
Triple Generative Self-Supervised Learning Method for Molecular Property Prediction


Abstract
1. Introduction
- (1)
- Although molecular representations based on SSL have been extensively studied, most methods focus on pre-training using sequence information or graph information only. The effective fusion of heterogeneous molecular information is important for enhancing the diversity of molecular representations. There are some methods that have considered this direction. Liu et al. [32] used 3D and 2D information for SSL, aiming to maximize the mutual information between 3D and 2D views of the same molecule. However, there is much less 3D molecular structural information than there is 2D and 1D information. Although there are some methods that could calculate 3D information about a molecule, the error accumulation could also result in the inaccuracy of predictions. Zhu et al. [33] used sequence and graph information to conduct SSL and proposed a pre-training algorithm that combined two molecular representations, including dual-view molecular pre-training (DMP), which maximized the consistency between molecular sequence and molecular graph representations. However, we believe that the generative model can reflect molecular information more accurately and effectively. Therefore, inspired by Liu’s work, this paper concentrates on how to use the generative SSL model to learn molecular representations from sequence and topological structural information from molecules.
- (2)
- The existing SSL models, whether generative or contrastive, generally only use a single or two different models. For example, in generative learning, the encoder and decoder are used to reconstruct features, and in contrastive learning, SSL is performed by minimizing the difference between the feature representation of two different types or sources of data. But there is currently no method to discuss the introduction of three or more models in SSL. We believe that, to a certain extent, more models participating in SSL can also improve the accuracy and generalization of the final feature representation.
- (3)
- After pre-training, multiple models are obtained for downstream tasks, and how to more effectively integrate multiple models is also a problem worth studying. Ensemble learning is widely used in the fusion of various models, but directly concatenating output features cannot effectively utilize the advantages of different models. Treating each output feature equally will also result in key information vanishing from multiple features. Therefore, how to design an effective fusion model, discover the important parts of different sources of features, and improve the accuracy of the prediction are also important issues in this paper.
2. Results
2.1. Datasets
- FreeSolv: the experiment and calculated hydration-free energies in water of 642 small neutral molecules.
- ESOL: 1128 compounds and their corresponding water solubility.
- Lipophilicity: the octanol/water partition coefficients of 4200 compounds.
- Classification dataset.
- BACE: the quantitative and qualitative binding results for a panel of human (BACE-1) inhibitors.
- BBBP: the permeability properties of 2039 compounds.
- HIV: more than 40,000 compounds with the ability to inhibit HIV replication, represented by inactivated and active tags.
- Tox21: the qualitative toxicity measurements of 12 different targets for 7831 compounds.
