
The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer


Abstract
1. Introduction
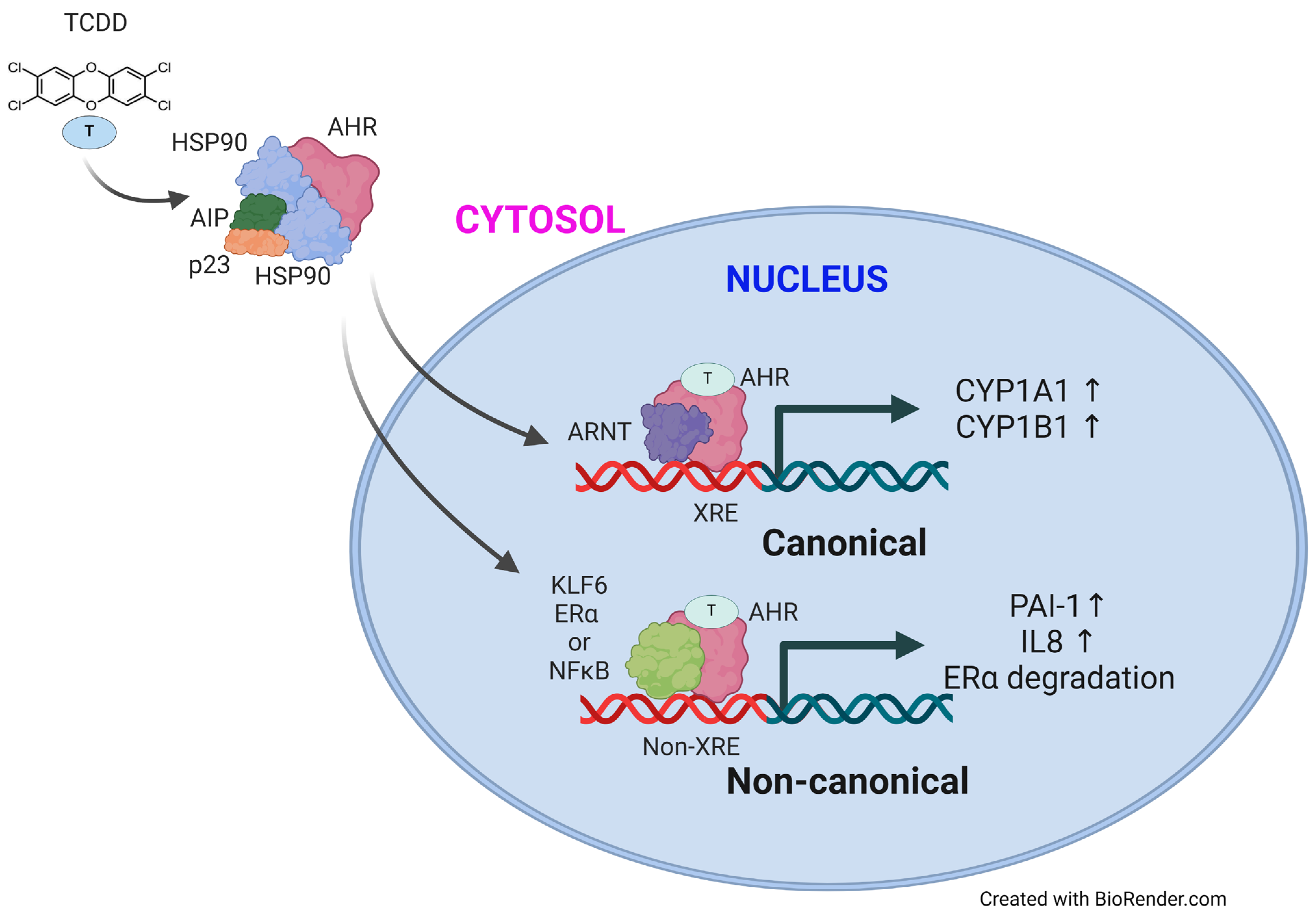
2. Aryl Hydrocarbon Receptor (AHR)
3. AHR Structure and Its Interactions with Various Ligands
4. EDCs from Environmental Pollutants and AHR
4.1. Dioxins and Dioxin-like Compounds
4.2. Polycyclic Aromatic Hydrocarbons (PAHs)
4.3. Hexachlorobenzene (HCB)
4.4. Bisphenol A (BPA)
4.5. Heavy Metals
5. Roles of EDC–AHR Interactions in the Pathogenesis of Pancreatic Diseases and Cancer
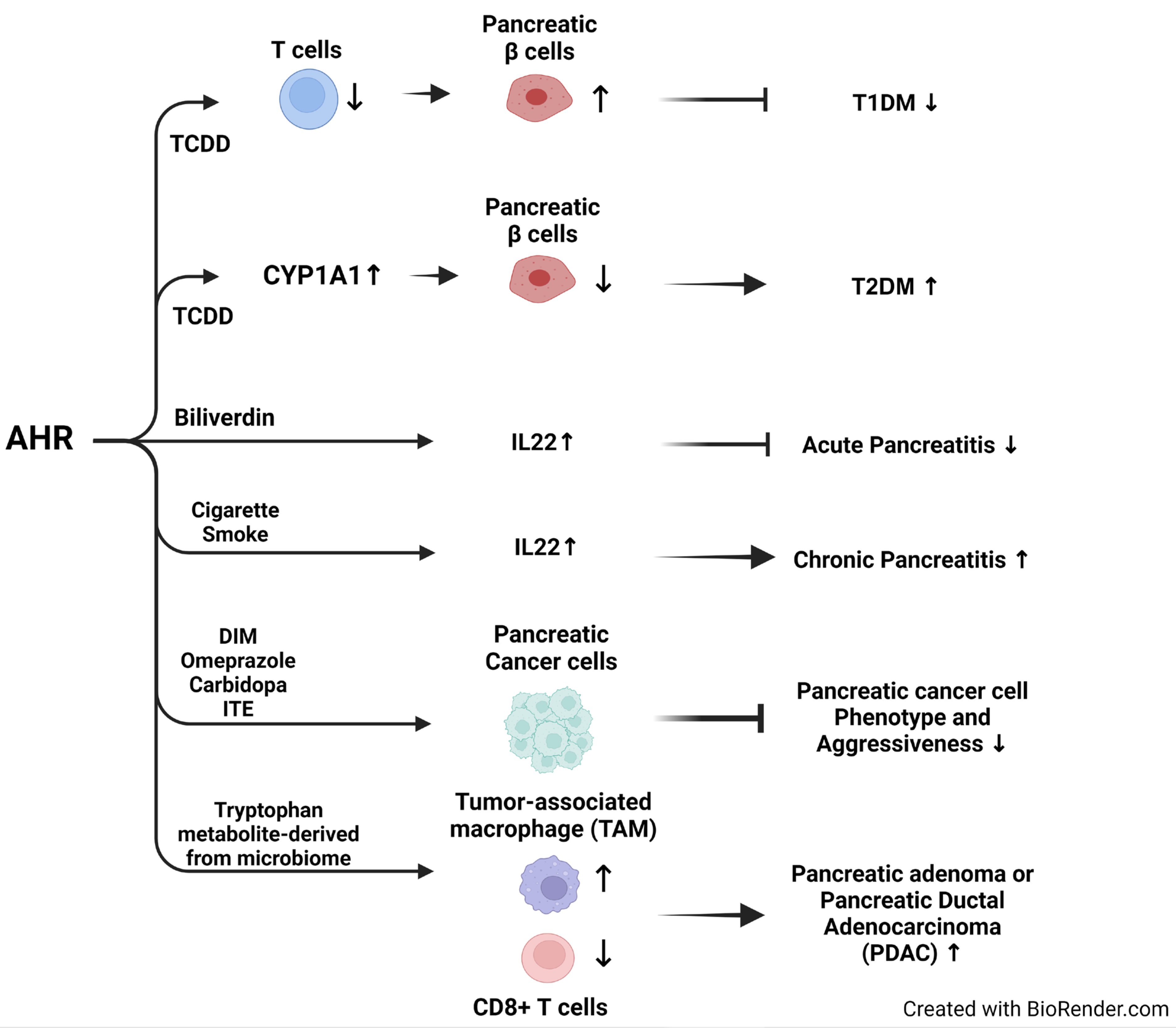
5.1. Role of EDC-Regulated AHR in Diabetes Mellitus
5.1.1. Type 1 Diabetes Mellitus (T1DM)
5.1.2. Type 2 Diabetes Mellitus (T2DM)
5.1.3. Role of EDC-Regulated AHR in Pancreatitis
5.1.4. Role of EDC-Regulated AHR in Pancreatic Cancer
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AHR | aryl hydrocarbon receptor |
| AHRR | aryl hydrocarbon receptor repressor |
| bHLH | helix-loop-helix |
| BPA | bisphenol A |
| DIM | diindolymethane |
| EDC | endocrine-disrupting chemical |
| HCB | hexachlorobenzene |
| LBP | ligand-binding pocket |
| PAHs | polycyclic aromatic hydrocarbons |
| PCBs | polychlorinated biphenyls |
| PCDF | polychlorinated dibenzofurans |
| PM | particulate matter |
| PDAC | pancreatic ductal adenocarcinoma |
| POPs | persistent organic pollutants |
| T1DM | type I diabetes mellitus |
| T2DM | type II diabetes mellitus |
| TAM | tumor-associated macrophages |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| 2,4-D | 2,4-Dichlorophenoxyacetic acid |
References
- Kahn, L.G.; Philippat, C.; Nakayama, S.F.; Slama, R.; Trasande, L. Endocrine-disrupting chemicals: Implications for human health. Lancet Diabetes Endocrinol. 2020, 8, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Kumar, S.; Kumar, V.; Lee, Y.M.; Kim, Y.S.; Kumar, V. Bisphenols as a Legacy Pollutant, and Their Effects on Organ Vulnerability. Int. J. Environ. Res. Public Health 2019, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, H. Phthalates and Their Impacts on Human Health. Healthcare 2021, 9, 603. [Google Scholar] [CrossRef]
- Kimbrough, R.D. Toxicity and health effects of selected organotin compounds: A review. Environ. Health Perspect. 1976, 14, 51–56. [Google Scholar] [CrossRef]
- Mnif, W.; Hassine, A.I.; Bouaziz, A.; Bartegi, A.; Thomas, O.; Roig, B. Effect of endocrine disruptor pesticides: A review. Int. J. Environ. Res. Public Health 2011, 8, 2265–2303. [Google Scholar] [CrossRef]
- Mukerjee, D. Health impact of polychlorinated dibenzo-p-dioxins: A critical review. J. Air Waste Manag. Assoc. 1998, 48, 157–165. [Google Scholar] [CrossRef]
- White, S.S.; Birnbaum, L.S. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 197–211. [Google Scholar] [CrossRef]
- Patel, A.B.; Shaikh, S.; Jain, K.R.; Desai, C.; Madamwar, D. Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and Remediation Approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef]
- Dishaw, L.V.; Macaulay, L.J.; Roberts, S.C.; Stapleton, H.M. Exposures, mechanisms, and impacts of endocrine-active flame retardants. Curr. Opin. Pharmacol. 2014, 19, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Acir, I.H.; Guenther, K. Endocrine-disrupting metabolites of alkylphenol ethoxylates—A critical review of analytical methods, environmental occurrences, toxicity, and regulation. Sci. Total Environ. 2018, 635, 1530–1546. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Terekeci, H.; Sandal, S.; Kelestimur, F. Endocrine disrupting chemicals: Exposure, effects on human health, mechanism of action, models for testing and strategies for prevention. Rev. Endocr. Metab. Disord. 2020, 21, 127–147. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, J.; Zheng, M.; Pan, K.; Yang, L.; Ma, L.; Wang, C.; Yu, J. Thyroid dysfunction caused by exposure to environmental endocrine disruptors and the underlying mechanism: A review. Chem. Biol. Interact. 2024, 391, 110909. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nat. Rev. Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. Exp. Suppl. 2012, 101, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Sall, M.L.; Diaw, A.K.D.; Gningue-Sall, D.; Efremova Aaron, S.; Aaron, J.J. Toxic heavy metals: Impact on the environment and human health, and treatment with conducting organic polymers, a review. Environ. Sci. Pollut. Res. Int. 2020, 27, 29927–29942. [Google Scholar] [CrossRef]
- Vogel, C.F.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox. Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, H.; Liao, C.; Diao, X.; Zhen, J.; Chen, L.; Xue, Q. Toxicology mechanism of the persistent organic pollutants (POPs) in fish through AhR pathway. Toxicol. Mech. Methods 2010, 20, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Qu, Y.; Wu, H.; Liao, C.; Zheng, J.; Diao, X.; Xue, Q. Molecular phylogenies and evolutionary behavior of AhR (aryl hydrocarbon receptor) pathway genes in aquatic animals: Implications for the toxicology mechanism of some persistent organic pollutants (POPs). Chemosphere 2010, 78, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Doan, T.Q.; Connolly, L.; Igout, A.; Nott, K.; Muller, M.; Scippo, M.L. In vitro profiling of the potential endocrine disrupting activities affecting steroid and aryl hydrocarbon receptors of compounds and mixtures prevalent in human drinking water resources. Chemosphere 2020, 258, 127332. [Google Scholar] [CrossRef]
