

Contrast Agents Based on Human Serum Albumin and Nitroxides for 1H-MRI and Overhauser-Enhanced MRI
 , , , and
, , , and 
Abstract
1. Introduction
2. Results and Discussion
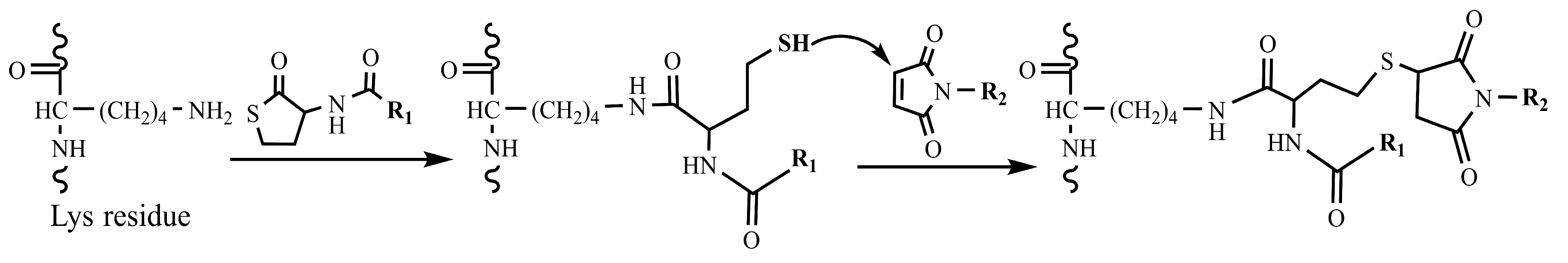
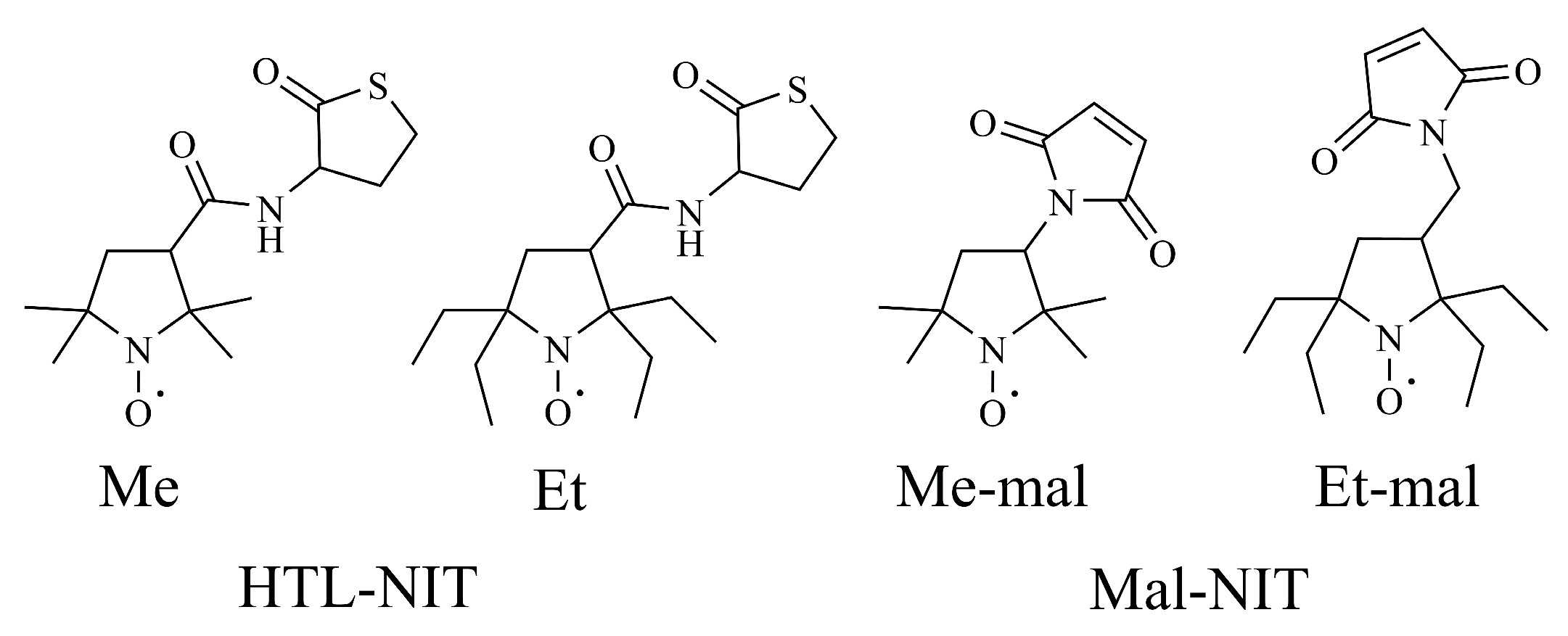
2.1. Design and Synthesis of HSA-NIT Conjugates
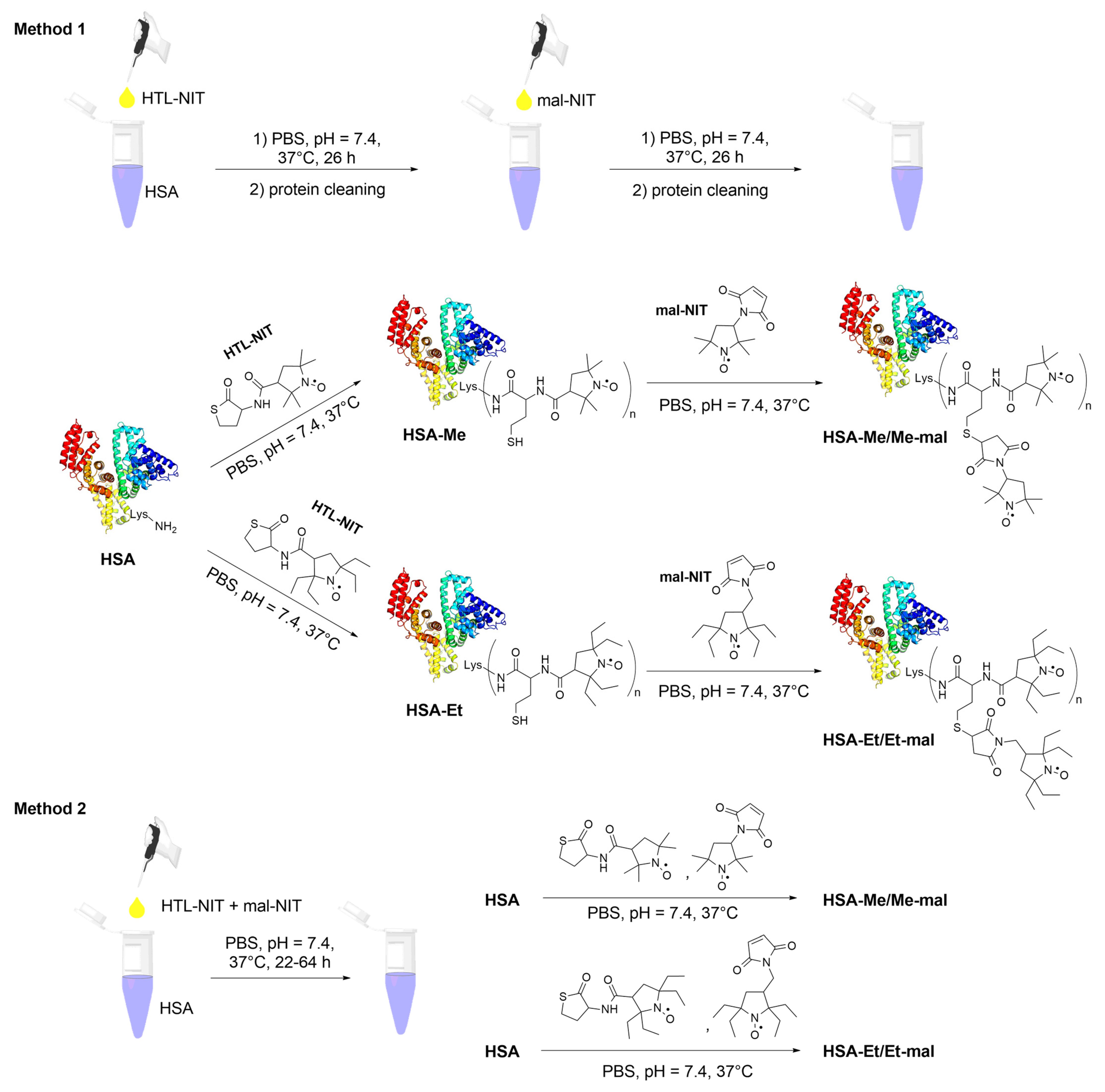
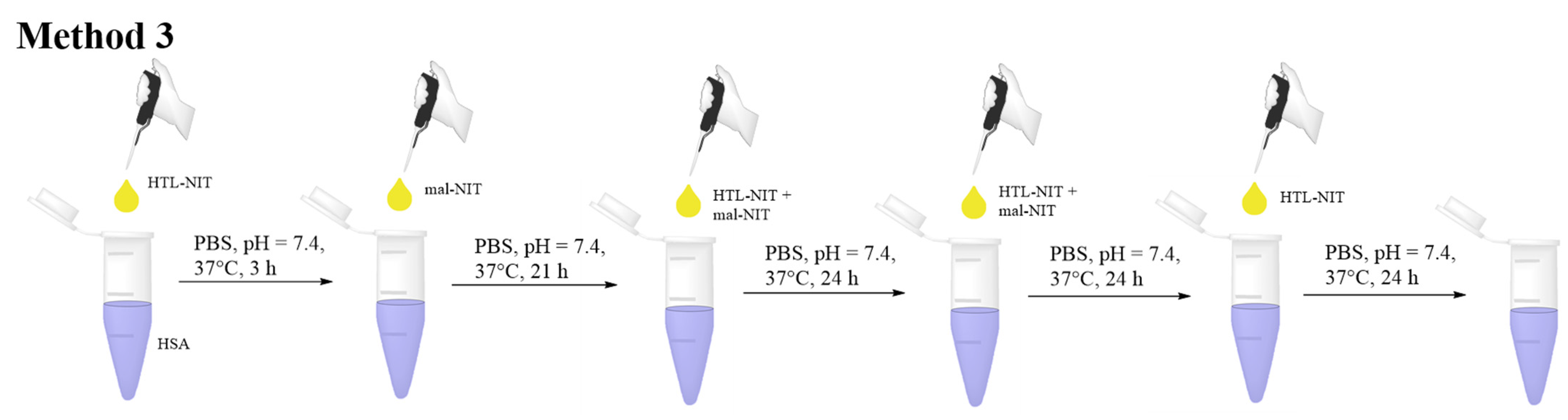
2.2. HSA-NIT Synthesis Method with Sequential and Simultaneous Addition of NIT Derivatives
2.3. Characterization of HSA-NIT Conjugates Synthesized by Method 3 Using SDS-PAGE, MALDI-ToF, Circular Dichroism, and EPR
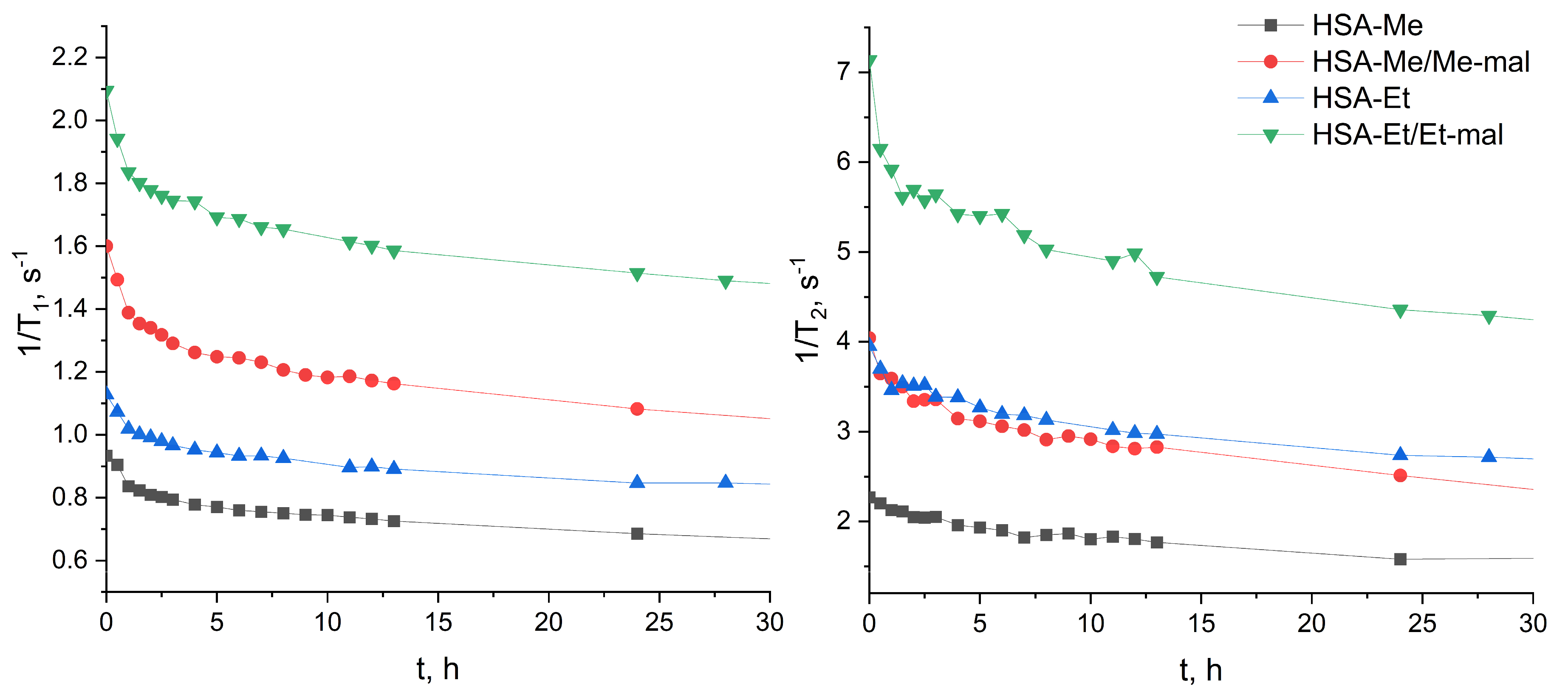
2.4. Reduction Constant Calculation of HSA-NIT
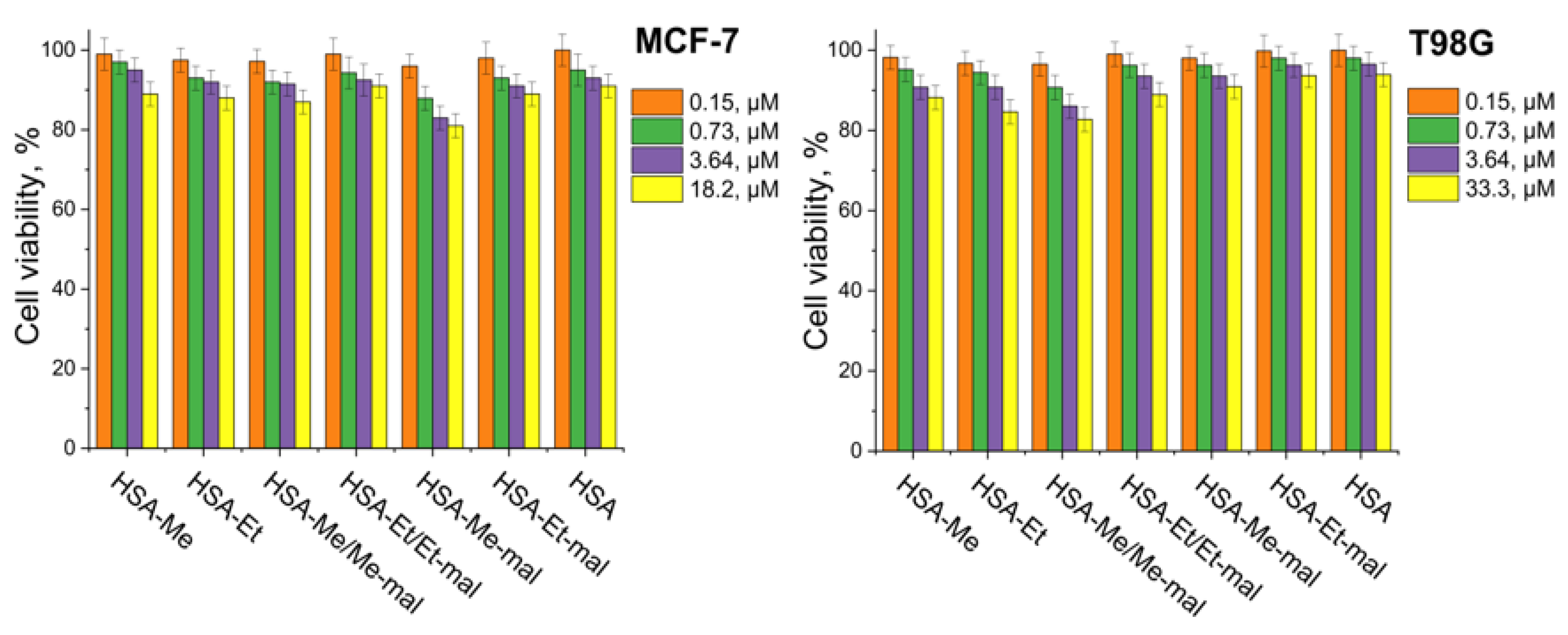
2.5. Cytotoxicity Assay
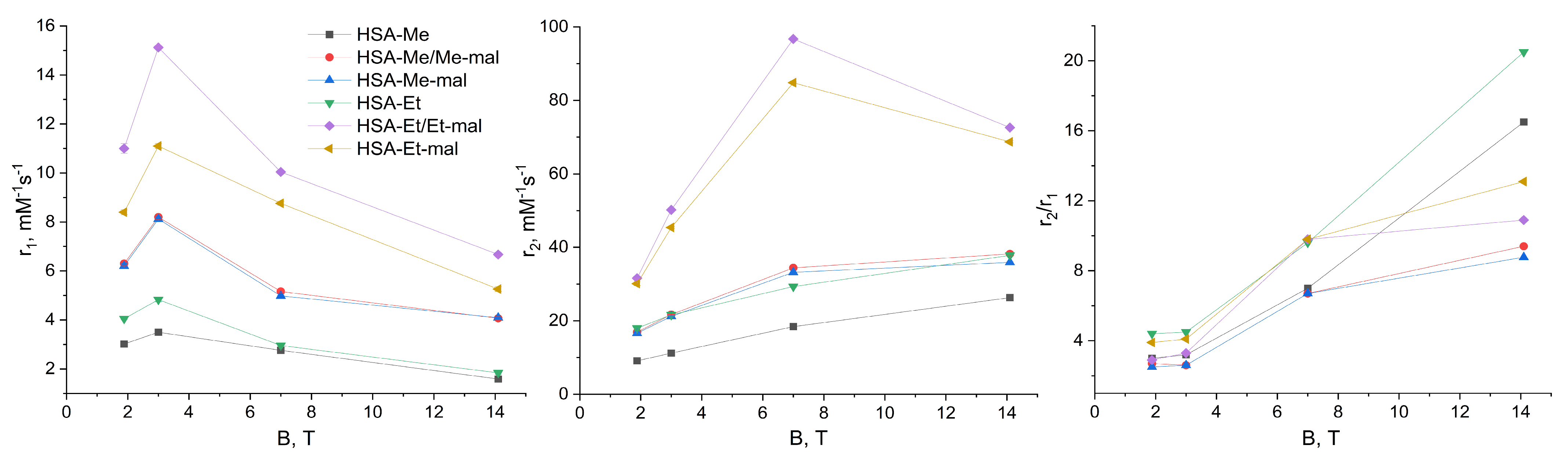
2.6. Relaxaxivities r1 and r2 of HSA-NIT
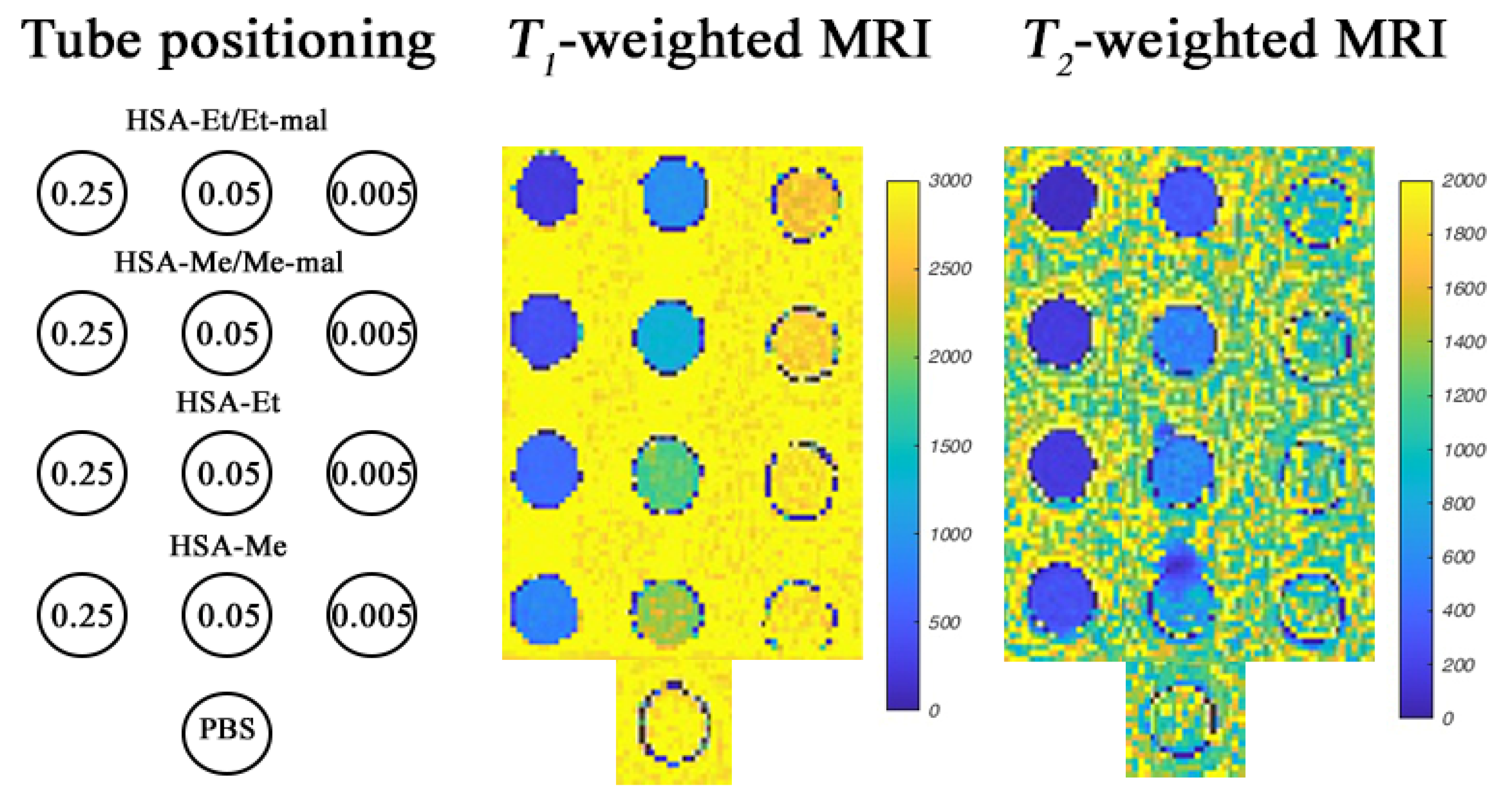
2.7. MRI Phantoms of HSA-NIT at 3 T and 14.1 T
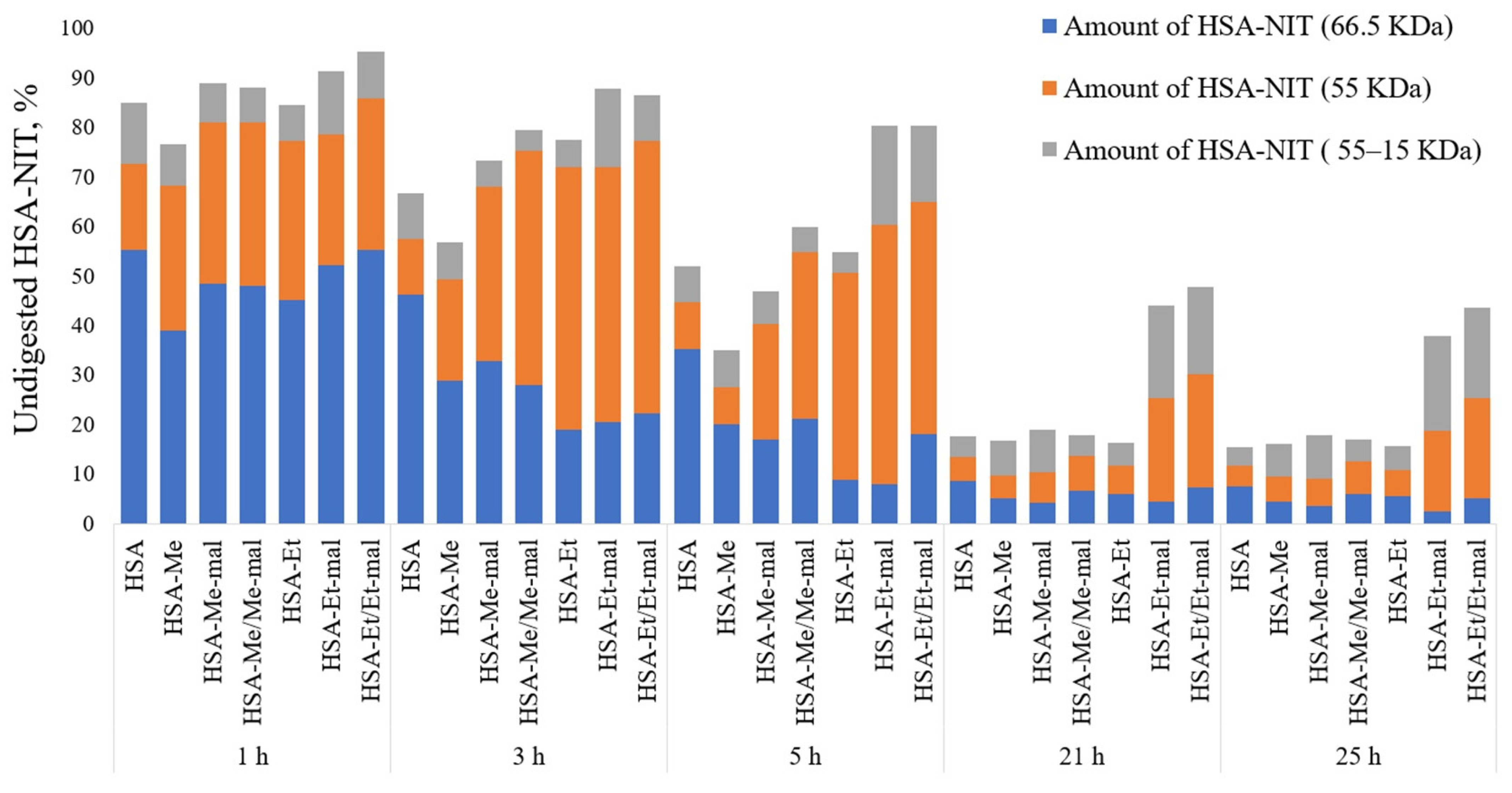
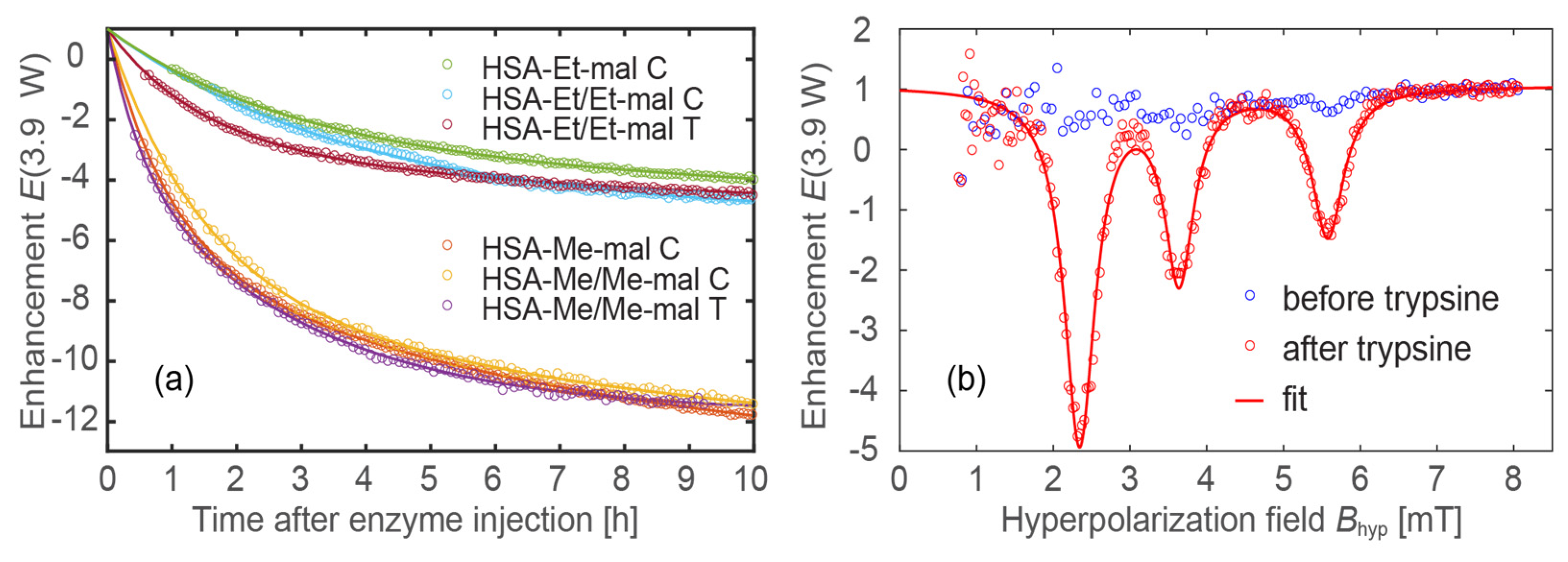
2.8. Trypsinolysis of HSA-NIT Conjugates and OMRI Application
3. Materials and Methods
3.1. Chemicals
3.2. Methods
3.3. Electron Paramagnetic Resonance (EPR)
3.4. Fractionation of HSA by Gel-Filtration Chromatography
3.5. HSA-NIT Conjugates Synthesis Methods
3.5.1. Method 1: HSA-NIT Synthesis Method with Sequential Addition of Nitroxide Derivatives
3.5.2. Method 2: HSA-NIT Synthesis Method with Simultaneous Addition of Nitroxide Derivatives
3.5.3. Method 3: HSA-NIT Synthesis Method with Simultaneous Addition of Nitroxide Derivatives Aliquots in Equal Time Intervals
3.6. Relaxivities (r1 and r2) of HSA-NIT Conjugates
3.7. Reduction of HSA-NIT Conjugates
3.7.1. Reduction Studies by T1-Relaxation Time Measuring
3.7.2. Reduction Studies by EPR
3.8. Trypsinolysis of HSA-NIT Conjugates
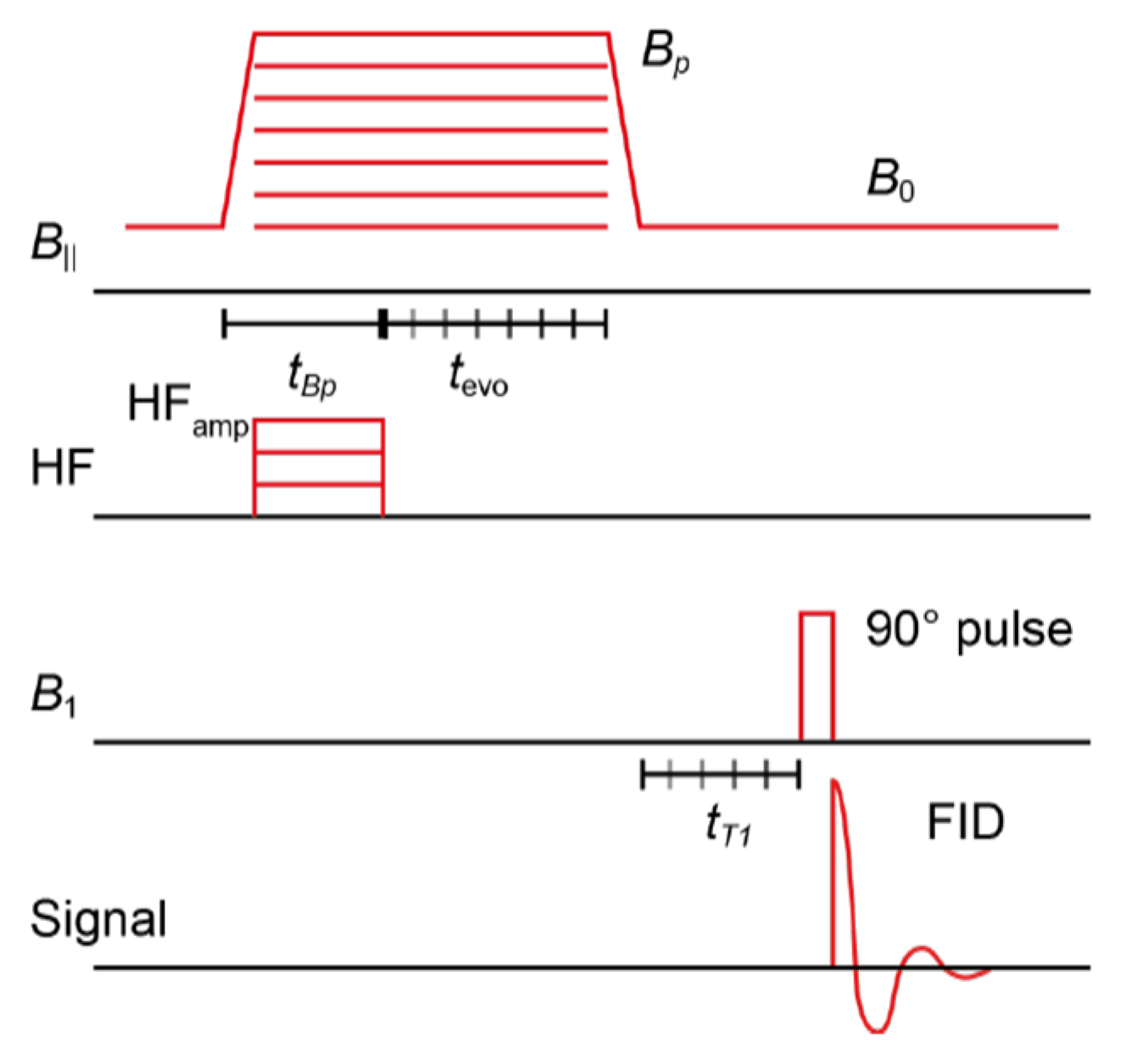
3.9. The Acquisition of OMRI Spectra of HSA-NIT Conjugates
3.10. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yousaf, T.; Dervenoulas, G.; Politis, M. Advances in MRI Methodology, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 141, ISBN 9780128154182. [Google Scholar]
- Kim, H.K.; Lee, G.H.; Chang, Y. Gadolinium as an MRI contrast agent. Future Med. Chem. 2018, 10, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Bartusik-Aebisher, D.; Bober, Z.; Zalejska-Fiolka, J.; Kawczyk-Krupka, A.; Aebisher, D. Multinuclear MRI in Drug Discovery. Molecules 2022, 27, 6493. [Google Scholar] [CrossRef]
- Caspani, S.; Magalhães, R.; Araújo, J.P.; Sousa, C.T. Magnetic nanomaterials as contrast agents for MRI. Materials 2020, 13, 2586. [Google Scholar] [CrossRef]
- Lv, J.; Roy, S.; Xie, M.; Yang, X.; Guo, B. Contrast Agents of Magnetic Resonance Imaging and Future Perspective. Nanomaterials 2023, 13, 2003. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, C.F.G.C.; Peters, J.A. MRI Contrast Agents in Glycobiology. Molecules 2022, 27, 8297. [Google Scholar] [CrossRef]
- Miloushev, V.Z.; Keshari, K.R.; Holodny, A.I. Hyperpolarization MRI preclinical models and potential applications in neuroradiology. Top. Magn. Reson. Imaging 2016, 25, 31–37. [Google Scholar] [CrossRef][Green Version]
- Chubarov, A.S. Serum Albumin for Magnetic Nanoparticles Coating. Magnetochemistry 2022, 8, 13. [Google Scholar] [CrossRef]
- Popova, V.; Dmitrienko, E.; Chubarov, A. Magnetic Nanocomposites and Imprinted Polymers for Biomedical Applications of Nucleic Acids. Magnetochemistry 2023, 9, 12. [Google Scholar] [CrossRef]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI contrast agents: Current challenges and new frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef]
- Xiao, Y.D.; Paudel, R.; Liu, J.; Ma, C.; Zhang, Z.S.; Zhou, S.K. MRI contrast agents: Classification and application (Review). Int. J. Mol. Med. 2016, 38, 1319–1326. [Google Scholar] [CrossRef]
- Bonnet, C.S.; Tóth, É. Metal-based environment-sensitive MRI contrast agents. Curr. Opin. Chem. Biol. 2021, 61, 154–169. [Google Scholar] [CrossRef]