- SIDER: 27 drug side effects labels for 1427 compounds.
2.2. Performance Comparison with Baselines
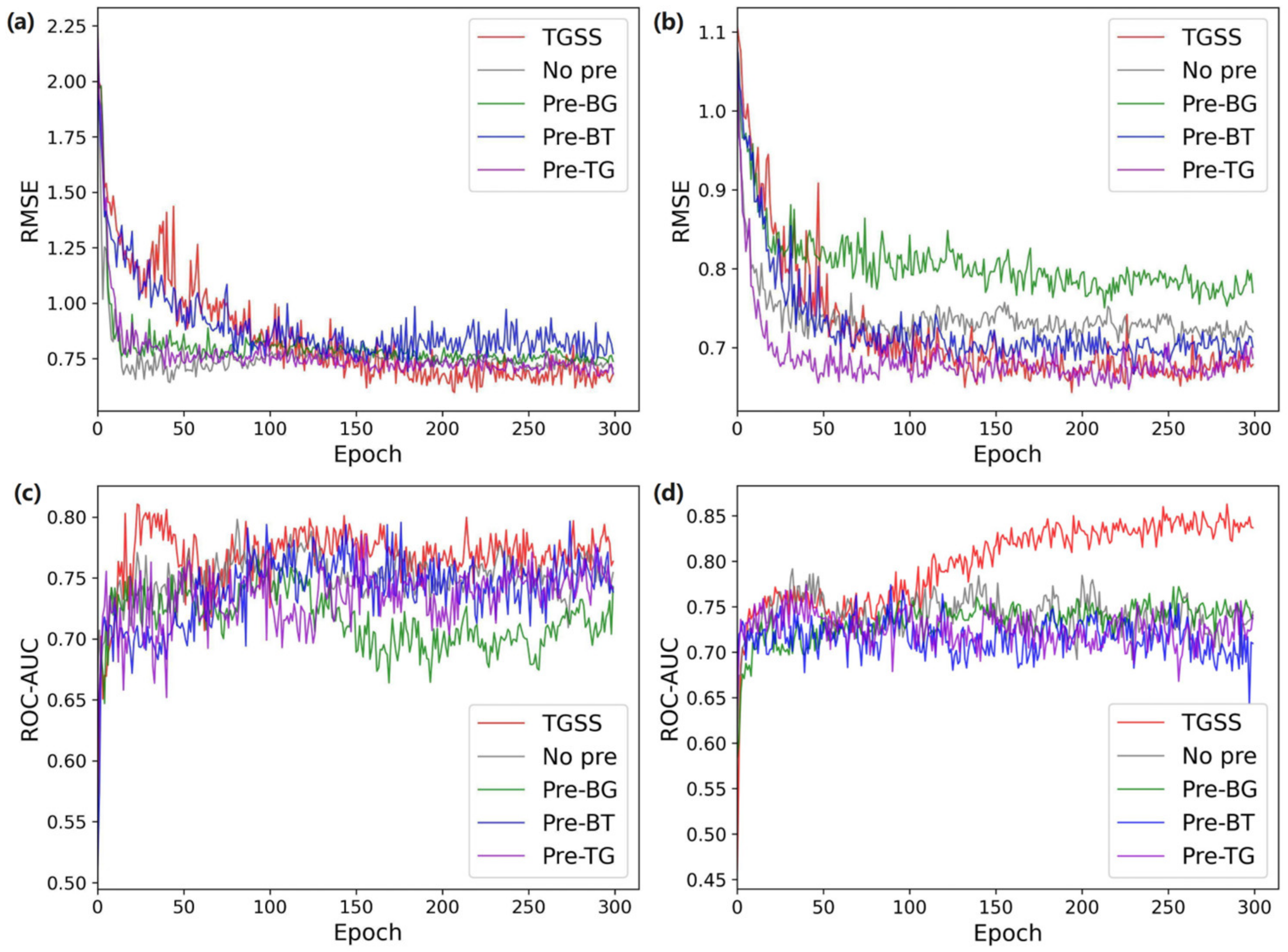
2.3. Ablation Experiments
2.3.1. Performance Comparison of Different Combinations of the Model in the Pre-Training Process
2.3.2. Performance Comparison of Different Sizes of Pre-Training Dataset
2.3.3. Performance Comparison of Different Combinations of Model in Downstream Tasks
2.3.4. Performance Comparison of Different Feature Fusion Methods
2.4. Feature Visualization
3. Discussion
4. Materials and Methods
4.1. Overview
4.2. Pre-Training Models
4.2.1. BiLSTM
4.2.2. Transformer
4.2.3. GAT
4.3. Molecular Representation Reconstruction
4.4. Downstream Task with Hierarchical Elem-Feature Fusion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gervasoni, S.; Manelfi, C.; Adobati, S.; Talarico, C.; Biswas, A.D.; Pedretti, A.; Vistoli, G.; Beccari, A.R. Target Prediction by Multiple Virtual Screenings: Analyzing the SARS-CoV-2 Phenotypic Screening by the Docking Simulations Submitted to the MEDIATE Initiative. Int. J. Mol. Sci. 2023, 25, 450. [Google Scholar] [CrossRef] [PubMed]
- Moschovou, K.; Antoniou, M.; Chontzopoulou, E.; Papavasileiou, K.D.; Melagraki, G.; Afantitis, A.; Mavromoustakos, T. Exploring the Binding Effects of Natural Products and Antihypertensive Drugs on SARS-CoV-2: An in Silico Investigation of Main Protease and Spike Protein. Int. J. Mol. Sci. 2023, 24, 15894. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Gonzalez, A.; Cabezon, A.; Seco-Gonzalez, A.; Conde-Torres, D.; Antelo-Riveiro, P.; Pineiro, A.; Garcia-Fandino, R. The Role of Ai in Drug Discovery: Challenges, Opportunities, and Strategies. Pharmaceuticals 2023, 16, 891. [Google Scholar] [CrossRef] [PubMed]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Aliev, T.A.; Belyaev, V.E.; Pomytkina, A.V.; Nesterov, P.V.; Shityakov, S.; Sadovnichii, R.V.; Novikov, A.S.; Orlova, O.Y.; Masalovich, M.S.; Skorb, E.V. Electrochemical Sensor to Detect Antibiotics in Milk Based on Machine Learning Algorithms. ACS Appl. Mater. Interfaces 2023, 15, 52010–52020. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, D.; Zhu, J.; Rodriguez-Paton, A.; Song, T. CSConv2d: A 2-D Structural Convolution Neural Network with a Channel and Spatial Attention Mechanism for Protein-Ligand Binding Affinity Prediction. Biomolecules 2021, 11, 643. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pan, S.; Xia, L.; Li, Z. Molecular Property Prediction by Combining LSTM and GAT. Biomolecules 2023, 13, 503. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Xu, L.; Pan, S.; Niu, D.; Zhang, B.; Li, Z. Drug-Target Binding Affinity Prediction Using Message Passing Neural Network and Self Supervised Learning. BMC Genom. 2023, 24, 557. [Google Scholar] [CrossRef]
- Pan, S.; Xia, L.; Xu, L.; Li, Z. SubMDTA: Drug Target Affinity Prediction Based on Substructure Extraction and Multi-Scale Features. BMC Bioinform. 2023, 24, 334. [Google Scholar] [CrossRef]
- Li, X.; Han, P.; Wang, G.; Chen, W.; Wang, S.; Song, T. SDNN-PPI: Self-Attention with Deep Neural Network Effect on Protein-Protein Interaction Prediction. BMC Genom. 2022, 23, 474. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Wieder, O.; Kohlbacher, S.; Kuenemann, M.; Garon, A.; Ducrot, P.; Seidel, T.; Langer, T. A Compact Review of Molecular Property Prediction with Graph Neural Networks. Drug Discov. Today Technol. 2020, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, S.; Bai, B.; Chan, H.C.S.; Yuan, S. Accurate Physical Property Predictions via Deep Learning. Molecules 2022, 27, 1668. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Shi, S.; Ueda, H.R. SMILES Transformer: Pre-Trained Molecular Fingerprint for Low Data Drug Discovery. arXiv 2019, arXiv:1911.04738. [Google Scholar]