- Poland, A.; Glover, E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: A potent inducer of -aminolevulinic acid synthetase. Science 1973, 179, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.H.; Mocarelli, P. Human health effects after exposure to 2,3,7,8-TCDD. Food Addit. Contam. 2000, 17, 303–316. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Population Health and Public Health Practice; Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides (Eleventh Biennial Update). Veterans and Agent Orange: Update 11 (2018); National Academies Press: Washington, DC, USA, 2018. [Google Scholar]
- Poland, A.; Kende, A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: Environmental contaminant and molecular probe. Fed. Proc. 1976, 35, 2404–2411. [Google Scholar]
- Mandal, P.K. Dioxin: A review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. B 2005, 175, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Holfelder, P.; Prentzell, M.T.; Trump, S. The complex biology of aryl hydrocarbon receptor activation in cancer and beyond. Biochem. Pharmacol. 2023, 216, 115798. [Google Scholar] [CrossRef]
- Puga, A.; Ma, C.; Marlowe, J.L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009, 77, 713–722. [Google Scholar] [CrossRef]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- McGuire, J.; Whitelaw, M.L.; Pongratz, I.; Gustafsson, J.A.; Poellinger, L. A cellular factor stimulates ligand-dependent release of hsp90 from the basic helix-loop-helix dioxin receptor. Mol. Cell. Biol. 1994, 14, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.R.; Poland, A. Binding of aryl hydrocarbon receptor (AhR) to AhR-interacting protein. The role of hsp90. J. Biol. Chem. 2000, 275, 36407–36414. [Google Scholar] [CrossRef]
- Nukaya, M.; Lin, B.C.; Glover, E.; Moran, S.M.; Kennedy, G.D.; Bradfield, C.A. The aryl hydrocarbon receptor-interacting protein (AIP) is required for dioxin-induced hepatotoxicity but not for the induction of the Cyp1a1 and Cyp1a2 genes. J. Biol. Chem. 2010, 285, 35599–35605. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.K.; Perdew, G.H. Characterization of the AhR-hsp90-XAP2 core complex and the role of the immunophilin-related protein XAP2 in AhR stabilization. Biochemistry 1999, 38, 8907–8917. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, J.; Yamakawa, K.; Tonda, E.; Nishitani, S.; Yoshinari, K.; Degawa, M.; Abe, I.; Noguchi, H.; Miwa, M. The induction of human UDP-glucuronosyltransferase 1A1 mediated through a distal enhancer module by flavonoids and xenobiotics. Biochem. Pharmacol. 2004, 67, 989–1000. [Google Scholar] [CrossRef]
- Auyeung, D.J.; Kessler, F.K.; Ritter, J.K. Mechanism of rat UDP-glucuronosyltransferase 1A6 induction by oltipraz: Evidence for a contribution of the Aryl hydrocarbon receptor pathway. Mol. Pharmacol. 2003, 63, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Münzel, P.A.; Schmohl, S.; Buckler, F.; Jaehrling, J.; Raschko, F.T.; Köhle, C.; Bock, K.W. Contribution of the Ah receptor to the phenolic antioxidant-mediated expression of human and rat UDP-glucuronosyltransferase UGT1A6 in Caco-2 and rat hepatoma 5L cells. Biochem. Pharmacol. 2003, 66, 841–847. [Google Scholar] [CrossRef]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef]
- Wormke, M.; Stoner, M.; Saville, B.; Walker, K.; Abdelrahim, M.; Burghardt, R.; Safe, S. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol. Cell. Biol. 2003, 23, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [PubMed]
- Oesch-Bartlomowicz, B.; Huelster, A.; Wiss, O.; Antoniou-Lipfert, P.; Dietrich, C.; Arand, M.; Weiss, C.; Bockamp, E.; Oesch, F. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: Divergent signaling pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 9218–9223. [Google Scholar] [CrossRef] [PubMed]
- Möglich, A.; Ayers, R.A.; Moffat, K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.A.; Denison, M.S. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef]
- Gruszczyk, J.; Grandvuillemin, L.; Lai-Kee-Him, J.; Paloni, M.; Savva, C.G.; Germain, P.; Grimaldi, M.; Boulahtouf, A.; Kwong, H.S.; Bous, J.; et al. Cryo-EM structure of the agonist-bound Hsp90-XAP2-AHR cytosolic complex. Nat. Commun. 2022, 13, 7010. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Ohe, N.; Suzuki, M.; Mimura, J.; Sogawa, K.; Ikawa, S.; Fujii-Kuriyama, Y. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J. Biol. Chem. 1994, 269, 27337–27343. [Google Scholar] [CrossRef]
- Poland, A.; Glover, E. Characterization and strain distribution pattern of the murine Ah receptor specified by the Ahd and Ahb-3 alleles. Mol. Pharmacol. 1990, 38, 306–312. [Google Scholar]