- Zhao, S.; Yu, X.; Qian, Y.; Chen, W.; Shen, J. Multifunctional magnetic iron oxide nanoparticles: An advanced platform for cancer theranostics. Theranostics 2020, 10, 6278–6309. [Google Scholar] [CrossRef]
- Biedenbänder, T.; Aladin, V.; Saeidpour, S.; Corzilius, B. Dynamic Nuclear Polarization for Sensitivity Enhancement in Biomolecular Solid-State NMR. Chem. Rev. 2022, 122, 9738–9794. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. European Medicines Agency EMA’s Final Opinion Confirms Restrictions on Use of Linear Gadolinium Agents in Body Scans. Recommendations Conclude EMA’s Scientific Review of Gadolinium Deposition; European Medicines Agency: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Ramalho, J.; Ramalho, M. Gadolinium Deposition and Chronic Toxicity. Magn. Reson. Imaging Clin. N. Am. 2017, 25, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Malikova, H.; Holesta, M. Gadolinium contrast agents—Are they really safe? J. Vasc. Access 2017, 18, s1–s7. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, L.; Napolitano, A.; Visconti, E.; Longo, D.; Romano, A.; Tomà, P.; Espagnet, M.C.R. Gadolinium-Based Contrast Agent-Related Toxicities. CNS Drugs 2018, 32, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Rogosnitzky, M.; Branch, S. Gadolinium-based contrast agent toxicity: A review of known and proposed mechanisms. BioMetals 2016, 29, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, J.C.; Rodby, R.A.; Yee, J.; Wang, C.L.; Fine, D.; McDonald, R.J.; Perazella, M.A.; Dillman, J.R.; Davenport, M.S. Use of intravenous gadolinium-based contrast media in patients with kidney disease: Consensus statements from the American college of radiology and the national kidney foundation. Radiology 2021, 298, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Yu, Y.; Xu, X.; Wang, Q.; Chang, Y.; Zhang, C.; Zhao, J.; Peng, H.; Whittaker, A.K. Functional polymers as metal-free magnetic resonance imaging contrast agents. Prog. Polym. Sci. 2020, 108, 101286. [Google Scholar] [CrossRef]
- Akakuru, O.U.; Iqbal, M.Z.; Saeed, M.; Liu, C.; Paunesku, T.; Woloschak, G.; Hosmane, N.S.; Wu, A. The Transition from Metal-Based to Metal-Free Contrast Agents for T1 Magnetic Resonance Imaging Enhancement. Bioconjug. Chem. 2019, 30, 2264–2286. [Google Scholar] [CrossRef]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef]
- Rajca, A.; Wang, Y.; Boska, M.; Paletta, J.T.; Olankitwanit, A.; Swanson, M.A.; Mitchell, D.G.; Eaton, S.S.; Eaton, G.R.; Rajca, S. Organic Radical Contrast Agents for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2012, 134, 15724–15727. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.V.T.; Detappe, A.; Gallagher, N.M.; Zhang, H.; Harvey, P.; Yan, C.; Mathieu, C.; Golder, M.R.; Jiang, Y.; Ottaviani, M.F.; et al. Triply Loaded Nitroxide Brush-Arm Star Polymers Enable Metal-Free Millimetric Tumor Detection by Magnetic Resonance Imaging. ACS Nano 2018, 12, 11343–11354. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.V.T.; Chen, Q.; Paletta, J.T.; Harvey, P.; Jiang, Y.; Zhang, H.; Boska, M.D.; Ottaviani, M.F.; Jasanoff, A.; Rajca, A.; et al. Nitroxide-Based Macromolecular Contrast Agents with Unprecedented Transverse Relaxivity and Stability for Magnetic Resonance Imaging of Tumors. ACS Cent. Sci. 2017, 3, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, Q.; Garcia-Hernandez, J.D.; Watanabe, L.K.; Rawson, J.M.; Rao, J.; Manners, I. Uniform and Length-Tunable, Paramagnetic Self-Assembled Nitroxide-Based Nanofibers for Magnetic Resonance Imaging. Macromolecules 2023, 56, 263–270. [Google Scholar] [CrossRef]