- Ma, H.; Bian, Y.; Rong, Y.; Huang, W.; Xu, T.; Xie, W.; Ye, G.; Huang, J. Multi-View Graph Neural Networks for Molecular Property Prediction. arXiv 2020, arXiv:2005.13607. [Google Scholar]
- Jiang, S.; Balaprakash, P. Graph Neural Network Architecture Search for Molecular Property Prediction. In Proceedings of the 2020 IEEE International Conference on Big Data (Big Data), Atlanta, GA, USA, 10 December 2020; IEEE: Piscataway, NJ, USA, 2020; pp. 1346–1353. [Google Scholar]
- Chen, J.; Zheng, S.; Song, Y.; Rao, J.; Yang, Y. Learning Attributed Graph Representations with Communicative Message Passing Transformer. arXiv 2021, arXiv:2107.08773. [Google Scholar]
- Song, Y.; Zheng, S.; Niu, Z.; Fu, Z.; Lu, Y.; Yang, Y. Communicative Representation Learning on Attributed Molecular Graphs. In Proceedings of the Twenty-Ninth International Joint Conference on Artificial Intelligence, Yokohama, Japan, 11–17 July 2020; International Joint Conferences on Artificial Intelligence Organization: California City, CA, USA, 2020; pp. 2831–2838. [Google Scholar]
- Shahab, M.; Zheng, G.; Khan, A.; Wei, D.; Novikov, A.S. Machine Learning-Based Virtual Screening and Molecular Simulation Approaches Identified Novel Potential Inhibitors for Cancer Therapy. Biomedicines 2023, 11, 2251. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, L.; Nie, J.; Wei, Z. Towards Adaptive Multi-Scale Intermediate Domain via Progressive Training for Unsupervised Domain Adaptation. IEEE Trans. Multimed. 2024, 26, 5054–5064. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, F.; Hou, Z.; Mian, L.; Wang, Z.; Zhang, J.; Tang, J. Self-Supervised Learning: Generative or Contrastive. IEEE Trans. Knowl. Data Eng. 2021, 35, 857–876. [Google Scholar] [CrossRef]
- Wang, J.; Guan, J.; Zhou, S. Molecular Property Prediction by Contrastive Learning with Attention-Guided Positive Sample Selection. Bioinformatics 2023, 39, btad258. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Huang, L.; Nie, J.; Wei, Z. Unsupervised Deep Hashing with Fine-Grained Similarity-Preserving Contrastive Learning for Image Retrieval. IEEE Trans. Circuits Syst. Video Technol. 2024. [Google Scholar] [CrossRef]
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models Are Few-Shot Learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901. [Google Scholar]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding. arXiv 2019, arXiv:1810.04805. [Google Scholar]
- Wen, N.; Liu, G.; Zhang, J.; Zhang, R.; Fu, Y.; Han, X. A Fingerprints Based Molecular Property Prediction Method Using the BERT Model. J. Cheminform. 2022, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Chen, Q.; Dong, Y.; Zhang, J.; Yang, H.; Ding, M.; Wang, K.; Tang, J. GCC: Graph Contrastive Coding for Graph Neural Network Pre-Training. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Virtual Event, CA, USA, 23 August 2020; ACM: New York, NY, USA, 2020; pp. 1150–1160. [Google Scholar]
