
Predicting the Structure of Enzymes with Metal Cofactors: The Example of [FeFe] Hydrogenases

 , ,
, ,  , and
, and
Abstract
1. Introduction
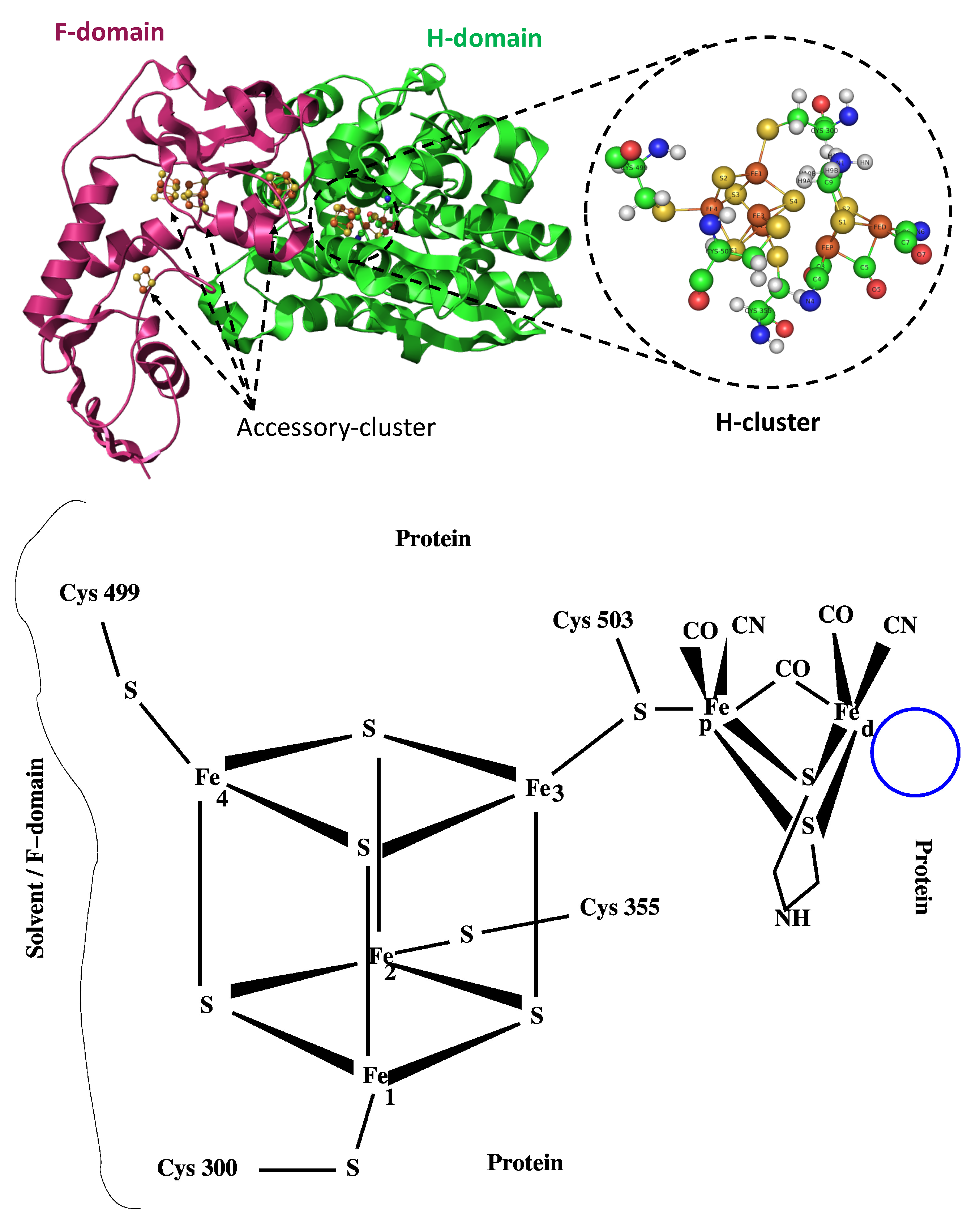
2. Results
2.1. Alignment and Gene Annotation
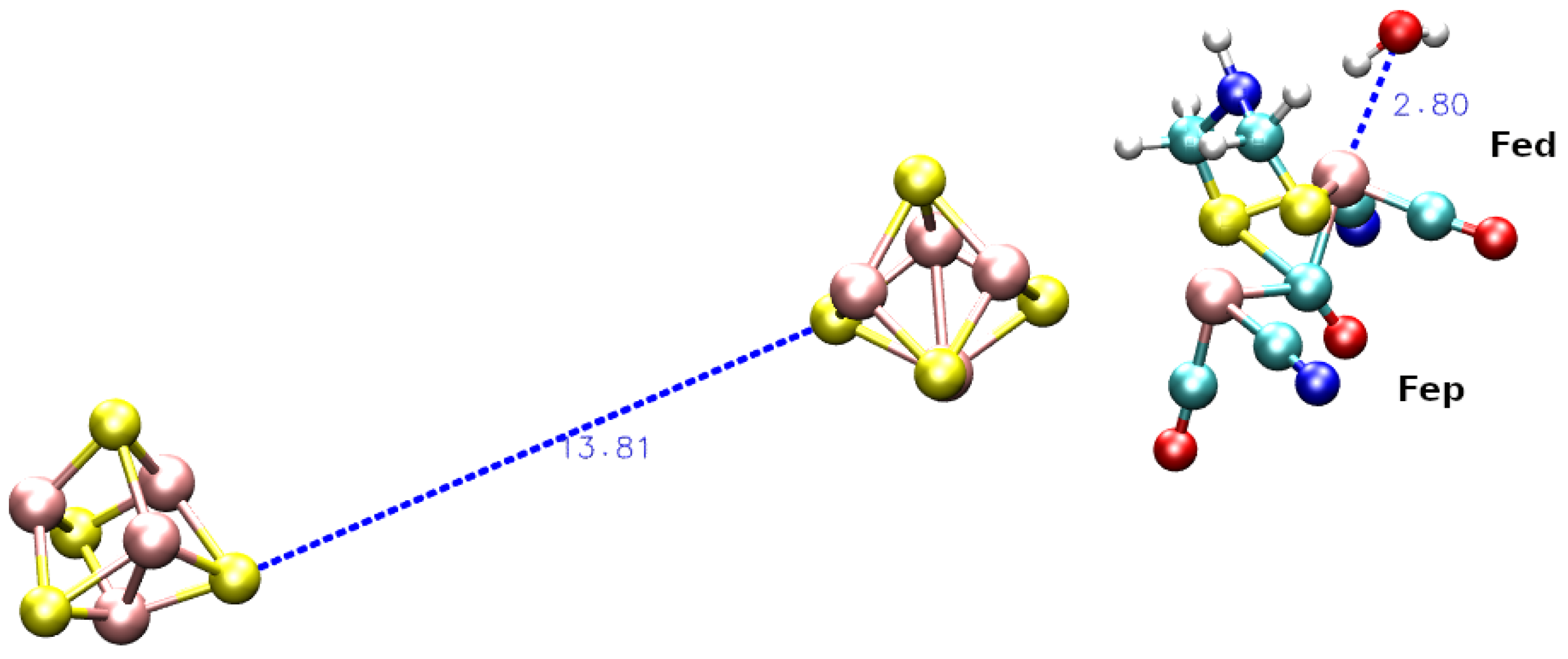
2.2. Structure Prediction
2.3. Structure Refinement
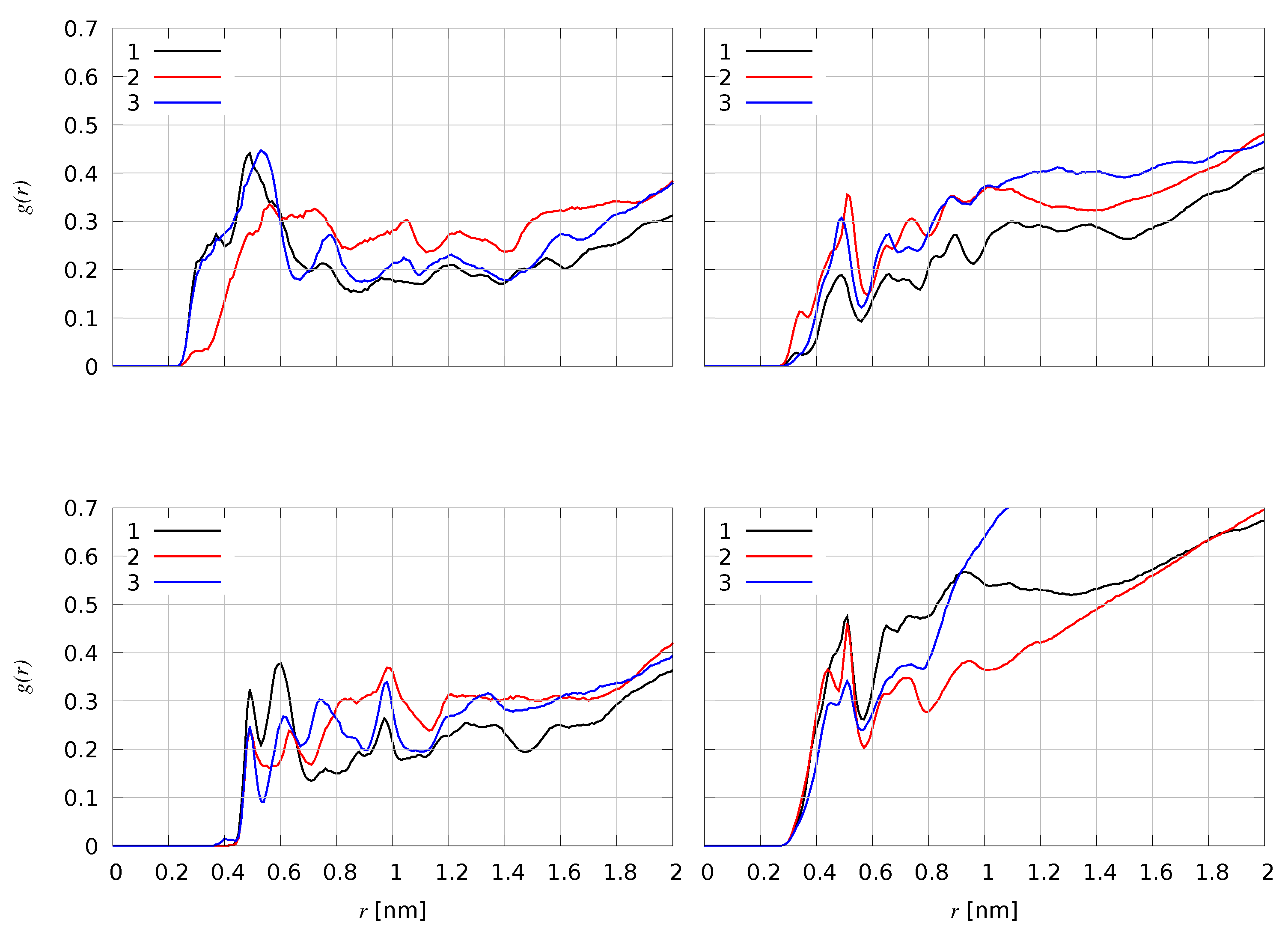
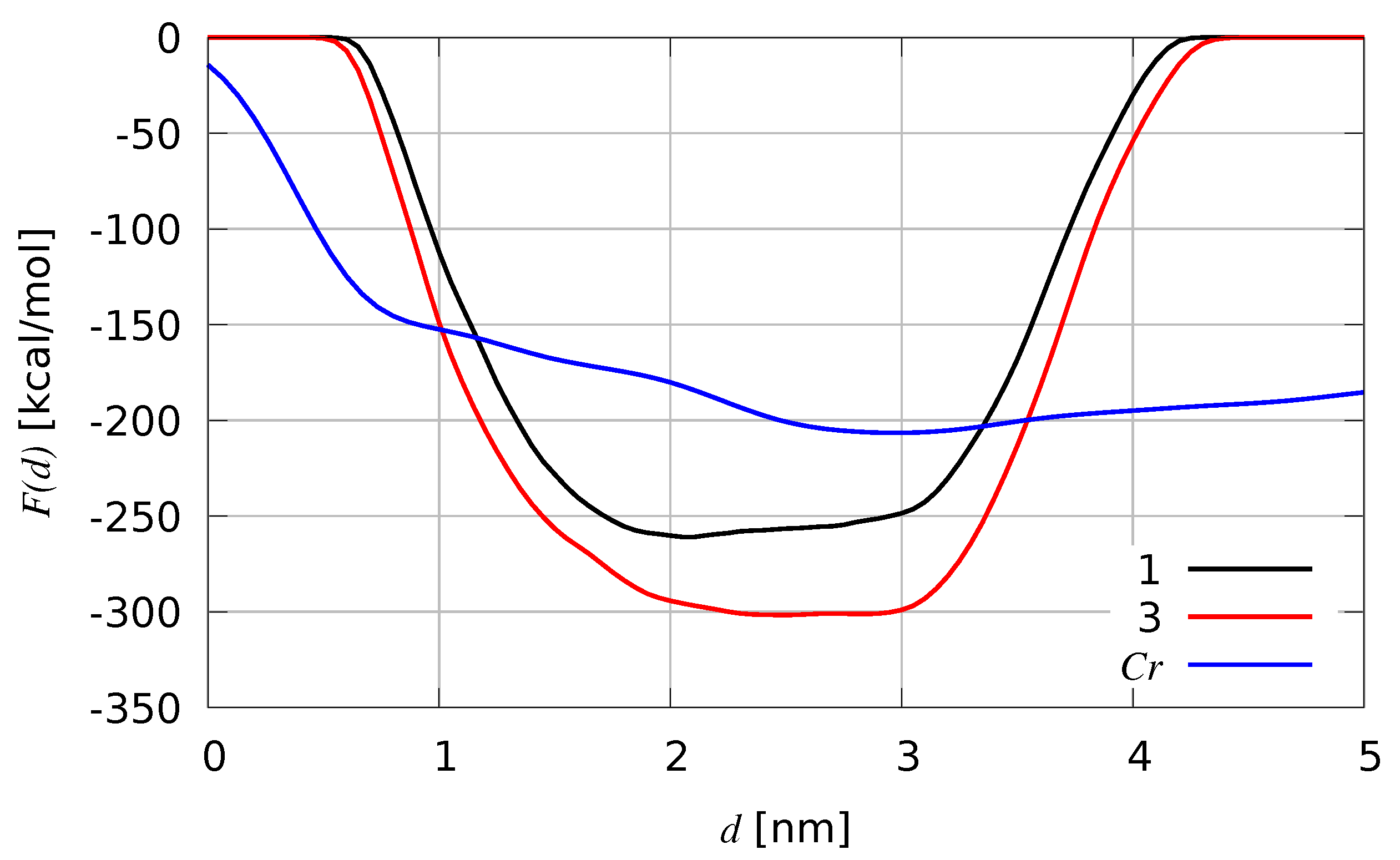
2.4. Enhancing Statistics
3. Discussion
4. Materials and Methods
4.1. Alignment and Gene Annotation
4.2. Structure Prediction
4.3. Structure Refinement
4.4. Enhancing Statistics with Well-Tempered Metadynamics
4.5. Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Hyd | [FeFe] hydrogenase |
| Cp | Clostridium pasteurianum |
| CpI | [FeFe] hydrogenase of Cp, gene CpI |
| Cv | Chlorella variabilis |
| Cv Hyd | [FeFe] hydrogenase of Cv, gene HydA1 |
| Cvu | Chlorella vulgaris 211/11P strain |
| Cvu Hyd | [FeFe] hydrogenase of Cvu, gene KAI3438965.1 |
| Cr | Chlamydomonas rheinardtii |
| Cr Hyd | [FeFe] hydrogenase of Cr, gene HydA1 |
| Dd | Desulfovibrio desulfuricans |
| Dd Hyd | [FeFe] hydrogenase of Dd HydAB |
| MD | molecular dynamics |
| MtD | molecular well-tempered metadynamics |
| AF | AlphaFold deep learning |
| SM | Supplementary Materials |
References
- Seibert, M.; Torzillo, G. (Eds.) Microalgal Hydrogen Production; Comprehensive Series in Photochemical & Photobiological Sciences; The Royal Society of Chemistry: Piccadilly, UK, 2018; pp. P001–P496. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Morra, S. Fantastic [FeFe]-Hydrogenases and Where to Find Them. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, J. Chapter 8—Production of biohydrogen. In Biofuels and Biorefining; Gómez Castro, F.I., Gutiérrez-Antonio, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 283–337. [Google Scholar] [CrossRef]