- Moriguchi, T.; Motohashi, H.; Hosoya, T.; Nakajima, O.; Takahashi, S.; Ohsako, S.; Aoki, Y.; Nishimura, N.; Tohyama, C.; Fujii-Kuriyama, Y.; et al. Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Qu, L.; Li, J.; Zhang, Y.; Jiang, L.; Wei, H.; Guo, M.; Chen, X.; Chen, Y. Structural insight into the ligand binding mechanism of aryl hydrocarbon receptor. Nat. Commun. 2022, 13, 6234. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Pandini, A.; Nagy, S.R.; Baldwin, E.P.; Bonati, L. Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 2002, 141, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Nukaya, M.; Satyshur, K.A.; Jiang, L.; Stanevich, V.; Korkmaz, E.N.; Burdette, L.; Kennedy, G.D.; Cui, Q.; Bradfield, C.A. Identification of the Ah-receptor structural determinants for ligand preferences. Toxicol. Sci. 2012, 129, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Murray, I.A.; Chiaro, C.R.; Perdew, G.H. Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol. Pharmacol. 2009, 75, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, R.; Breivik, K.; Dachs, J.; Muir, D. Global fate of POPs: Current and future research directions. Environ. Pollut. 2007, 150, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.A. Persistent organic pollutants (POPs): A global issue, a global challenge. Environ. Sci. Pollut. Res. Int. 2017, 24, 4223–4227. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. Aryl hydrocarbon receptor (AHR)-mediated inflammation and resolution: Non-genomic and genomic signaling. Biochem. Pharmacol. 2020, 182, 114220. [Google Scholar] [CrossRef]
- Piwarski, S.A.; Salisbury, T.B. The effects of environmental aryl hydrocarbon receptor ligands on signaling and cell metabolism in cancer. Biochem. Pharmacol. 2023, 216, 115771. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, H.Q.; Li, Y.; Zhou, M.; Zhou, Z.; Wang, R.; Hahn, M.E.; Zhao, B. The aryl hydrocarbon receptor: A predominant mediator for the toxicity of emerging dioxin-like compounds. J. Hazard. Mater. 2022, 426, 128084. [Google Scholar] [CrossRef] [PubMed]
- Hites, R.A. Dioxins: An overview and history. Environ. Sci. Technol. 2011, 45, 16–20. [Google Scholar] [CrossRef]
- Behnisch, P.A.; Hosoe, K.; Sakai, S. Bioanalytical screening methods for dioxins and dioxin-like compounds a review of bioassay/biomarker technology. Environ. Int. 2001, 27, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Pirkle, J.L.; Wolfe, W.H.; Patterson, D.G.; Needham, L.L.; Michalek, J.E.; Miner, J.C.; Peterson, M.R.; Phillips, D.L. Estimates of the half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Vietnam Veterans of Operation Ranch Hand. J. Toxicol. Environ. Health 1989, 27, 165–171. [Google Scholar] [CrossRef]
- Kerger, B.D.; Leung, H.W.; Scott, P.; Paustenbach, D.J.; Needham, L.L.; Patterson, D.G., Jr.; Gerthoux, P.M.; Mocarelli, P. Age- and concentration-dependent elimination half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Seveso children. Environ. Health Perspect. 2006, 114, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Saurat, J.H.; Kaya, G.; Saxer-Sekulic, N.; Pardo, B.; Becker, M.; Fontao, L.; Mottu, F.; Carraux, P.; Pham, X.C.; Barde, C.; et al. The cutaneous lesions of dioxin exposure: Lessons from the poisoning of Victor Yushchenko. Toxicol. Sci. 2012, 125, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Pelclová, D.; Urban, P.; Preiss, J.; Lukás, E.; Fenclová, Z.; Navrátil, T.; Dubská, Z.; Senholdová, Z. Adverse health effects in humans exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Rev. Environ. Health 2006, 21, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Tsuji, G. Chloracne and Hyperpigmentation Caused by Exposure to Hazardous Aryl Hydrocarbon Receptor Ligands. Int. J. Environ. Res. Public Health 2019, 16, 4864. [Google Scholar] [CrossRef]
- Cole, P.; Trichopoulos, D.; Pastides, H.; Starr, T.; Mandel, J.S. Dioxin and cancer: A critical review. Regul. Toxicol. Pharmacol. 2003, 38, 378–388. [Google Scholar] [CrossRef]
- Steenland, K.; Bertazzi, P.; Baccarelli, A.; Kogevinas, M. Dioxin revisited: Developments since the 1997 IARC classification of dioxin as a human carcinogen. Environ. Health Perspect. 2004, 112, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H. Polycyclic aromatic hydrocarbons in the diet. Mutat. Res. 1999, 443, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, G.; Fadda, E.; Marzia, V. Polycyclic aromatic hydrocarbons and cancer in man. Environ. Health Perspect. 1996, 104, 1166–1170. [Google Scholar] [CrossRef]
- Li, Z.; Romanoff, L.; Bartell, S.; Pittman, E.N.; Trinidad, D.A.; McClean, M.; Webster, T.F.; Sjödin, A. Excretion profiles and half-lives of ten urinary polycyclic aromatic hydrocarbon metabolites after dietary exposure. Chem. Res. Toxicol. 2012, 25, 1452–1461. [Google Scholar] [CrossRef]