- Li, H.; Zhang, R.; Min, Y.; Ma, D.; Zhao, D.; Zeng, J. A Knowledge-Guided Pre-Training Framework for Improving Molecular Representation Learning. Nat. Commun. 2023, 14, 7568. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, Z.; Subramonian, A.; Sun, Y. Motif-Driven Contrastive Learning of Graph Representations. IEEE Trans. Knowl. Data Eng. 2024. [Google Scholar] [CrossRef]
- Zang, X.; Zhao, X.; Tang, B. Hierarchical Molecular Graph Self-Supervised Learning for Property Prediction. Commun. Chem. 2023, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, H.; Liu, W.; Lasenby, J.; Guo, H.; Tang, J. Pre-Training Molecular Graph Representation with 3D Geometry. arXiv 2022, arXiv:2110.07728. [Google Scholar]
- Zhu, J.; Xia, Y.; Wu, L.; Xie, S.; Zhou, W.; Qin, T.; Li, H.; Liu, T.-Y. Dual-View Molecular Pre-Training. In Proceedings of the 29th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, Long Beach, CA, USA, 6 August 2023; ACM: New York, NY, USA, 2023; pp. 3615–3627. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Cao, Z.; Farimani, A.B. Molecular Contrastive Learning of Representations via Graph Neural Networks. Nat. Mach. Intell. 2022, 4, 279–287. [Google Scholar] [CrossRef]
- You, Y.; Chen, T.; Sui, Y.; Chen, T.; Wang, Z.; Shen, Y. Graph Contrastive Learning with Augmentations. Adv. Neural Inf. Process. Syst. 2020, 33, 5812–5823. [Google Scholar]
- Liu, M.; Yang, Y.; Gong, X.; Liu, L.; Liu, Q. HierMRL: Hierarchical Structure-Aware Molecular Representation Learning for Property Prediction. In Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Las Vegas, NV, USA, 6 December 2022; IEEE: Piscataway, NJ, USA, 2022; pp. 386–389. [Google Scholar]
- Xu, M.; Wang, H.; Ni, B.; Guo, H.; Tang, J. Self-Supervised Graph-Level Representation Learning with Local and Global Structure. In Proceedings of the International Conference on Machine Learning, Virtual Event, 18–24 July 2021; PMLR. pp. 11548–11558. [Google Scholar]
- Hou, Z. GraphMAE: Self-Supervised Masked Graph Autoencoders. In Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, Washington, DC, USA, 14–18 August 2022; pp. 594–604. [Google Scholar]
- Fang, Y.; Zhang, Q.; Zhang, N.; Chen, Z.; Zhuang, X.; Shao, X.; Fan, X.; Chen, H. Knowledge Graph-Enhanced Molecular Contrastive Learning with Functional Prompt. Nat. Mach. Intell. 2023, 5, 542–553. [Google Scholar] [CrossRef]
- Li, X.; Fourches, D. Inductive Transfer Learning for Molecular Activity Prediction: Next-Gen QSAR Models with MolPMoFiT. J. Cheminform. 2020, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Fabian, B.; Edlich, T.; Gaspar, H.; Segler, M.; Meyers, J.; Fiscato, M.; Ahmed, M. Molecular Representation Learning with Language Models and Domain-Relevant Auxiliary Tasks. arXiv 2020, arXiv:2011.13230. [Google Scholar]
- Gasteiger, J.; Groß, J.; Günnemann, S. Directional Message Passing for Molecular Graphs. arXiv 2022, arXiv:2003.03123. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2020, 63, 8749–8760. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Lei, X. A Deep Learning Framework for Predicting Molecular Property Based on Multi-Type Features Fusion. Comput. Biol. Med. 2024, 169, 107911. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Guan, Q.; Luo, W.; Fang, L.; Lai, Z.-R.; Wang, J. Molecular Substructure Graph Attention Network for Molecular Property Identification in Drug Discovery. Pattern Recognit. 2022, 128, 108659. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; Available online: https://proceedings.neurips.cc/paper_files/paper/2017/file/3f5ee243547dee91fbd053c1c4a845aa-Paper.pdf (accessed on 18 February 2024).