- Cecchin, M.; Marcolungo, L.; Rossato, M.; Girolomoni, L.; Cosentino, E.; Cuine, S.; Li-Beisson, Y.; Delledonne, M.; Ballottari, M. Chlorella vulgaris genome assembly and annotation reveals the molecular basis for metabolic acclimation to high light conditions. Plant J. 2019, 100, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Masojídek, J.; Karolína, R.; Rearte, T.A.; Celis Plá, P.S.; Torzillo, G.; Silva Benavides, A.M.; Neori, A.; Gómez, C.; Álvarez-Gómez, F.; Lukeš, M.; et al. Changes in photosynthesis, growth and biomass composition in outdoor Chlorella g120 culture during the metabolic shift from heterotrophic to phototrophic cultivation regime. Algal Res. 2021, 56, 102303. [Google Scholar] [CrossRef]
- Touloupakis, E.; Faraloni, C.; Silva Benavides, A.M.; Masojídek, J.; Torzillo, G. Sustained photobiological hydrogen production by Chlorella vulgaris without nutrient starvation. Int. J. Hydrogen Energy 2021, 46, 3684–3694. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H. Hydrogenases, Structure and Function. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 562–567. [Google Scholar] [CrossRef]
- Birrell, J.A.; Rodríguez-Maciá, P.; Reijerse, E.J.; Martini, M.A.; Lubitz, W. The catalytic cycle of [FeFe] hydrogenase: A tale of two sites. Coord. Chem. Rev. 2021, 449, 214191. [Google Scholar] [CrossRef]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Meuser, J.E.; Boyd, E.S.; Ananyev, G.; Karns, D.; Radakovits, R.; Narayana Murthy, U.M.; Ghirardi, M.L.; Dismukes, G.C.; Peters, J.W.; Posewitz, M.C. Evolutionary significance of an algal gene encoding an [FeFe]-hydrogenase with F-domain homology and hydrogenase activity in Chlorella variabilis NC64A. Planta 2011, 234, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Artz, J.H.; Zadvornyy, O.A.; Mulder, D.W.; Keable, S.M.; Cohen, A.E.; Ratzloff, M.W.; Williams, S.G.; Ginovska, B.; Kumar, N.; Song, J.; et al. Tuning Catalytic Bias of Hydrogen Gas Producing Hydrogenases. J. Am. Chem. Soc. 2020, 142, 1227–1235. [Google Scholar] [CrossRef]
- Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C.E.; Fontecilla-Camps, J.C. Desulfovibrio desulfuricans iron hydrogenase: The structure shows unusual coordination to an active site Fe binuclear center. Structure 1999, 7, 13–23. [Google Scholar] [CrossRef]
- Mulder, D.W.; Boyd, E.S.; Sarma, R.; Lange, R.K.; Endrizzi, J.A.; Broderick, J.B.; Peters, J.W. Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydA Delta EFG. Nature 2010, 465, 248–251. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.M.; Billoud, B. Occurrence, Classification, and Biological Function of Hydrogenases: An Overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.D.; Ratzloff, M.W.; Mulder, D.W.; Artz, J.H.; Ghose, S.; Hoffman, A.; White, S.; Zadvornyy, O.A.; Broderick, J.B.; Bothner, B.; et al. [FeFe]-Hydrogenase Oxygen Inactivation Is Initiated at the H Cluster 2Fe Subcluster. J. Am. Chem. Soc. 2015, 137, 1809–1816. [Google Scholar] [CrossRef]
- Kamp, C.; Silakov, A.; Winkler, M.; Reijerse, E.J.; Lubitz, W.; Happe, T. Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žıdek, A.; Hassabis, D.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Binbay, F.A.; Rathod, D.C.; George, A.A.P.; Imhof, D. Quality Assessment of Selected Protein Structures Derived from Homology Modeling and AlphaFold. Pharmaceuticals 2023, 16, 1662. [Google Scholar] [CrossRef]
- Liu, D.; Yuan, C.; Guo, C.; Huang, M.; Lin, D. Structural and Functional Insights into the Stealth Protein CpsY of Mycobacterium tuberculosis. Biomolecules 2023, 13, 1611. [Google Scholar] [CrossRef]
- Nagy, V.; Podmaniczki, A.; Vidal-Meireles, A.; Tengölics, R.; Kovács, L.; Rákhely, G.; Scoma, A.; Tóth, S.Z. Water-splitting-based, sustainable and efficient H2 production in green algae as achieved by substrate limitation of the Calvin-Benson-Bassham cycle. Biotechnol. Biofuels 2018, 11, 69. [Google Scholar] [CrossRef]