- Motorykin, O.; Santiago-Delgado, L.; Rohlman, D.; Schrlau, J.E.; Harper, B.; Harris, S.; Harding, A.; Kile, M.L.; Massey Simonich, S.L. Metabolism and excretion rates of parent and hydroxy-PAHs in urine collected after consumption of traditionally smoked salmon for Native American volunteers. Sci. Total Environ. 2015, 514, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Cecinato, A.; Bacaloni, A.; Romagnoli, P.; Perilli, M.; Balducci, C. Molecular signatures of organic particulates as tracers of emission sources. Environ. Sci. Pollut. Res. Int. 2022, 29, 65904–65923. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, H.; Zhang, X.; Xing, W.; Wang, Y.; Bai, P.; Zhang, L.; Hayakawa, K.; Toriba, A.; Tang, N. Exposure to Atmospheric Particulate Matter-Bound Polycyclic Aromatic Hydrocarbons and Their Health Effects: A Review. Int. J. Environ. Res. Public Health 2021, 18, 2177. [Google Scholar] [CrossRef]
- Møller, M.; Alfheim, I. Mutagenicity and PAH-analysis of airborne particulate matter. Atmos. Environ. 1980, 14, 83–88. [Google Scholar] [CrossRef]
- Sjaastad, A.K.; Jørgensen, R.B.; Svendsen, K. Exposure to polycyclic aromatic hydrocarbons (PAHs), mutagenic aldehydes and particulate matter during pan frying of beefsteak. Occup. Environ. Med. 2010, 67, 228–232. [Google Scholar] [CrossRef]
- Anderson, K.E.; Kadlubar, F.F.; Kulldorff, M.; Harnack, L.; Gross, M.; Lang, N.P.; Barber, C.; Rothman, N.; Sinha, R. Dietary intake of heterocyclic amines and benzo(a)pyrene: Associations with pancreatic cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2261–2265. [Google Scholar] [CrossRef]
- Andreotti, G.; Silverman, D.T. Occupational risk factors and pancreatic cancer: A review of recent findings. Mol. Carcinog. 2012, 51, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Alguacil, J.; Porta, M.; Kauppinen, T.; Malats, N.; Kogevinas, M.; Carrato, A. PANKRAS II Study Group. Occupational exposure to dyes, metals, polycyclic aromatic hydrocarbons and other agents and K-ras activation in human exocrine pancreatic cancer. Int. J. Cancer 2003, 107, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Starek-Świechowicz, B.; Budziszewska, B.; Starek, A. Hexachlorobenzene as a persistent organic pollutant: Toxicity and molecular mechanism of action. Pharmacol. Rep. 2017, 69, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Gocmen, A.; Peters, H.A.; Cripps, D.J.; Bryan, G.T.; Morris, C.R. Hexachlorobenzene episode in Turkey. Biomed. Environ. Sci. 1989, 2, 36–43. [Google Scholar] [PubMed]
- Miret, N.V.; Pontillo, C.A.; Zárate, L.V.; Kleiman de Pisarev, D.; Cocca, C.; Randi, A.S. Impact of endocrine disruptor hexachlorobenzene on the mammary gland and breast cancer: The story thus far. Environ. Res. 2019, 173, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Hoppin, J.A.; Tolbert, P.E.; Holly, E.A.; Brock, J.W.; Korrick, S.A.; Altshul, L.M.; Zhang, R.H.; Bracci, P.M.; Burse, V.W.; Needham, L.L. Pancreatic cancer and serum organochlorine levels. Cancer Epidemiol. Biomark. Prev. 2000, 9, 199–205. [Google Scholar]
- Bosch de Basea, M.; Porta, M.; Alguacil, J.; Puigdomènech, E.; Gasull, M.; Garrido, J.A.; López, T.; PANKRAS II Study Group. Relationships between occupational history and serum concentrations of organochlorine compounds in exocrine pancreatic cancer. Occup. Environ. Med. 2011, 68, 332–338. [Google Scholar] [CrossRef]
- Porta, M.; Bosch de Basea, M.; Benavides, F.G.; López, T.; Fernandez, E.; Marco, E.; Alguacil, J.; Grimalt, J.O.; Puigdomènech, E.; PANKRAS II Study Group. Differences in serum concentrations of organochlorine compounds by occupational social class in pancreatic cancer. Environ. Res. 2008, 108, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Abraham, A.; Chakraborty, P. A review on sources and health impacts of bisphenol, A. Rev. Environ. Health 2020, 35, 201–210. [Google Scholar] [CrossRef]
- Teeguarden, J.G.; Waechter, J.M., Jr.; Clewell, H.J., 3rd; Covington, T.R.; Barton, H.A. Evaluation of oral and intravenous route pharmacokinetics, plasma protein binding, and uterine tissue dose metrics of bisphenol A: A physiologically based pharmacokinetic approach. Toxicol. Sci. 2005, 85, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Völkel, W.; Bittner, N.; Dekant, W. Quantitation of bisphenol A and bisphenol A glucuronide in biological samples by high performance liquid chromatography-tandem mass spectrometry. Drug Metab. Dispos. 2005, 33, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, F.; Aquilina, A.; Vassallo, J.; Pace, N.P. Bisphenol A and Type 2 Diabetes Mellitus: A Review of Epidemiologic, Functional, and Early Life Factors. Int. J. Environ. Res. Public Health 2021, 18, 716. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lim, J.E.; Choi, Y.; Jee, S.H. Bisphenol A exposure and type 2 diabetes mellitus risk: A meta-analysis. BMC Endocr. Disord. 2018, 18, 81. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pinna, J.; Marroqui, L.; Hmadcha, A.; Lopez-Beas, J.; Soriano, S.; Villar-Pazos, S.; Alonso-Magdalena, P.; Dos Santos, R.S.; Quesada, I.; Martin, F.; et al. Oestrogen receptor β mediates the actions of bisphenol-A on ion channel expression in mouse pancreatic beta cells. Diabetologia 2019, 62, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Boronat-Belda, T.; Ferrero, H.; Al-Abdulla, R.; Quesada, I.; Gustafsson, J.A.; Nadal, Á.; Alonso-Magdalena, P. Bisphenol-A exposure during pregnancy alters pancreatic β-cell division and mass in male mice offspring: A role for ERβ. Food Chem. Toxicol. 2020, 145, 111681. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Ropero, A.B.; Carrera, M.P.; Cederroth, C.R.; Baquié, M.; Gauthier, B.R.; Nef, S.; Stefani, E.; Nadal, A. Pancreatic insulin content regulation by the estrogen receptor ER alpha. PLoS ONE 2008, 3, e2069. [Google Scholar] [CrossRef]
