (HQK-1001) | A phase II study of efficacy and safety of the oral fetal globin inducer, HQK-1001 | Reid et al., 2014 [63] |
| Valproate | Activation of p38 MAPK pathway, HDAC inhibition | Rönndahl et al., 2006 [64] |
| Thalidomide derivatives | Activation of p38 MAPK pathway | Aerbajinai et al., 2007 [65] Moutouh-De Parseval et al., 2008 [66] |
| Trichostatin A | HDAC inhibition | Marianna et al., 2001 [67] |
| Oridonin | Activation of p38 MAPK signaling | Guo L et al., 2020 [68] |
| Rapamycin (sirolimus) | mTOR inhibitor | Fibach et al., 2006 [69] Zuccato et al., 2022 [70] |
| Everolimus | mTOR inhibitor | Zuccato et al., 2007 [71] |
| Mithramycin | Inhibition of Sp1/DNA interactions | Fibach et al., 2003 [72] |
| Resveratrol | Activation of p38 MAPK signaling | Fibach et al., 2012 [73] |
| Angelicin | Induction of γ-globin expression via NRF2/ARE stress response pathway | Lampronti et al., 2003 [74] |
| Cinchonidine, Quinidine and Cinchonine | Cinchona alkaloids are potent inducers of the expression of γ-globin genes in erythroid cells | Iftikhar et al., 2019 [75] Zuccato et al., 2021 [76] |
| Tranylcypromine | Lysine-specific demethylase 1 (LSD1) inhibition | Shi et al., 2013 [77] |
| Vorinostat | HDAC inhibition reduces the expression of α-globin, induces γ-globin expression | Mettananda et al., 2019 [78] |
| Vasicinol and Vasicine | Induction of γ-globin genes in a pre-clinical study of HbF inducers isolated from Adhatoda vasica | Iftikhar et al., 2022 [79] |
| Hydroxyurea | Clinical trials NCT00001958 and NCT03183375 | Yasara et al., 2022 [80] |
| HQK-1001 | Clinical trial NCT00790127 | Reid et al. [63] |
| Thalidomide | Clinical trial NCT05132270 | Ansari et al. [81] |
| Sirolimus | Clinical trials NCT03877809 and NCT04247750 | Gamberini et al., 2021 [82] |
| Non-Coding RNA | Target mRNA | Comments | Reference |
|---|---|---|---|
| miR-34a | STAT3 | Stable expression mediates fetal hemoglobin induction | Ward et al., 2016 [138] |
| miR-210-3p | BCL11A | High expression associated with high γ-globin mRNA content; down-regulated BCL11A in erythroid precursor cells induced with mithramycin | Gasparello et al., 2017 [143] |
| miR-92a-3p | BCL11A | Upregulates γ-globin expression and inhibits oxidative stress and apoptosis in erythroid precursor cells | Li H et al., 2022 [144] |
| miR-30a | BCL11A | Regulation of γ-globin gene expression through targeting BCL11A | Gholampour et al., 2020 [145] |
| miR-2355-5p | KLF6 | Upregulates γ-globin gene expression | Cheng et al., 2021 [146] |
| miR-144 | NRF2 | Regulates oxidative stress tolerance of thalassemic erythroid cells | Srinoun et al., 2019 [147] |
| miR-326 | EKLF | Regulates HbF synthesis by targeting EKLF in human erythroid cells | Li et al., 2018 [148] |
| miR-155 | BACH1 | Enhances phagocytic activity of β-thalassemia/HbE monocytes | Srinoun et al., 2017 [149] |
| miR-26b | MYB 3′UTR | Upregulates HbF expression in erythroleukemic K-562 cell line. | Pule et al., 2016 [150] |
| miR-29b-3p | Sp1 | Mediates epigenetic mechanisms of HBG gene activation | Starlard-Davenport A 2019 [151] |
| miR-486-3p | BCL11A | Regulation of γ-globin gene expression through direct modulation of BCL11A | Lulli et al., 2013 [152] |
| miR-15a and miR-16-1 | MYB | Elevates fetal hemoglobin expression | Sankaran et al., 2011 [153] |
| miR-190b-5p | BCL11A | Regulates BCL11A expression | Chen et al., 2023 [154] |