- Bye, N.; Hutt, O.E.; Hinton, T.M.; Acharya, D.P.; Waddington, L.J.; Moffat, B.A.; Wright, D.K.; Wang, H.X.; Mulet, X.; Muir, B.W. Nitroxide-loaded hexosomes provide MRI contrast in vivo. Langmuir 2014, 30, 8898–8906. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.M.; Mitchell, J.B.; Krishna, M.C. Nitroxides as Cancer Imaging Agents. Anticancer Agents Med. Chem. 2012, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Soikkeli, M.; Horkka, K.; Moilanen, J.O.; Timonen, M.; Kavakka, J.; Heikkinen, S. Synthesis, stability and relaxivity of teepo-met: An organic radical as a potential tumour targeting contrast agent for magnetic resonance imaging. Molecules 2018, 23, 1034. [Google Scholar] [CrossRef] [PubMed]
- Soikkeli, M.; Sievänen, K.; Peltonen, J.; Kaasalainen, T.; Timonen, M.; Heinonen, P.; Rönkkö, S.; Lehto, V.P.; Kavakka, J.S.; Heikkinen, S. Synthesis and in vitro phantom NMR and MRI studies of fully organic free radicals, TEEPO-glucose and TEMPO-glucose, potential contrast agents for MRI. RSC Adv. 2015, 5, 15507–15510. [Google Scholar] [CrossRef]
- Chan, H.C.; Sun, K.Q.; Magin, R.L.; Swartz, H.M. Potential of Albumin Labeled with Nitroxides as a Contrast Agent for Magnetic Resonance Imaging and Spectroscopy. Bioconjug. Chem. 1990, 1, 32–36. [Google Scholar] [CrossRef]
- Lee, H.; Shahrivarkevishahi, A.; Lumata, J.L.; Luzuriaga, M.A.; Hagge, L.M.; Benjamin, C.E.; Brohlin, O.R.; Parish, C.R.; Firouzi, H.R.; Nielsen, S.O.; et al. Chemical Science enhancement of metal-free magnetic resonance imaging contrast agents. Chem. Sci. 2020, 11, 2045–2050. [Google Scholar] [CrossRef]
- Eisele, K.; Gropeanu, R.; Musante, A.; Glasser, G.; Li, C.; Muellen, K.; Weil, T. Tailored Albumin-based Copolymers for Receptor-Mediated Delivery of Perylenediimide Guest Molecules. Macromol. Rapid Commun. 2010, 31, 1501–1508. [Google Scholar] [CrossRef]
- Alvaradejo, G.G.; Nguyen, H.V.-T.; Harvey, P.; Gallagher, N.M.; Le, D.; Ottaviani, M.F.; Jasanoff, A.; Delaittre, G.; Johnson, J.A. Polyoxazoline-Based Bottlebrush and Brush-Arm Star Polymers via ROMP: Syntheses and Applications as Organic Radical Contrast Agents. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Nagura, K.; Takemoto, Y.; Yoshino, F.; Bogdanov, A.; Chumakova, N.; Vorobiev, A.K.; Imai, H.; Matsuda, T.; Shimono, S.; Kato, T.; et al. Magnetic mixed micelles composed of a non-ionic surfactant and nitroxide radicals containing a D-glucosamine unit: Preparation, stability, and biomedical application. Pharmaceutics 2019, 11, 42. [Google Scholar] [CrossRef]
- Guo, S.; Wang, X.; Li, Z.; Pan, D.; Dai, Y.; Ye, Y.; Tian, X.; Gu, Z.; Gong, Q.; Zhang, H.; et al. A nitroxides-based macromolecular MRI contrast agent with an extraordinary longitudinal relaxivity for tumor imaging via clinical T1WI SE sequence. J. Nanobiotechnol. 2021, 19, 244. [Google Scholar] [CrossRef]
- Zhang, S.; Lloveras, V.; Lope-piedra, S.; Calero-p, P.; Wu, S.; Candiota, A.P.; Vidal-gancedo, J. Metal-Free Radical Dendrimers as MRI Contrast Agents for Glioblastoma Diagnosis: Ex Vivo and In Vivo Approaches. Biomacromolecules 2022, 23, 2767–2777. [Google Scholar] [CrossRef]
- Guo, S.; Wang, X.; Dai, Y.; Dai, X.; Li, Z.; Luo, Q.; Zheng, X.; Gu, Z.; Zhang, H.; Gong, Q.; et al. Enhancing the Efficacy of Metal-Free MRI Contrast Agents via Conjugating Nitroxides onto PEGylated Cross-Linked Poly(Carboxylate Ester). Adv. Sci. 2020, 7, 2000467. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.F.; Lloveras, V.; Zhang, S.; Liko, F.; Veciana, J.; Muñoz-Gómez, J.L.; Vidal-Gancedo, J. Fully Water-Soluble Polyphosphorhydrazone-Based Radical Dendrimers Functionalized with Tyr-PROXYL Radicals as Metal-Free MRI T1 Contrast Agents. ACS Appl. Bio Mater. 2020, 3, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Gokuden, R.; Watanabe, K.; Mori, T.; Naganuma, T.; Utsumi, H.; Ichikawa, K.; Katayama, Y. Nitroxyl radicals-modified dendritic poly(l-lysine) as a contrast agent for Overhauser-enhanced MRI. J. Biomater. Sci. Polym. Ed. 2014, 25, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Jannin, S.; Dumez, J.N.; Giraudeau, P.; Kurzbach, D. Application and methodology of dissolution dynamic nuclear polarization in physical, chemical and biological contexts. J. Magn. Reson. 2019, 305, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Eills, J.; Budker, D.; Cavagnero, S.; Chekmenev, E.Y.; Elliott, S.J.; Jannin, S.; Lesage, A.; Matysik, J.; Meersmann, T.; Prisner, T.; et al. Spin Hyperpolarization in Modern Magnetic Resonance. Chem. Rev. 2023, 123, 1417–1551. [Google Scholar] [CrossRef]