- Soriano, S.; Alonso-Magdalena, P.; García-Arévalo, M.; Novials, A.; Muhammed, S.J.; Salehi, A.; Gustafsson, J.A.; Quesada, I.; Nadal, A. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: Role of estrogen receptor β. PLoS ONE 2012, 7, e31109. [Google Scholar] [CrossRef]
- Ziv-Gal, A.; Craig, Z.R.; Wang, W.; Flaws, J.A. Bisphenol A inhibits cultured mouse ovarian follicle growth partially via the aryl hydrocarbon receptor signaling pathway. Reprod. Toxicol. 2013, 42, 58–67. [Google Scholar] [CrossRef]
- Nishizawa, H.; Imanishi, S.; Manabe, N. Effects of exposure in utero to bisphenol a on the expression of aryl hydrocarbon receptor, related factors, and xenobiotic metabolizing enzymes in murine embryos. J. Reprod. Dev. 2005, 51, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Donini, C.F.; El Helou, M.; Wierinckx, A.; Győrffy, B.; Aires, S.; Escande, A.; Croze, S.; Clezardin, P.; Lachuer, J.; Diab-Assaf, M.; et al. Long-Term Exposure of Early-Transformed Human Mammary Cells to Low Doses of Benzo[a]pyrene and/or Bisphenol A Enhances Their Cancerous Phenotype via an AhR/GPR30 Interplay. Front. Oncol. 2020, 10, 712. [Google Scholar] [CrossRef]
- Banerjee, O.; Singh, S.; Prasad, S.K.; Bhattacharjee, A.; Seal, T.; Mandal, J.; Sinha, S.; Banerjee, A.; Maji, B.K.; Mukherjee, S. Exploring aryl hydrocarbon receptor (AhR) as a target for Bisphenol-A (BPA)-induced pancreatic islet toxicity and impaired glucose homeostasis: Protective efficacy of ethanol extract of Centella asiatica. Toxicology 2023, 500, 153693. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Witkowska, D.; Słowik, J.; Chilicka, K. Heavy Metals and Human Health: Possible Exposure Pathways and the Competition for Protein Binding Sites. Molecules 2021, 26, 6060. [Google Scholar] [CrossRef]
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176. [Google Scholar] [CrossRef]
- Jan, A.T.; Azam, M.; Siddiqui, K.; Ali, A.; Choi, I.; Haq, Q.M. Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants. Int. J. Mol. Sci. 2015, 16, 29592–29630. [Google Scholar] [CrossRef] [PubMed]
- Elbekai, R.H.; El-Kadi, A.O. Modulation of aryl hydrocarbon receptor-regulated gene expression by arsenite, cadmium, and chromium. Toxicology 2004, 202, 249–269. [Google Scholar] [CrossRef]
- Kann, S.; Huang, M.Y.; Estes, C.; Reichard, J.F.; Sartor, M.A.; Xia, Y.; Puga, A. Arsenite-induced aryl hydrocarbon receptor nuclear translocation results in additive induction of phase I genes and synergistic induction of phase II genes. Mol. Pharmacol. 2005, 68, 336–346. [Google Scholar] [CrossRef]
- Albores, A.; Cebrián, M.E.; Bach, P.H.; Connelly, J.C.; Hinton, R.H.; Bridges, J.W. Sodium arsenite induced alterations in bilirubin excretion and heme metabolism. J. Biochem. Toxicol. 1989, 4, 73–78. [Google Scholar] [CrossRef]
- Kou, Z.; Yang, R.; Lee, E.; Cuddapah, S.; Choi, B.H.; Dai, W. Oxidative stress modulates expression of immune checkpoint genes via activation of AhR signaling. Toxicol. Appl. Pharmacol. 2022, 457, 116314. [Google Scholar] [CrossRef] [PubMed]
- Anwar-Mohamed, A.; Elbekai, R.H.; El-Kadi, A.O. Regulation of CYP1A1 by heavy metals and consequences for drug metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, P.; Xie, J.; Lin, H.P.; Kumagai, K.; Harkema, J.; Yang, C. Arsenic and benzo[a]pyrene co-exposure acts synergistically in inducing cancer stem cell-like property and tumorigenesis by epigenetically down-regulating SOCS3 expression. Environ Int. 2020, 137, 105560. [Google Scholar] [CrossRef] [PubMed]
- Antwi, S.O.; Eckert, E.C.; Sabaque, C.V.; Leof, E.R.; Hawthorne, K.M.; Bamlet, W.R.; Chaffee, K.G.; Oberg, A.L.; Petersen, G.M. Exposure to environmental chemicals and heavy metals, and risk of pancreatic cancer. Cancer Causes Control 2015, 26, 1583–1591. [Google Scholar] [CrossRef]
- Djordjevic, V.R.; Wallace, D.R.; Schweitzer, A.; Boricic, N.; Knezevic, D.; Matic, S.; Grubor, N.; Kerkez, M.; Radenkovic, D.; Bulat, Z.; et al. Environmental cadmium exposure and pancreatic cancer: Evidence from case control, animal and in vitro studies. Environ. Int. 2019, 128, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, P.E.; Hentz, J.G.; Gordon, G.; Morgan, J.L.; Raimondo, M.; Anbar, A.D.; Miller, L.J. Distinctive heavy metal composition of pancreatic juice in patients with pancreatic carcinoma. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2656–2663. [Google Scholar] [CrossRef] [PubMed]