- Tamayo-Ordoñez, Y.d.J.; Ayil-Gutiérrez, B.A.; Moreno-Davila, I.M.M.; Tamayo-Ordoñez, F.A.; Córdova-Quiroz, A.V.; Poot-Poot, W.A.; Damas-Damas, S.; Villanueva-Alonzo, H.d.J.; Tamayo-Ordoñez, M.C. Bioinformatic analysis and relative expression of hyd and fdx during H2 production in microalgae. Phycol. Res. 2023, 71, 37–55. [Google Scholar] [CrossRef]
- Mészáros, L.S.; Ceccaldi, P.; Lorenzi, M.; Redman, H.J.; Pfitzner, E.; Heberle, J.; Senger, M.; Stripp, S.T.; Berggren, G. Spectroscopic investigations under whole-cell conditions provide new insight into the metal hydride chemistry of [FeFe]-hydrogenase. Chem. Sci. 2020, 11, 4608–4617. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, M.; Ceccaldi, P.; Rodríguez-Maciá, P.; Redman, H.J.; Zamader, A.; Birrell, J.A.; Mészáros, L.S.; Berggren, G. Stability of the H-cluster under whole-cell conditions—Formation of an Htrans-like state and its reactivity towards oxygen. J. Biol. Inorg. Chem. 2022, 27, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Chien, L.F.; Kuo, T.T.; Liu, B.H.; Lin, H.D.; Feng, T.Y.; Huang, C.C. Solar-to-bioH2 production enhanced by homologous overexpression of hydrogenase in green alga Chlorella sp. DT. Int. J. Hydrogen Energy 2012, 37, 17738–17748. [Google Scholar] [CrossRef]
- Greening, C.; Biswas, A.; Carere, C.R.; Jackson, C.J.; Taylor, M.C.; Stott, M.B.; Cook, G.M.; Morales, S.E. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 2016, 10, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A web tool for hydrogenase classification and analysis. Sci. Rep. 2016, 6, 34212–34218. [Google Scholar] [CrossRef]
- Balk, J.; Pierik, A.J.; Netz, D.J.A.; Mühlenhoff, U.; Lill, R. The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron–sulphur proteins. EMBO J. 2004, 23, 2105–2115. [Google Scholar] [CrossRef]