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph Attention Networks. arXiv 2018, arXiv:1710.10903. [Google Scholar]
- Hua, Y.; Song, X.; Feng, Z.; Wu, X. MFR-DTA: A Multi-Functional and Robust Model for Predicting Drug–Target Binding Affinity and Region. Bioinformatics 2023, 39, btad056. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Task | Task Type | #Molecule | Splits | Metric |
|---|---|---|---|---|---|
| FreeSolv | 1 | Regression | 642 | Random | RMSE |
| ESOL | 1 | Regression | 1128 | Random | RMSE |
| Lipophilicity | 1 | Regression | 4200 | Random | RMSE |
| BACE | 1 | Classification | 1513 | Scaffold | ROC-AUC |
| BBBP | 1 | Classification | 2039 | Scaffold | ROC-AUC |
| HIV | 1 | Classification | 41127 | Scaffold | ROC-AUC |
| Tox21 | 12 | Classification | 7831 | Scaffold | ROC-AUC |
| SIDER | 27 | Classification | 1427 | Scaffold | ROC-AUC |
| Dataset | BACE | BBBP | HIV | Tox21 | SIDER | |
|---|---|---|---|---|---|---|
| Supervised learning | D-MPNN | 0.809 (0.006) | 0.710 (0.003) | 0.771 (0.005) | 0.759 (0.007) | 0.570 (0.007) |
| AttentionFP | 0.784 (0.022) | 0.643 (0.018) | 0.757 (0.014) | 0.761 (0.005) | 0.606 (0.032) | |
| MSSGAT | 0.881 | 0.726 | 0.787 | - | 0.617 | |
| Self-Supervised learning | MolCLR | 0.828 (0.007) | 0.733 (0.010) | 0.774 (0.006) | 0.741 (0.053) | 0.612 (0.036) |
| GraphCL | 0.754 (0.014) | 0.697 (0.007) | 0.698 (0.027) | 0.739 (0.007) | 0.605 (0.009) | |
| HierMRL | 0.877 (0.017) | 0.745 (0.016) | 0.782 (0.011) | 0.792 (0.006) | 0.686 (0.011) | |
| GraphLoG | 0.835 (0.012) | 0.657 (0.014) | 0.778 (0.008) | 0.757 (0.006) | 0.612 (0.011) | |
| GraphMVP | 0.768 (0.011) | 0.685 (0.002) | 0.748 (0.014) | 0.745 (0.004) | 0.623 (0.016) | |
| GraphMAE | 0.831 (0.009) | 0.720 (0.006) | 0.772 (0.010) | 0.755 (0.006) | 0.603 (0.011) | |
| TGSS | 0.810(0.004) | 0.790 (0.068) | 0.789 (0.041) | 0.754 (0.005) | 0.721 (0.004) | |
| Dataset | FreeSolv | ESOL | Lipophilicity | |
|---|---|---|---|---|
| Supervised learning | D-MPNN | 2.082 (0.082) | 1.050 (0.008) | 0.683 (0.016) |
| DimeNet | 2.094 (0.118) | 0.878 (0.023) | 0.727 (0.019) | |
| DLF-MFF | 1.849 | 0.747 | 0.772 | |
| Self-Supervised learning | KEMPNN | 1.188 (0.158) | 0.703 (0.024) | 0.563 (0.011) |
| MolPMoFiT | 1.197 (0.127) | - | 0.565 (0.037) | |
| MolBERT | 1.523 (0.660) | 0.552 (0.070) | 0.602 (0.010) | |
| FP-BERT | 1.140 (0.006) | 0.670 (0.004) | 0.660 (0.002) | |
| SMILES Transformer | 1.650 | 0.720 | 0.921 | |
| TGSS | 0.960 (0.065) | 0.645 (0.075) | 0.652 (0.009) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Xia, L.; Pan, S.; Li, Z. Triple Generative Self-Supervised Learning Method for Molecular Property Prediction. Int. J. Mol. Sci. 2024, 25, 3794. https://doi.org/10.3390/ijms25073794
Xu L, Xia L, Pan S, Li Z. Triple Generative Self-Supervised Learning Method for Molecular Property Prediction. International Journal of Molecular Sciences. 2024; 25(7):3794. https://doi.org/10.3390/ijms25073794
Chicago/Turabian StyleXu, Lei, Leiming Xia, Shourun Pan, and Zhen Li. 2024. "Triple Generative Self-Supervised Learning Method for Molecular Property Prediction" International Journal of Molecular Sciences 25, no. 7: 3794. https://doi.org/10.3390/ijms25073794
APA StyleXu, L., Xia, L., Pan, S., & Li, Z. (2024). Triple Generative Self-Supervised Learning Method for Molecular Property Prediction. International Journal of Molecular Sciences, 25(7), 3794. https://doi.org/10.3390/ijms25073794