- Corzilius, B. High-Field Dynamic Nuclear Polarization. Annu. Rev. Phys. Chem. 2020, 71, 143–170. [Google Scholar] [CrossRef]
- Gunther, U.L. Topics in Current Chemistry 335 Modern NMR Methodology. In Modern NMR Methodology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 23–69. ISBN 9783642379901. [Google Scholar]
- Fehling, P.; Buckenmaier, K.; Dobrynin, S.A.; Morozov, D.A.; Polienko, Y.F.; Khoroshunova, Y.V.; Borozdina, Y.; Mayer, P.; Engelmann, J.; Scheffler, K.; et al. The effects of nitroxide structure upon 1H Overhauser dynamic nuclear polarization efficacy at ultralow-field. J. Chem. Phys. 2021, 155, 144203. [Google Scholar] [CrossRef]
- Zamora, P.L.; Villamena, F.A.; Zamora, P.L.; Villamena, F.A. Measuring Oxidants and Oxidative Stress in Biological Systems; Springer: Berlin/Heidelberg, Germany, 2020; Volume 34, ISBN 9783030473174. [Google Scholar]
- Takakusagi, Y.; Kobayashi, R.; Saito, K.; Kishimoto, S.; Krishna, M.C.; Murugesan, R.; Matsumoto, K.I. EPR and Related Magnetic Resonance Imaging Techniques in Cancer Research. Metabolites 2023, 13, 69. [Google Scholar] [CrossRef]
- Koonjoo, N.; Parzy, E.; Massot, P.; Lepetit-Coiffé, M.; Marque, S.R.A.; Franconi, J.M.; Thiaudiere, E.; Mellet, P. In vivo Overhauser-enhanced MRI of proteolytic activity. Contrast Media Mol. Imaging 2014, 9, 363–371. [Google Scholar] [CrossRef]
- Parzy, E.; Bouchaud, V.; Massot, P.; Voisin, P.; Koonjoo, N.; Moncelet, D.; Franconi, J.M.; Thiaudière, E.; Mellet, P. Overhauser-Enhanced MRI of Elastase Activity from In Vitro Human Neutrophil Degranulation. PLoS ONE 2013, 8, e57946. [Google Scholar] [CrossRef]
- Jugniot, N.; Duttagupta, I.; Rivot, A.; Massot, P.; Cardiet, C.; Pizzoccaro, A.; Jean, M.; Vanthuyne, N.; Franconi, J.M.; Voisin, P.; et al. An elastase activity reporter for Electronic Paramagnetic Resonance (EPR) and Overhauser-enhanced Magnetic Resonance Imaging (OMRI) as a line-shifting nitroxide. Free Radic. Biol. Med. 2018, 126, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Buckenmaier, K.; Rudolph, M.; Fehling, P.; Steffen, T.; Back, C.; Bernard, R.; Pohmann, R.; Bernarding, J.; Kleiner, R.; Koelle, D.; et al. Mutual benefit achieved by combining ultralow-field magnetic resonance and hyperpolarizing techniques. Rev. Sci. Instrum. 2018, 89, 125103. [Google Scholar] [CrossRef]
- Waddington, D.E.J.; Sarracanie, M.; Salameh, N.; Herisson, F.; Ayata, C.; Rosen, M.S. An Overhauser-enhanced-MRI platform for dynamic free radical imaging in vivo. NMR Biomed. 2018, 31, e3896. [Google Scholar] [CrossRef] [PubMed]
- Bennati, M.; Orlando, T. Overhauser DNP in liquids on 13C nuclei. eMagRes 2019, 8, 11. [Google Scholar] [CrossRef]
- Shankar Palani, R.; Mardini, M.; Quan, Y.; Griffin, R.G. Dynamic nuclear polarization with trityl radicals. J. Magn. Reson. 2023, 349, 107411. [Google Scholar] [CrossRef]
- Ichikawa, K.; Yamato, M.; Naganuma, T. Biomedical Overhauser Magnetic Resonance Imaging (OMRI): Noninvasive Imaging of Redox Processes; Springer: Berlin/Heidelberg, Germany, 2020; pp. 221–229. [Google Scholar] [CrossRef]
- Kosem, N.; Naganuma, T.; Ichikawa, K.; Phumala Morales, N.; Yasukawa, K.; Hyodo, F.; Yamada, K.I.; Utsumi, H. Whole-body kinetic image of a redox probe in mice using Overhauser-enhanced MRI. Free Radic. Biol. Med. 2012, 53, 328–336. [Google Scholar] [CrossRef]
- Spada, A.; Emami, J.; Tuszynski, J.A.; Lavasanifar, A. The Uniqueness of Albumin as a Carrier in Nanodrug Delivery. Mol. Pharm. 2021, 18, 1862–1894. [Google Scholar] [CrossRef]