- Pothuraju, R.; Rachagani, S.; Junker, W.M.; Chaudhary, S.; Saraswathi, V.; Kaur, S.; Batra, S.K. Pancreatic cancer associated with obesity and diabetes: An alternative approach for its targeting. J. Exp. Clin. Cancer Res. 2018, 37, 319. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Claude Mbanya, J.; et al. Erratum to “IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045” [Diabetes Res. Clin. Pract. 183 (2022) 109119]. Diabetes Res. Clin. Pract. 2023, 204, 110945. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.M.; Dong, X.W.; Han, X.; Ling, Z.; Lu, G.T.; Sun, Y.Y.; Yin, X.D. Pancreatitis and Pancreatic Cancer Risk. Technol. Cancer Res. Treat. 2023, 22, 15330338231164875. [Google Scholar] [CrossRef]
- Bodin, J.; Stene, L.C.; Nygaard, U.C. Can exposure to environmental chemicals increase the risk of diabetes type 1 development? Biomed. Res. Int. 2015, 2015, 208947. [Google Scholar] [CrossRef]
- Lim, C.C.; Thurston, G.D. Air Pollution, Oxidative Stress, and Diabetes: A Life Course Epidemiologic Perspective. Curr. Diabetes Rep. 2019, 19, 58. [Google Scholar] [CrossRef]
- Hao, N.; Whitelaw, M.L. The emerging roles of AhR in physiology and immunity. Biochem. Pharmacol. 2013, 86, 561–570. [Google Scholar] [CrossRef]
- Kerkvliet, N.I.; Steppan, L.B.; Vorachek, W.; Oda, S.; Farrer, D.; Wong, C.P.; Pham, D.; Mourich, D.V. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Wang, X.; Rohlman, D.; Punj, S.; Löhr, C.V.; Newman, M.T.; Kolluri, S.K.; Kerkvliet, N.I. Activation of the Aryl Hydrocarbon Receptor by 10-Cl-BBQ Prevents Insulitis and Effector T Cell Development Independently of Foxp3+ Regulatory T Cells in Nonobese Diabetic Mice. J. Immunol. 2016, 196, 264–273. [Google Scholar] [CrossRef]
- Kelishadi, R.; Hovsepian, S.; Amin, M.M.; Mozafarian, N.; Sedaghat, S.; Hashemipour, M. Association of Polycyclic Aromatic Hydrocarbons Urine Metabolites with Type 1 Diabetes. J. Diabetes Res. 2023, 2023, 6692810. [Google Scholar] [CrossRef]
- Ou, K.; Song, J.; Zhang, S.; Fang, L.; Lin, L.; Lan, M.; Chen, M.; Wang, C. Prenatal exposure to a mixture of PAHs causes the dysfunction of islet cells in adult male mice: Association with type 1 diabetes mellitus. Ecotoxicol. Environ. Saf. 2022, 239, 113695. [Google Scholar] [CrossRef]
- Tosirisuk, N.; Sakorn, N.; Jantarat, C.; Nosoongnoen, W.; Aroonpakmongkol, S.; Supornsilchai, V. Increased bisphenol A levels in Thai children and adolescents with type 1 diabetes mellitus. Pediatr. Int. 2022, 64, e14944. [Google Scholar] [CrossRef]
- Bodin, J.; Bølling, A.K.; Becher, R.; Kuper, F.; Løvik, M.; Nygaard, U.C. Transmaternal bisphenol A exposure accelerates diabetes type 1 development in NOD mice. Toxicol. Sci. 2014, 137, 311–323. [Google Scholar] [CrossRef]
- Cetkovic-Cvrlje, M.; Thinamany, S.; Bruner, K.A. Bisphenol A (BPA) aggravates multiple low-dose streptozotocin-induced Type 1 diabetes in C57BL/6 mice. J. Immunotoxicol. 2017, 14, 160–168. [Google Scholar] [CrossRef]
- Bodin, J.; Kocbach Bølling, A.; Wendt, A.; Eliasson, L.; Becher, R.; Kuper, F.; Løvik, M.; Nygaard, U.C. Exposure to bisphenol, A. but not phthalates, increases spontaneous diabetes type 1 development in NOD mice. Toxicol. Rep. 2015, 2, 99–110. [Google Scholar] [CrossRef]
- Chafe, R.; Aslanov, R.; Sarkar, A.; Gregory, P.; Comeau, A.; Newhook, L.A. Association of type 1 diabetes and concentrations of drinking water components in Newfoundland and Labrador, Canada. BMJ Open Diabetes Res. Care 2018, 6, e000466. [Google Scholar] [CrossRef]
- Grau-Pérez, M.; Kuo, C.C.; Spratlen, M.; Thayer, K.A.; Mendez, M.A.; Hamman, R.F.; Dabelea, D.; Adgate, J.L.; Knowler, W.C.; Bell, R.A.; et al. The Association of Arsenic Exposure and Metabolism With Type 1 and Type 2 Diabetes in Youth: The SEARCH Case-Control Study. Diabetes Care 2017, 40, 46–53. [Google Scholar] [CrossRef]
- Dávila-Esqueda, M.E.; Morales, J.M.; Jiménez-Capdeville, M.E.; De la Cruz, E.; Falcón-Escobedo, R.; Chi-Ahumada, E.; Martin-Pérez, S. Low-level subchronic arsenic exposure from prenatal developmental stages to adult life results in an impaired glucose homeostasis. Exp. Clin. Endocrinol. Diabetes 2011, 119, 613–617. [Google Scholar] [CrossRef]
- Henriksen, G.L.; Ketchum, N.S.; Michalek, J.E.; Swaby, J.A. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology 1997, 8, 252–258. [Google Scholar] [CrossRef]
- Remillard, R.B.; Bunce, N.J. Linking dioxins to diabetes: Epidemiology and biologic plausibility. Environ. Health Perspect. 2002, 110, 853–858. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, I.K.; Song, K.; Steffes, M.; Toscano, W.; Baker, B.A.; Jacobs, D.R., Jr. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: Results from the National Health and Examination Survey 1999-2002. Diabetes Care 2006, 29, 1638–1644. [Google Scholar] [CrossRef]
- Novelli, M.; Piaggi, S.; De Tata, V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets. Toxicol. Lett. 2005, 156, 307–314. [Google Scholar] [CrossRef]
- Piaggi, S.; Novelli, M.; Martino, L.; Masini, M.; Raggi, C.; Orciuolo, E.; Masiello, P.; Casini, A.; De Tata, V. Cell death and impairment of glucose-stimulated insulin secretion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the beta-cell line INS-1E. Toxicol. Appl. Pharmacol. 2007, 220, 333–340. [Google Scholar] [CrossRef]
