

Gut Microbiome and Transcriptomic Changes in Cigarette Smoke-Exposed Mice Compared to COPD and CD Patient Datasets
 , , and
, , and 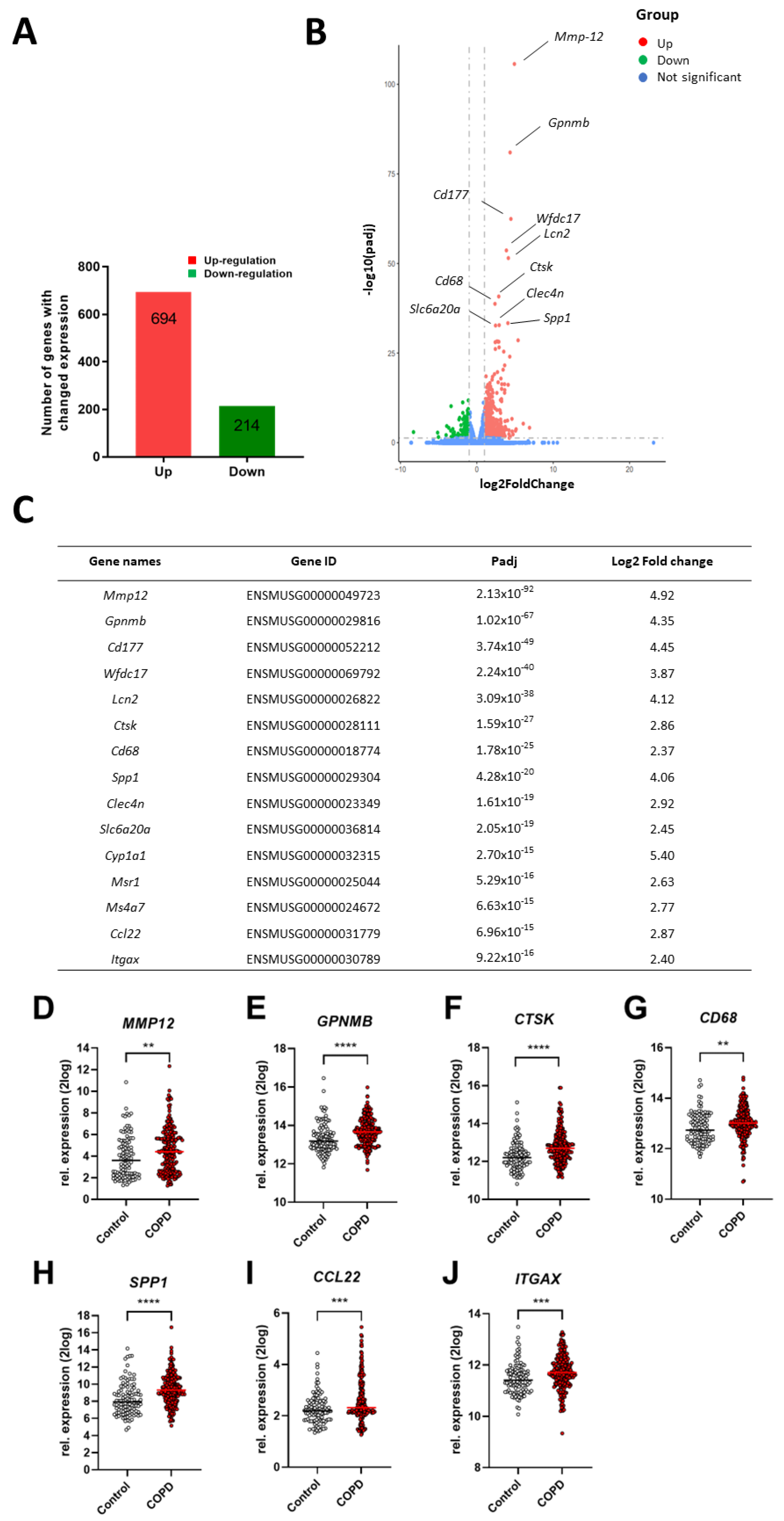{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Differential Changes in