- Chubarov, A.; Spitsyna, A.; Krumkacheva, O.; Mitin, D.; Suvorov, D.; Tormyshev, V.; Fedin, M.; Bowman, M.K.; Bagryanskaya, E. Reversible Dimerization of Human Serum Albumin. Molecules 2021, 26, 108. [Google Scholar] [CrossRef]
- Ishima, Y.; Maruyama, T.; Otagiri, M.; Ishida, T. Drug delivery system for refractory cancer therapy via an endogenous albumin transport system. Chem. Pharm. Bull. 2020, 68, 583–588. [Google Scholar] [CrossRef]
- Lamichhane, S.; Lee, S. Albumin nanoscience: Homing nanotechnology enabling targeted drug delivery and therapy. Arch. Pharm. Res. 2020, 43, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Chubarov, A.S.; Zakharova, O.D.; Koval, O.A.; Romaschenko, A.V.; Akulov, A.E.; Zavjalov, E.L.; Razumov, I.A.; Koptyug, I.V.; Knorre, D.G.; Godovikova, T.S. Design of protein homocystamides with enhanced tumor uptake properties for 19F magnetic resonance imaging. Bioorg. Med. Chem. 2015, 23, 6943–6954. [Google Scholar] [CrossRef]
- Lisitskiy, V.A.; Khan, H.; Popova, T.V.; Chubarov, A.S.; Zakharova, O.D.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Koptyug, I.V.; Moshkin, M.P.; et al. Multifunctional human serum albumin-therapeutic nucleotide conjugate with redox and pH-sensitive drug release mechanism for cancer theranostics. Bioorg. Med. Chem. Lett. 2017, 27, 3925–3930. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. [Google Scholar] [CrossRef] [PubMed]
- Asanbaeva, N.B.; Sukhanov, A.A.; Diveikina, A.A.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Maryasov, A.G.; Genaev, A.M.; Shernyukov, A.V.; et al. Application of W-band 19F electron nuclear double resonance (ENDOR) spectroscopy to distance measurement using a trityl spin probe and a fluorine label. Phys. Chem. Chem. Phys. 2022, 24, 5982–6001. [Google Scholar] [CrossRef] [PubMed]
- Chubarov, A.S.; Shakirov, M.M.; Koptyug, I.V.; Sagdeev, R.Z.; Knorre, D.G.; Godovikova, T.S. Synthesis and characterization of fluorinated homocysteine derivatives as potential molecular probes for 19F magnetic resonance spectroscopy and imaging. Bioorg. Med. Chem. Lett. 2011, 21, 4050–4053. [Google Scholar] [CrossRef]
- Nishi, K.; Yamasaki, K.; Otagiri, M. Serum Albumin, Lipid and Drug Binding; Springer: Berlin/Heidelberg, Germany, 2020; Volume 94, ISBN 9783030417680. [Google Scholar]
- Vusse, G.J. van der Albumin as Fatty Acid Transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef]
- Otagiri, M.; Giam Chuang, V.T. Albumin in Medicine: Pathological and Clinical Applications; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–277. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Kragh-Hansen, U. Albumin in Medicine; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 978-981-10-2115-2. [Google Scholar]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. Methods Mol. Biol. 2017, 1601, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Torricella, F.; Pierro, A.; Mileo, E.; Belle, V.; Bonucci, A. Nitroxide spin labels and EPR spectroscopy: A powerful association for protein dynamics studies. Biochim. Biophys. Acta-Proteins Proteom. 2021, 1869, 140653. [Google Scholar] [CrossRef]
- Ackermann, K.; Chapman, A.; Bode, B.E. A comparison of cysteine-conjugated nitroxide spin labels for pulse dipolar epr spectroscopy. Molecules 2021, 26, 7534. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Waudby, C.A.; Bernardo-Gancedo, A.; De Genst, E.; Dhulesia, A.; Salvatella, X.; Christodoulou, J.; Dobson, C.M.; Kumita, J.R. Application of Lysine-specific Labeling to Detect Transient Interactions Present during Human Lysozyme Amyloid Fibril Formation. Sci. Rep. 2017, 7, 15018. [Google Scholar] [CrossRef]
- Dharmarwardana, M.; Martins, A.F.; Chen, Z.; Palacios, P.M.; Nowak, C.M.; Welch, R.P.; Li, S.; Luzuriaga, M.A.; Bleris, L.; Pierce, B.S.; et al. Nitroxyl Modified Tobacco Mosaic Virus as a Metal-Free High-Relaxivity MRI and EPR Active Superoxide Sensor. Mol. Pharm. 2018, 15, 2973–2983. [Google Scholar] [CrossRef]