- Urzica, E.; Pierik, A.J.; Mühlenhoff, U.; Lill, R. Crucial Role of Conserved Cysteine Residues in the Assembly of Two Iron-Sulfur Clusters on the CIA Protein Nar1. Biochemistry 2009, 48, 4946–4958. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure—Pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Furlan, C.; Chongdar, N.; Gupta, P.; Lubitz, W.; Ogata, H.; Blaza, J.N.; Birrell, J.A. Structural insight on the mechanism of an electron-bifurcating [FeFe] hydrogenase. eLife 2022, 11, e79361. [Google Scholar] [CrossRef] [PubMed]
- Puthenkalathil, R.C.; Ensing, B. Fast Proton Transport in FeFe Hydrogenase via a Flexible Channel and a Proton Hole Mechanism. J. Phys. Chem. B 2022, 126, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Beratan, D.N.; Onuchic, J.N.; Winkler, J.R.; Gray, H.B. Electron-Tunneling Pathways in Proteins. Science 1992, 258, 1740–1741. [Google Scholar] [CrossRef]
- Kaila, V.R.I. Long-range proton-coupled electron transfer in biological energy conversion: Towards mechanistic understanding of respiratory complex I. J. R. Soc. Interface 2018, 15, 20170916. [Google Scholar] [CrossRef]
- Winkler, M.; Duan, J.; Rutz, A.; Felbek, C.; Scholtysek, L.; Lampret, O.; Jaenecke, J.; Apfel, U.P.; Gilardi, G.; Valetti, F.; et al. A safety cap protects hydrogenase from oxygen attack. Nat. Commun. 2021, 12, 756. [Google Scholar] [CrossRef]
- Rutz, A.; Das, C.K.; Fasano, A.; Jaenecke, J.; Yadav, S.; Apfel, U.P.; Engelbrecht, V.; Fourmond, V.; Léger, C.; Schäfer, L.V.; et al. Increasing the O2 Resistance of the [FeFe]-Hydrogenase CbA5H through Enhanced Protein Flexibility. ACS Catal. 2023, 13, 856–865. [Google Scholar] [CrossRef]
- Stripp, S.T.; Goldet, G.; Brandmayr, C.; Sanganas, O.; Vincent, K.A.; Haumann, M.; Armstrong, F.A.; Happe, T. How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc. Natl. Acad. Sci. USA 2009, 106, 17331–17336. [Google Scholar] [CrossRef]