| circ-0008102 | tbd (*) | May serve as a novel clinical biomarker in beta-thalassemia | Chen et al., 2024 [155] |
| lncRNA H19 | tbd (*) | Leads to upregulation of γ-globin gene expression | Xie et al., 2024 [156] |
| lncRNA HBBP1 | ELK1 | Leads to upregulation of γ-globin gene expression | A et al., 2021 [157] |
| lncRNA HMI | tbd(*) | Regulates human fetal hemoglobin expression | Morrison et al., 2018 [158] |
| Gene/ Biomarker | Drug | Drug RxNorm ID | Medical Intervention (Established or Proposed) | FDA Labeling Sections | CPIC Guideline Classification of Recommendation | Reference from the Literature |
|---|---|---|---|---|---|---|
| CYP2D6 | Codeine | 2670 | Pain management | Boxed Warning, Warnings and Precautions, Use in Specific Populations, Patient Counseling Information | https://cpicpgx.org/guidelines/guideline-for-codeine-and-cyp2d6/ (accessed on 25 March 2024) | Gammal et al. (2016) [194] |
| CYP2C9 | Ibuprofen | 5640 | Anti-inflammatory analgesics | n.a. (*) | https://cpicpgx.org/guidelines/cpic-guideline-for-nsaids-based-on-cyp2c9-genotype/ (accessed on 25 March 2024) | Cho et al. (2016) [204] |
| CYP3A4 | Rosuvastatin | 301542 | Reduction in serum cholesterol and cardiovascular complications | Clinical Pharmacology | https://cpicpgx.org/guidelines/cpic-guideline-for-statins/ (accessed on 25 March 2024) | Adam et al. (2013) [205] |
| OTC, POLG | Valproic acid | 11118 | Fetal hemoglobin induction | Boxed Warning, Contraindications, Warnings and Precautions; Testing required | n.a. (*) | Selby et al. (1997) [206] |
| CYP3A5 | Sirolimus | 35302 | Fetal hemoglobin induction | n.a. (*) | Provisional (accessed on 25 March 2024). | Zuccato et al. (2022) [70] |
| ACTP2, ERCC1, TPMT | Cisplatin | 2555 | Fetal hemoglobin induction, Bone marrow transplantation | Adverse Reactions | Provisional (accessed on 25 March 2024). | Lampronti et al. (2006) [207] |
| CYP2D6 | Quinidine | 9068 | Fetal hemoglobin induction | Warnings and Precautions, Clinical Pharmacology | Provisional (accessed on 25 March 2024). | Zuccato et al. (2021) [76] |
| CYP2D6 | Quinine | 9071 | Fetal hemoglobin induction | Drug Interactions, Warnings and Precautions | Provisional (accessed on 25 March 2024). | Zuccato et al. (2021) [76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambari, R.; Waziri, A.D.; Goonasekera, H.; Peprah, E. Pharmacogenomics of Drugs Used in β-Thalassemia and Sickle-Cell Disease: From Basic Research to Clinical Applications. Int. J. Mol. Sci. 2024, 25, 4263. https://doi.org/10.3390/ijms25084263
Gambari R, Waziri AD, Goonasekera H, Peprah E. Pharmacogenomics of Drugs Used in β-Thalassemia and Sickle-Cell Disease: From Basic Research to Clinical Applications. International Journal of Molecular Sciences. 2024; 25(8):4263. https://doi.org/10.3390/ijms25084263
Chicago/Turabian StyleGambari, Roberto, Aliyu Dahiru Waziri, Hemali Goonasekera, and Emmanuel Peprah. 2024. "Pharmacogenomics of Drugs Used in β-Thalassemia and Sickle-Cell Disease: From Basic Research to Clinical Applications" International Journal of Molecular Sciences 25, no. 8: 4263. https://doi.org/10.3390/ijms25084263
APA StyleGambari, R., Waziri, A. D., Goonasekera, H., & Peprah, E. (2024). Pharmacogenomics of Drugs Used in β-Thalassemia and Sickle-Cell Disease: From Basic Research to Clinical Applications. International Journal of Molecular Sciences, 25(8), 4263. https://doi.org/10.3390/ijms25084263