- Petrov, K.D.; Chubarov, A.S. Magnetite Nanoparticles for Biomedical Applications. Encyclopedia 2022, 2, 1811–1828. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids 2014, 46, 235–244. [Google Scholar] [CrossRef]
- Marczak, L.; Sikora, M.; Stobiecki, M.; Jakubowski, H. Analysis of site-specific N-homocysteinylation of human serum albumin in vitro and in vivo using MALDI-ToF and LC-MS/MS mass spectrometry. J. Proteom. 2011, 74, 967–974. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Twardowski, T.; Stobiecki, M.; Jakubowski, H. Direct monitoring of albumin lysine-525 N-homocysteinylation in human serum by liquid chromatography/mass spectrometry. Anal. Biochem. 2010, 405, 132–134. [Google Scholar] [CrossRef]
- Popova, T.V.; Krumkacheva, O.A.; Burmakova, A.S.; Spitsyna, A.S.; Zakharova, O.D.; Lisitskiy, V.A.; Kirilyuk, I.A.; Silnikov, V.N.; Bowman, M.K.; Bagryanskaya, E.G.; et al. Protein modification by thiolactone homocysteine chemistry: A multifunctionalized human serum albumin theranostic. RSC Med. Chem. 2020, 11, 1314–1325. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. Role of Glycation in Amyloid: Effect on the Aggregation Process and Cytotoxicity. In Exploring New Findings on Amyloidosis; Intechopen: London, UK, 2016. [Google Scholar] [CrossRef]
- Watanabe, H.; Imafuku, T.; Otagiri, M.; Maruyama, T. Clinical Implications Associated With the Posttranslational Modification–Induced Functional Impairment of Albumin in Oxidative Stress–Related Diseases. J. Pharm. Sci. 2017, 106, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Yiannios, C.N.; Karabinos, J.V. Oxidation of Thiols by Dimethyl Sulfoxide. J. Org. Chem. 1963, 28, 3246–3248. [Google Scholar] [CrossRef]
- Papanyan, Z.K. Interaction of L-cysteine with dimethyl sulfoxide in mild conditions. Proc. Yerevan State Univ. 2013, 2, 11–14. [Google Scholar]
- Sterling, H.J.; Prell, J.S.; Cassou, C.A.; Williams, E.R. Protein conformation and supercharging with DMSO from aqueous solution. J. Am. Soc. Mass Spectrom. 2011, 22, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Taratayko, A.I.; Glazachev, Y.I.; Eltsov, I.V.; Chernyak, E.I.; Kirilyuk, I.A. 3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains. Molecules 2022, 27, 1922. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.F.; Riehm, J.P. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal. Biochem. 1967, 18, 248–255. [Google Scholar] [CrossRef]
- Cornell, C.N.; Chang, R.; Kaplan, L.J. The environment of the sulfhydryl group in human plasma albumin as determined by spin labeling. Arch. Biochem. Biophys. 1981, 209, 1–6. [Google Scholar] [CrossRef]
- Perczel, A.; Hollósi, M.; Tusnády, G.; Fasman, G.D. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Protein Eng. 1991, 4, 669–679. [Google Scholar] [CrossRef]
- Varkovitzky, R.L. Assimilation, accommodation, and overaccommodation: An examination of information processing styles in female victims of adolescent and adult sexual assault. ProQuest Diss. Theses 2012, 1, 218. [Google Scholar] [CrossRef]
- Sowiński, M.P.; Gahlawat, S.; Lund, B.A.; Warnke, A.L.; Hopmann, K.H.; Lovett, J.E.; Haugland, M.M. Conformational tuning improves the stability of spirocyclic nitroxides with long paramagnetic relaxation times. Commun. Chem. 2023, 6, 4–11. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.-I.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and reduction kinetics of sterically shielded pyrrolidine nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef]
- Bobko, A.A.; Kirilyuk, I.A.; Grigor’ev, I.A.; Zweier, J.L.; Khramtsov, V.V. Reversible reduction of nitroxides to hydroxylamines: Roles for ascorbate and glutathione. Free Radic. Biol. Med. 2007, 42, 404–412. [Google Scholar] [CrossRef]
- Kirilyuk, I.A.; Polienko, Y.F.; Krumkacheva, O.A.; Strizhakov, R.K.; Gatilov, Y.V.; Grigor’ev, I.A.; Bagryanskaya, E.G. Synthesis of 2,5-bis(spirocyclohexane)-substituted nitroxides of pyrroline and pyrrolidine series, including thiol-specific spin label: An analogue of MTSSL with long relaxation time. J. Org. Chem. 2012, 77, 8016–8027. [Google Scholar] [CrossRef]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef]