- Hwang, J.H.; Kim, H.C.; Choi, J.A.; Abou-Shanab, R.; Dempsey, B.A.; Regan, J.M.; Kim, J.R.; Song, H.; Nam, I.H.; Kim, S.N.; et al. Photoautotrophic hydrogen production by eukaryotic microalgae under aerobic conditions. Nat. Commun. 2014, 5, 3234. [Google Scholar] [CrossRef]
- Duan, J.; Senger, M.; Esselborn, J.; Engelbrecht, V.; Wittkamp, F.; Apfel, U.P.; Hofmann, E.; Stripp, S.T.; Happe, T.; Winkler, M. Crystallographic and spectroscopic assignment of the proton transfer pathway in [FeFe]-hydrogenases. Nat. Commun. 2018, 9, 4726–4736. [Google Scholar] [CrossRef]
- Outten, F.W. Iron-sulfur clusters as oxygen-responsive molecular switches. Nat. Chem. Biol. 2007, 3, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2021, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.; Schwartz, R.M.; Orcutt, B. Chapter: A Model of Evolutionary Change in Proteins. In Atlas of Protein Sequence and Structure; National Biomedical Research Foundation: Washington, DC, USA, 1978; Volume 5, pp. 345–352. [Google Scholar]
- Schwarz, R.; Dayhoff, M. Chapter: Matrices for Detecting Distant Relationships. In Atlas of Protein Sequence and Structure; National Biomedical Research Foundation: Washington, DC, USA, 1979; pp. 353–358. [Google Scholar]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Kim, K. Density Functional Theory Calculation of Bonding and Charge Parameters for Molecular Dynamics Studies on [FeFe] Hydrogenases. J. Chem. Theory Comput. 2009, 5, 1137–1145. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone phi, psi and Side-Chain chi1 and chi2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N-log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hošek, P.; Kříž, P.; Toulcová, D.; Spiwok, V. Multisystem altruistic metadynamics- Well-tempered variant. J. Chem. Phys. 2017, 146, 125103. [Google Scholar] [CrossRef] [PubMed]