- Kubi, J.A.; Chen, A.C.H.; Fong, S.W.; Lai, K.P.; Wong, C.K.C.; Yeung, W.S.B.; Lee, K.F.; Lee, Y.L. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the differentiation of embryonic stem cells towards pancreatic lineage and pancreatic beta cell function. Environ. Int. 2019, 130, 104885. [Google Scholar] [CrossRef]
- Novelli, M.; Beffy, P.; Masini, M.; Vantaggiato, C.; Martino, L.; Marselli, L.; Marchetti, P.; De Tata, V. Selective beta-cell toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin on isolated pancreatic islets. Chemosphere 2021, 265, 129103. [Google Scholar] [CrossRef]
- Ibrahim, M.; MacFarlane, E.M.; Matteo, G.; Hayek, M.P.; Rick, K.R.C.; Farokhi, S.; Copley, C.M.; O’Dwyer, S.; Bruin, J.E. Functional cytochrome P450 1A enzymes are induced in mouse and human islets following pollutant exposure. Diabetologia 2020, 63, 162–178. [Google Scholar] [CrossRef]
- Kuzgun, G.; Başaran, R.; Arıoğlu İnan, E.; Can Eke, B. Effects of insulin treatment on hepatic CYP1A1 and CYP2E1 activities and lipid peroxidation levels in streptozotocin-induced diabetic rats. J. Diabetes Metab. Disord. 2020, 19, 1157–1164. [Google Scholar] [CrossRef]
- Wang, C.; Xu, C.X.; Krager, S.L.; Bottum, K.M.; Liao, D.F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency enhances insulin sensitivity and reduces PPAR-α pathway activity in mice. Environ. Health Perspect. 2011, 119, 1739–1744. [Google Scholar] [CrossRef]
- Hoyeck, M.P.; Blair, H.; Ibrahim, M.; Solanki, S.; Elsawy, M.; Prakash, A.; Rick, K.R.C.; Matteo, G.; O’Dwyer, S.; Bruin, J.E. Long-term metabolic consequences of acute dioxin exposure differ between male and female mice. Sci. Rep. 2020, 10, 1448. [Google Scholar] [CrossRef]
- Lee, Y.M.; Ha, C.M.; Kim, S.A.; Thoudam, T.; Yoon, Y.R.; Kim, D.J.; Kim, H.C.; Moon, H.B.; Park, S.; Lee, I.K.; et al. Low-Dose Persistent Organic Pollutants Impair Insulin Secretory Function of Pancreatic β-Cells: Human and In Vitro Evidence. Diabetes 2017, 66, 2669–2680. [Google Scholar] [CrossRef]
- Pérez-Bermejo, M.; Mas-Pérez, I.; Murillo-Llorente, M.T. The Role of the Bisphenol A in Diabetes and Obesity. Biomedicines 2021, 9, 666. [Google Scholar] [CrossRef]
- Stallings-Smith, S.; Mease, A.; Johnson, T.M.; Arikawa, A.Y. Exploring the association between polycyclic aromatic hydrocarbons and diabetes among adults in the United States. Environ. Res. 2018, 166, 588–594. [Google Scholar] [CrossRef]
- Wu, H.; Bertrand, K.A.; Choi, A.L.; Hu, F.B.; Laden, F.; Grandjean, P.; Sun, Q. Persistent organic pollutants and type 2 diabetes: A prospective analysis in the nurses’ health study and meta-analysis. Environ. Health Perspect. 2013, 121, 153–161. [Google Scholar] [CrossRef]
- Ji, J.H.; Jin, M.H.; Kang, J.H.; Lee, S.I.; Lee, S.; Kim, S.H.; Oh, S.Y. Relationship between heavy metal exposure and type 2 diabetes: A large-scale retrospective cohort study using occupational health examinations. BMJ Open 2021, 11, e039541. [Google Scholar] [CrossRef]
- Khan, A.R.; Awan, F.R. Metals in the pathogenesis of type 2 diabetes. J. Diabetes Metab. Disord. 2014, 13, 16. [Google Scholar] [CrossRef]
- Weiss, F.U.; Laemmerhirt, F.; Lerch, M.M. Etiology and Risk Factors of Acute and Chronic Pancreatitis. Visc. Med. 2019, 35, 73–81. [Google Scholar] [CrossRef]
- Kasai, A.; Hiramatsu, N.; Hayakawa, K.; Yao, J.; Maeda, S.; Kitamura, M. High levels of dioxin-like potential in cigarette smoke evidenced by in vitro and in vivo biosensing. Cancer Res. 2006, 66, 7143–7150. [Google Scholar] [CrossRef]
- Kitamura, M.; Kasai, A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007, 252, 184–194. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, K.B.; Han, J.H.; Kim, N.; Kang, T.U.; Swan, H.; Kim, H.J. Incidence and risk of pancreatic cancer in patients with acute or chronic pancreatitis: A population-based cohort study. Sci. Rep. 2023, 13, 18930. [Google Scholar] [CrossRef]
- Umans, D.S.; Hoogenboom, S.A.; Sissingh, N.J.; Lekkerkerker, S.J.; Verdonk, R.C.; van Hooft, J.E. Pancreatitis and pancreatic cancer: A case of the chicken or the egg. World J. Gastroenterol. 2021, 27, 3148–3157. [Google Scholar] [CrossRef]
- Xue, J.; Nguyen, D.T.; Habtezion, A. Aryl hydrocarbon receptor regulates pancreatic IL-22 production and protects mice from acute pancreatitis. Gastroenterology 2012, 143, 1670–1680. [Google Scholar] [CrossRef]
- Ghosh, J.; Chowdhury, A.R.; Srinivasan, S.; Chattopadhyay, M.; Bose, M.; Bhattacharya, S.; Raza, H.; Fuchs, S.Y.; Rustgi, A.K.; Gonzalez, F.J.; et al. Cigarette Smoke Toxins-Induced Mitochondrial Dysfunction and Pancreatitis Involves Aryl Hydrocarbon Receptor Mediated Cyp1 Gene Expression: Protective Effects of Resveratrol. Toxicol. Sci. 2018, 166, 428–440. [Google Scholar] [CrossRef]
- Lee, J.E.; Cho, S.G.; Ko, S.G.; Ahrmad, S.A.; Puga, A.; Kim, K. Regulation of a long noncoding RNA MALAT1 by aryl hydrocarbon receptor in pancreatic cancer cells and tissues. Biochem. Biophys. Res. Commun. 2020, 532, 563–569. [Google Scholar] [CrossRef]