- Koyanagi, M.; Kawakabe, S.; Arimura, Y. A comparative study of colorimetric cell proliferation assays in immune cells. Cytotechnology 2016, 68, 1489–1498. [Google Scholar] [CrossRef]
- Chou, C.; Abdesselem, M.; Bouzigues, C.; Chu, M.; Guiga, A.; Huang, T.; Ferrage, F.; Gacoin, T.; Alexandrou, A. Ultra-wide range field-dependent measurements of the relaxivity of Gd1−xEuxVO4 nanoparticle contrast agents using a mechanical sample-shuttling relaxometer. Sci. Rep. 2017, 7, 44770. [Google Scholar] [CrossRef]
- Hagberg, G.E.; Schef, K. Effect of r 1 and r 2 relaxivity of gadolinium-based contrast agents on the T 1 -weighted MR signal at increasing magnetic fi eld strengths. Contrast Media Mol. Imaging 2013, 8, 456–465. [Google Scholar] [CrossRef]
- Ta, H.T.; Li, Z.; Wu, Y.; Cowin, G.; Zhang, S.; Yago, A.; Whittaker, A.K. Effects of magnetic field strength and particle aggregation on relaxivity of ultra-small dual contrast iron oxide nanoparticles. Mater. Res. Express 2017, 4, 116105. [Google Scholar] [CrossRef]
- Blockley, N.P.; Jiang, L.; Gardener, A.G.; Ludman, C.N.; Francis, S.T.; Gowland, P.A. Field strength dependence of R1 and R2* relaxivities of human whole blood to ProHance, vasovist, and deoxyhemoglobin. Magn. Reson. Med. 2008, 60, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Pintaske, J.; Martirosian, P.; Graf, H.; Erb, G.; Lodemann, K.-P.; Claussen, C.D.; Schick, F. Relaxivity of Gadopentetate Dimeglumine (Magnevist), Gadobutrol (Gadovist), and Gadobenate Dimeglumine (MultiHance) in human blood plasma at 0.2, 1.5, and 3 Tesla. Investig. Radiol. 2006, 41, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Ludwig, K.; Adamsom, E.; Eliceiri, K.; Fain, S. An open source, 3D printed preclinical MRI phantom for repeated measures of contrast agents and reference standards. Biomed. Phys. Eng. Express 2018, 4, 027005. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Miao, J.; Soond, S.M.; Rudzińska, M.; Zamyatnin, A.A. Albumin nanovectors in cancer therapy and imaging. Biomolecules 2019, 9, 218. [Google Scholar] [CrossRef]
- Mitin, D.E.; Chubarov, A.S. Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe. Molecules 2023, 28, 1695. [Google Scholar] [CrossRef]
- Mei, J.; Tian, H. Most recent advances on enzyme-activatable optical probes for bioimaging. Aggregate 2021, 2, e32. [Google Scholar] [CrossRef]
- Solomon, M.; Berezin, Y.; Achilefu, S. Optical Imaging in Cancer Research: Basic Principles, Tumor Detectio, and Therapeutic Monitoring. Med. Princ. Pract. 2011, 63110, 397–415. [Google Scholar] [CrossRef]
- Vizovisek, M.; Ristanovic, D.; Menghini, S.; Christiansen, M.G.; Schuerle, S. The Tumor Proteolytic Landscape: A Challenging Frontier in Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 22, 2514. [Google Scholar] [CrossRef]
- Sleep, D.; Sleep, D. Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 793–812. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Dharmasivam, M.; Richardson, D.R. The role of extracellular proteases in tumor progression and the development of innovative metal ion chelators that inhibit their activity. Int. J. Mol. Sci. 2020, 21, 6805. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, L.; Zheng, Y.; Li, A.; Lin, H.; Gao, J. Nanoparticle-Based Activatable MRI Probes for Disease Imaging and Monitoring. Chem. Biomed. Imaging 2023, 1, 192–204. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. 2018, 130, 1380–1384. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series. Molecules 2021, 26, 5761. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, T.H.J.; Strohalm, M. mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. PLoS ONE 2012, 7, e44913. [Google Scholar] [CrossRef] [PubMed]