- Nobili, G.; Botticelli, S.; La Penna, G.; Morante, S.; Rossi, G.; Salina, G. Probing protein stability: Towards a computational atomistic, reliable, affordable, and improvable model. Front. Mol. Biosci. 2023, 10, 1122269. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Laio, A.; Tiwary, P. Metadynamics: A Unified Framework for Accelerating Rare Events and Sampling Thermodynamics and Kinetics. In Handbook of Materials Modeling: Methods: Theory and Modeling; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–31. [Google Scholar] [CrossRef]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E 2002, 65, 031910-10. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The Double Cubic Lattice Method: Efficient Approaches to Numerical Integration of Surface Area and Volume and to Dot Surface Contouring of Molecular Assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. J. Comput. Phys. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Vermaas, J.V.; Hardy, D.J.; Stone, J.E.; Tajkhorshid, E.; Kohlmeyer, A. TopoGromacs: Automated Topology Conversion from CHARMM to GROMACS within VMD. J. Chem. Inform. Model. 2016, 56, 1112–1116. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | F-domain | H-domain | C-terminus |
|---|---|---|---|
| Aux. clusters | H-cluster | ||
| CpI | 1–209 | 210–568 | 569–574 |
| 157, 190, 193, 196 | 300, 355, 499, 503 | ||
| 147, 150, 153, 200 | |||
| 94, 98, 101, 107 | |||
| 36, 46, 49, 64 | |||
| Dd Hyd | 1–86 | 87–392 | 393–421 |
| 45, 66, 69, 72 | 178, 234, 378, 372 | ||
| 35, 38, 41, 76 | |||
| Cr Hyd | 1–64 | 65–487 | 488–497 |
| 170, 225, 417, 421 | |||
| Cvu Hyd | 1–88 | 89–530 | 531–549 |
| 21, 72, 75, 78 | 224, 280, 465, 469 |
| Reference | CpI | Dd | Cr | Tm | Cvu |
|---|---|---|---|---|---|
| Target | Hyd | Hyd | HydABC | Hyd1 | |
| CpI | 0 | ||||
| Dd Hyd | 1.7/1.8/0.8 | 0 | |||
| Cr Hyd | 3.2/3.6/0.7 | 3.0/3.4/0.9 | 0 | ||