- Kamata, K.; Hara, A.; Minaga, K.; Yoshikawa, T.; Kurimoto, M.; Sekai, I.; Okai, N.; Omaru, N.; Masuta, Y.; Otsuka, Y.; et al. Activation of the aryl hydrocarbon receptor inhibits the development of experimental autoimmune pancreatitis through IL-22-mediated signaling pathways. Clin. Exp. Immunol. 2023, 212, 171–183. [Google Scholar] [CrossRef]
- Xue, J.; Zhao, Q.; Sharma, V.; Nguyen, L.P.; Lee, Y.N.; Pham, K.L.; Edderkaoui, M.; Pandol, S.J.; Park, W.; Habtezion, A. Aryl Hydrocarbon Receptor Ligands in Cigarette Smoke Induce Production of Interleukin-22 to Promote Pancreatic Fibrosis in Models of Chronic Pancreatitis. Gastroenterology 2016, 151, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, L.; Chen, Y.; Lian, G.; Qian, C.; Chen, S.; Li, J.; Huang, K. Pancreatic Cancer Epidemiology, Detection, and Management. Gastroenterol. Res. Pract. 2016, 2016, 8962321. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Gasull, M.; Pumarega, J.; Kiviranta, H.; Rantakokko, P.; Raaschou-Nielsen, O.; Bergdahl, I.A.; Sandanger, T.M.; Agudo, A.; Rylander, C.; et al. Plasma concentrations of persistent organic pollutants and pancreatic cancer risk. Int. J. Epidemiol. 2022, 51, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Helou, K.; Harmouche-Karaki, M.; Karake, S.; Narbonne, J.F. A review of organochlorine pesticides and polychlorinated biphenyls in Lebanon: Environmental and human contaminants. Chemosphere 2019, 231, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Nyska, A.; Jokinen, M.P.; Brix, A.E.; Sells, D.M.; Wyde, M.E.; Orzech, D.; Haseman, J.K.; Flake, G.; Walker, N.J. Exocrine pancreatic pathology in female Harlan Sprague-Dawley rats after chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin and dioxin-like compounds. Environ. Health Perspect. 2004, 112, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Koliopanos, A.; Kleeff, J.; Xiao, Y.; Safe, S.; Zimmermann, A.; Büchler, M.W.; Friess, H. Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene 2002, 21, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Kim, S.B.; Safe, S. Omeprazole Inhibits Pancreatic Cancer Cell Invasion through a Nongenomic Aryl Hydrocarbon Receptor Pathway. Chem. Res. Toxicol. 2015, 28, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Korac, K.; Rajasekaran, D.; Sniegowski, T.; Schniers, B.K.; Ibrahim, A.F.; Bhutia, Y.D. Carbidopa, an activator of aryl hydrocarbon receptor, suppresses IDO1 expression in pancreatic cancer and decreases tumor growth. Biochem. J. 2022, 479, 1807–1824. [Google Scholar] [CrossRef]
- Stukas, D.; Jasukaitiene, A.; Bartkeviciene, A.; Matthews, J.; Maimets, T.; Teino, I.; Jaudzems, K.; Gulbinas, A.; Dambrauskas, Z. Targeting AHR Increases Pancreatic Cancer Cell Sensitivity to Gemcitabine through the ELAVL1-DCK Pathway. Int. J. Mol. Sci. 2023, 24, 13155. [Google Scholar] [CrossRef]
- Cheng, J.; Li, W.; Kang, B.; Zhou, Y.; Song, J.; Dan, S.; Yang, Y.; Zhang, X.; Li, J.; Yin, S.; et al. Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat. Commun. 2015, 6, 7209. [Google Scholar] [CrossRef] [PubMed]
- Hezaveh, K.; Shinde, R.S.; Klötgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e8. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Jin, U.H.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl Hydrocarbon Receptor (AHR) Ligands as Selective AHR Modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| EDC | Regulation of AHR Signaling | Epidemiological Studies Relevant to Pancreatic Diseases or Cancer | Mechanistic Role of AHR in Pancreatic Diseases and Cancer |
|---|---|---|---|
| Dioxin and dioxin-like compounds | AHR agonists [60,61,62] | See 5. Roles of EDC–AHR Interactions in the Pathogenesis of Pancreatic Diseases and Cancer | See 5. Roles of EDC–AHR Interactions in the Pathogenesis of Pancreatic Diseases and Cancer |
| Polycyclic aromatic hydrocarbons | AHR agonists and oxidative stress inducers [73,74,75,76,77,78,79,80] | [81,82,83] | Unknown |
| Hexachlorobenzene | Weak AHR agonist [86] | [87,88,89] | Unknown |
| Bisphenol A | Weak AHR agonist [101,102,103,104] | [94,95,96,97,98,99,100] | Unknown |
| Heavy metals | AHR agonists and oxidative stress inducers [109,110,111,112,113,114] | [115,116,117] | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K. The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer. Int. J. Mol. Sci. 2024, 25, 3818. https://doi.org/10.3390/ijms25073818
Kim K. The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer. International Journal of Molecular Sciences. 2024; 25(7):3818. https://doi.org/10.3390/ijms25073818
Chicago/Turabian StyleKim, Kyounghyun. 2024. "The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer" International Journal of Molecular Sciences 25, no. 7: 3818. https://doi.org/10.3390/ijms25073818
APA StyleKim, K. (2024). The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer. International Journal of Molecular Sciences, 25(7), 3818. https://doi.org/10.3390/ijms25073818