- Strohalm, M.; Kavan, D.; Novák, P.; Volný, M.; Havlíček, V. MMass 3: A cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 2010, 82, 4648–4651. [Google Scholar] [CrossRef]
- Marinas, M.; Sa, E.; Rojas, M.M.; Moalem, M.; Urbano, F.J.; Guillou, C.; Rallo, L. A nuclear magnetic resonance (1H and 13C) and isotope ratio mass spectrometry (δ13C, δ2H and δ18O) study of Andalusian olive oils. Rapid Commun. Mass Spectrom. 2010, 24, 1457–1466. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin: Simulating cw ESR spectra. Biol. Magn. Reson. 2007, 27, 299–321. [Google Scholar]
- Krumkacheva, O.A.; Timofeev, I.O.; Politanskaya, L.V.; Polienko, Y.F.; Tretyakov, E.V.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Bagryanskaya, E.G.; et al. Triplet Fullerenes as Prospective Spin Labels for Nanoscale Distance Measurements by Pulsed Dipolar EPR. Angew. Chem. Int. Ed. 2019, 58, 13271–13275. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | NIT Residue Per HSA Molecule | CD Spectral Analysis | |
|---|---|---|---|
| α-Helix, % | β-Sheet, % | ||
| Monomer HSA | - | 55.0 ± 1.0 | 4.8 ± 1.0 |
| HSA-Me | 13.7 ± 0.7 | 49.2 ± 1.5 | 5.9 ± 0.6 |
| HSA-Me-mal | 16.4 ± 0.9 | 44.3 ± 1.7 | 8.1 ± 0.6 |
| HSA-Me/Me-mal | 17.1 ± 1.0 | 43.7 ± 2.0 | 7.8 ± 0.8 |
| HSA-Et | 13.1 ± 0.2 | 48.8 ± 1.4 | 6.4 ± 0.6 |
| HSA-Et-mal | 21.9 ± 1.0 | 44.1 ± 1.7 | 10.9 ± 0.7 |
| HSA-Et/Et-mal | 20.8 ± 1.1 | 43.5 ± 2.0 | 9.7 ± 0.8 |
| Sample | Method | kinit, M−1s−1 | k“free” NIT, M−1s−1 |
|---|---|---|---|
| HSA-Me | EPR | 0.044 ± 0.006 | 0.097 [100], 0.063 [98] |
| T1-meas. | 0.027 ± 0.005 | ||
| HSA-Me-Mal | EPR | 0.041 ± 0.006 | |
| HSA-Me/Me-Mal | EPR | 0.043 ± 0.008 | |
| T1-meas. | 0.033 ± 0.006 | ||
| HSA-Et | EPR | 0.0019 ± 0.0005 | 0.001 [98], 0.002 [101] |
| T1-meas. | 0.0015 ± 0.0001 | ||
| HSA-Et-Mal | EPR | 0.0016 ± 0.0004 | |
| HSA-Et/Et-Mal | EPR | 0.0015 ± 0.0004 | |
| T1-meas. | 0.0007 ± 0.0001 |
| Sample | A1 | A2 | τ1 [1/h] | τ2 [1/h] | P1/2 [W] | |Emax| |
|---|---|---|---|---|---|---|
| HSA-Me-mal C * | −7.4 ± 0.1 | −7.2 ± 0.1 | 6.9 ± 0.2 | 0.9 ± 0.1 | 10.8 ± 1.2 | 54 ± 3 |
| HSA-Me/Me-mal C | −5.7 ± 0.3 | −8.4 ± 0.3 | 8.3 ± 0.9 | 1.4 ± 0.1 | 10.3 ± 1.2 | 50 ± 3 |
| HSA-Me/Me-mal T | −8.7 ± 0.4 | −4.0 ± 0.5 | 2.8 ± 0.2 | 0.5 ± 0.1 | 10.9 ± 1.1 | 47 ± 2 |
| HSA-Et-mal C | −2.8 ± 0.1 | −3.9 ± 0.3 | 18.2 ± 7.4 | 2.7 ± 0.3 | 22.6 ± 3.9 | 45 ± 5 |
| HSA-Et/Et-mal C | −6.2 ± 0.1 | – | 3.9 ± 0.1 | – | 21.2 ± 4.0 | 39 ± 5 |
| HSA-Et/Et-mal T | −2.4 ± 0.1 | −3.6 ± 0.1 | 6.9 ± 0.6 | 1.4 ± 0.1 | 24.3 ± 3.1 | 42 ± 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitin, D.; Bullinger, F.; Dobrynin, S.; Engelmann, J.; Scheffler, K.; Kolokolov, M.; Krumkacheva, O.; Buckenmaier, K.; Kirilyuk, I.; Chubarov, A. Contrast Agents Based on Human Serum Albumin and Nitroxides for 1H-MRI and Overhauser-Enhanced MRI. Int. J. Mol. Sci. 2024, 25, 4041. https://doi.org/10.3390/ijms25074041
Mitin D, Bullinger F, Dobrynin S, Engelmann J, Scheffler K, Kolokolov M, Krumkacheva O, Buckenmaier K, Kirilyuk I, Chubarov A. Contrast Agents Based on Human Serum Albumin and Nitroxides for 1H-MRI and Overhauser-Enhanced MRI. International Journal of Molecular Sciences. 2024; 25(7):4041. https://doi.org/10.3390/ijms25074041
Chicago/Turabian StyleMitin, Dmitry, Friedemann Bullinger, Sergey Dobrynin, Jörn Engelmann, Klaus Scheffler, Mikhail Kolokolov, Olesya Krumkacheva, Kai Buckenmaier, Igor Kirilyuk, and Alexey Chubarov. 2024. "Contrast Agents Based on Human Serum Albumin and Nitroxides for 1H-MRI and Overhauser-Enhanced MRI" International Journal of Molecular Sciences 25, no. 7: 4041. https://doi.org/10.3390/ijms25074041
APA StyleMitin, D., Bullinger, F., Dobrynin, S., Engelmann, J., Scheffler, K., Kolokolov, M., Krumkacheva, O., Buckenmaier, K., Kirilyuk, I., & Chubarov, A. (2024). Contrast Agents Based on Human Serum Albumin and Nitroxides for 1H-MRI and Overhauser-Enhanced MRI. International Journal of Molecular Sciences, 25(7), 4041. https://doi.org/10.3390/ijms25074041