| Tm HydABC | 2.2/2.3/1.6 | 2.3/2.4/1.6 | 3.7/4.2/1.5 | 0 | |
| Cvu Hyd 1 | 22.2/4.5/2.0 | 22.2/4.2/2.2 | 22.8/5.4/1.7 | 22.4/4.8/2.5 | 0 |
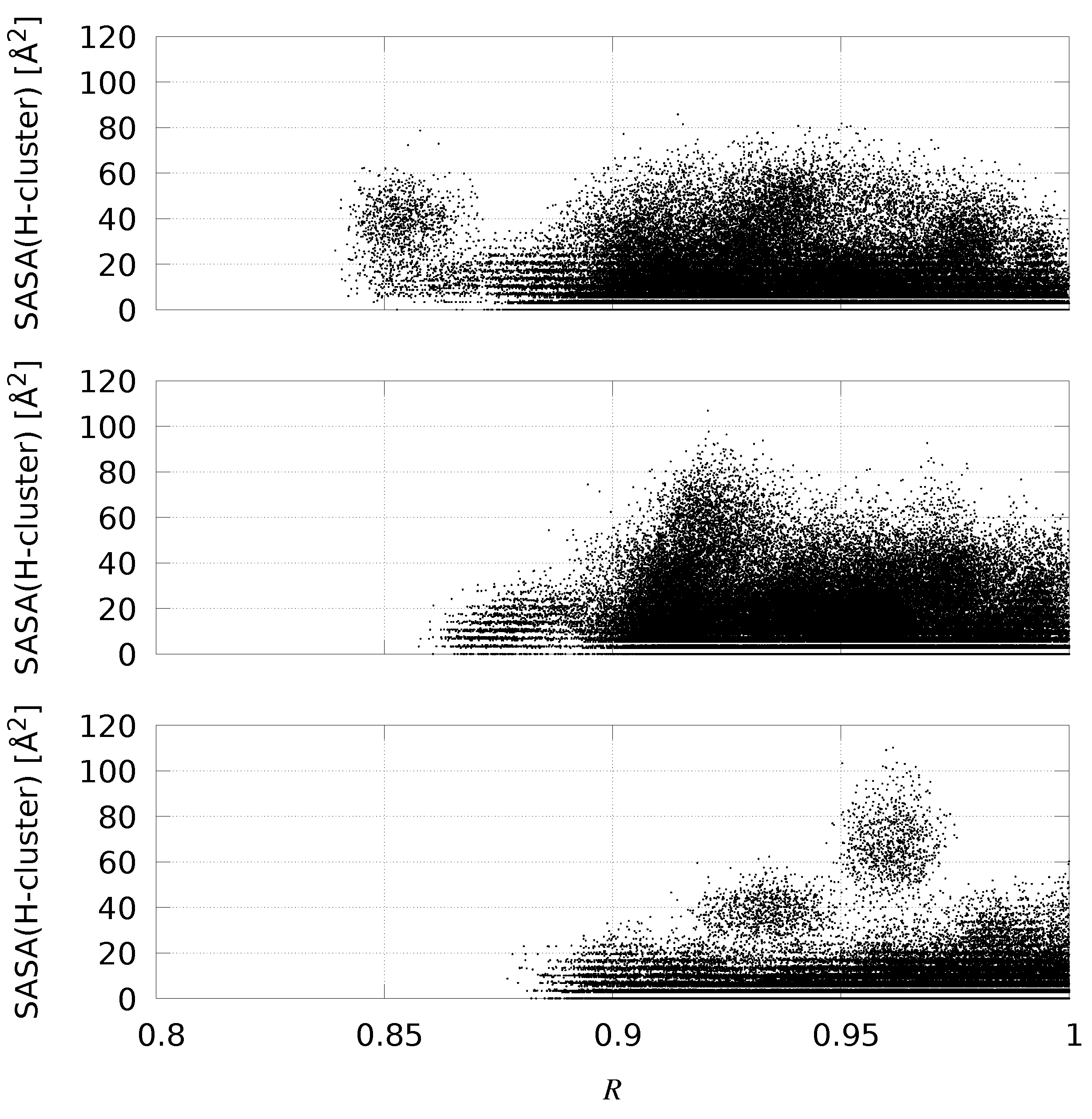
| Configurations | 〈RMSD〉 (Å) | Samples in Set | 〈SASA(H-Cluster)〉 | Compact Samples (%) |
|---|---|---|---|---|
| 1/w1 | 3.6 | 31,217 | 1.8 ± 1.6 | 18 |
| 1/w2 | 3.5 | 6733 | 1.7 ± 1.1 | 4 |
| 1/w3 | 3.4 | 4220 | 1.3 ± 1.4 | 1 |
| 3/w1 | 4.0 | 43,305 | 1.6 ± 1.5 | 31 |
| 3/w2 | 3.9 | 4410 | 1.0 ± 0.7 | 2 |
| 3/w3 | 3.7 | 240 | 1.9 ± 0.7 | 0 |
| Cr/w1 | - | 0 | - | - |
| Cr/w2 | 2.7 | 14 | 1.7 ± 1.0 | 0 |
| Cr/w3 | 2.8 | 1751 | 0.9 ± 0.9 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botticelli, S.; La Penna, G.; Minicozzi, V.; Stellato, F.; Morante, S.; Rossi, G.; Faraloni, C. Predicting the Structure of Enzymes with Metal Cofactors: The Example of [FeFe] Hydrogenases. Int. J. Mol. Sci. 2024, 25, 3663. https://doi.org/10.3390/ijms25073663
Botticelli S, La Penna G, Minicozzi V, Stellato F, Morante S, Rossi G, Faraloni C. Predicting the Structure of Enzymes with Metal Cofactors: The Example of [FeFe] Hydrogenases. International Journal of Molecular Sciences. 2024; 25(7):3663. https://doi.org/10.3390/ijms25073663
Chicago/Turabian StyleBotticelli, Simone, Giovanni La Penna, Velia Minicozzi, Francesco Stellato, Silvia Morante, Giancarlo Rossi, and Cecilia Faraloni. 2024. "Predicting the Structure of Enzymes with Metal Cofactors: The Example of [FeFe] Hydrogenases" International Journal of Molecular Sciences 25, no. 7: 3663. https://doi.org/10.3390/ijms25073663
APA StyleBotticelli, S., La Penna, G., Minicozzi, V., Stellato, F., Morante, S., Rossi, G., & Faraloni, C. (2024). Predicting the Structure of Enzymes with Metal Cofactors: The Example of [FeFe] Hydrogenases. International Journal of Molecular Sciences, 25(7), 3663. https://doi.org/10.3390/ijms25073663