Lung Tissue Transcriptome of Cigarette Smoke-Exposed Mice
2.2. Comparison of Lung Gene Expression Profiles of Cigarette Smoke Exposed-Mice and COPD Patients
2.3. Differential Changes in Ileum Transcriptome of Cigarette Smoke-Exposed Mice
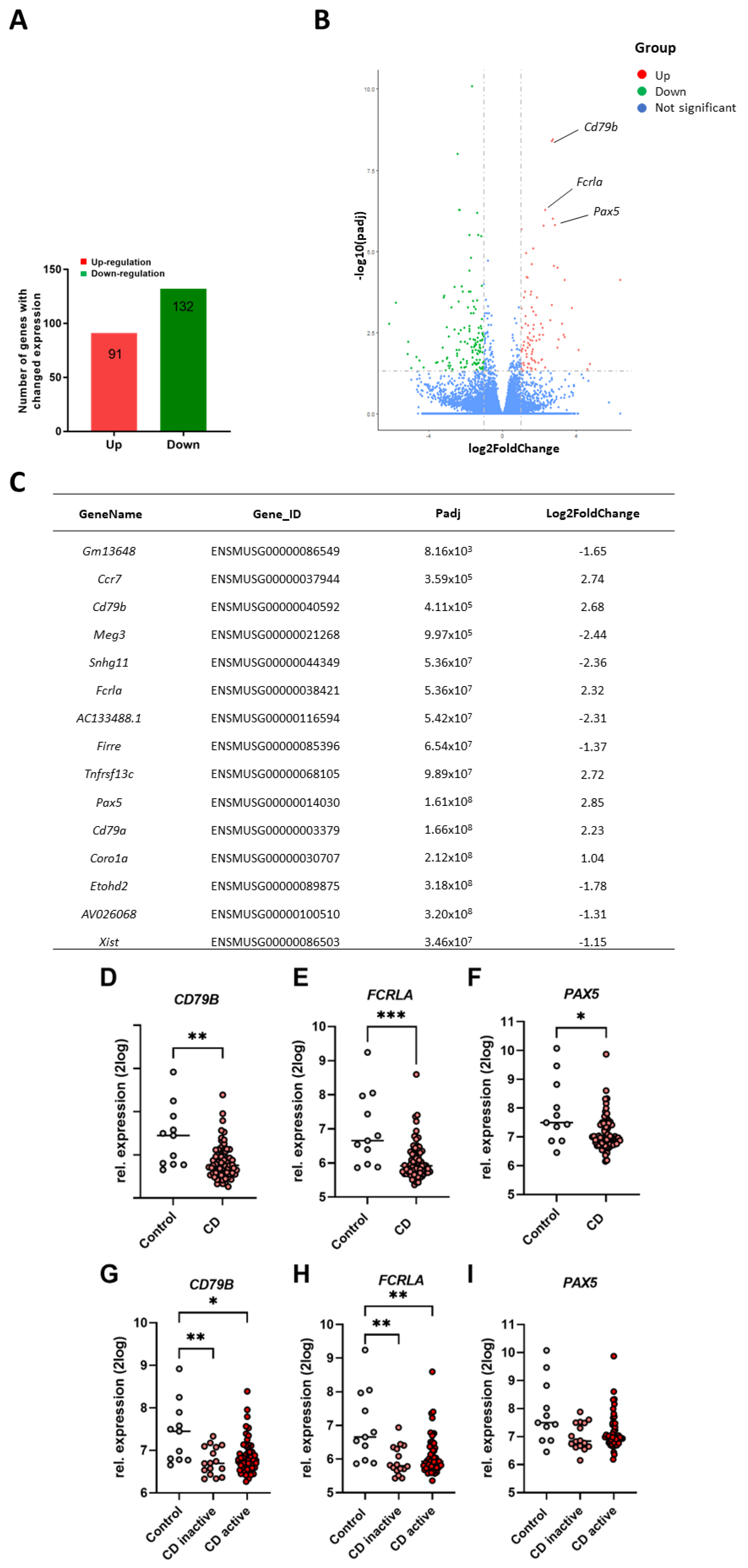
2.4. Comparison of Murine Ileum Gene Expression Profiles versus Ileum Gene Expression Levels of CD Patients
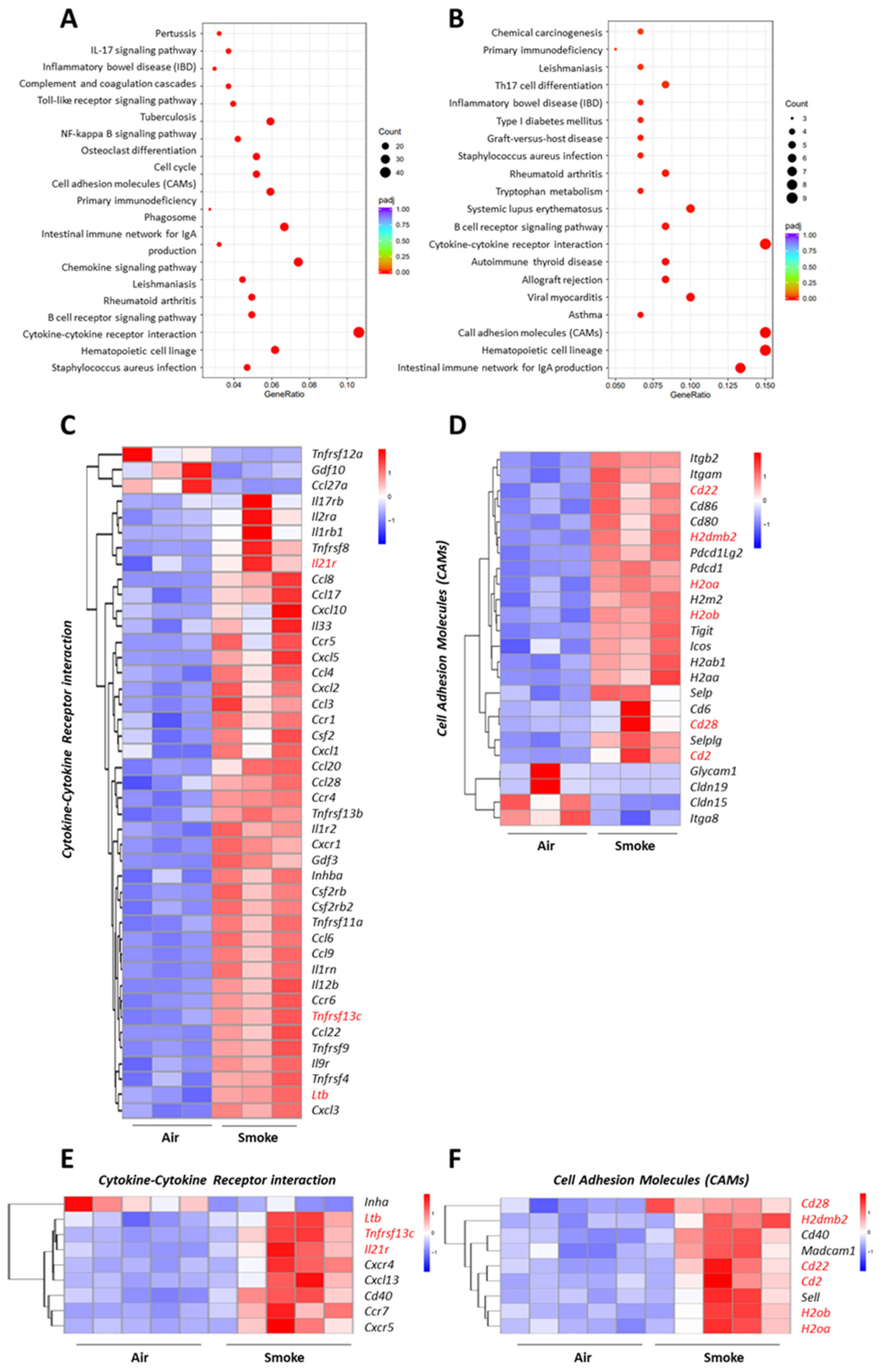
2.5. Overlapping Pathways in the Lung and Ileum of Cigarette Smoke-Exposed Mice
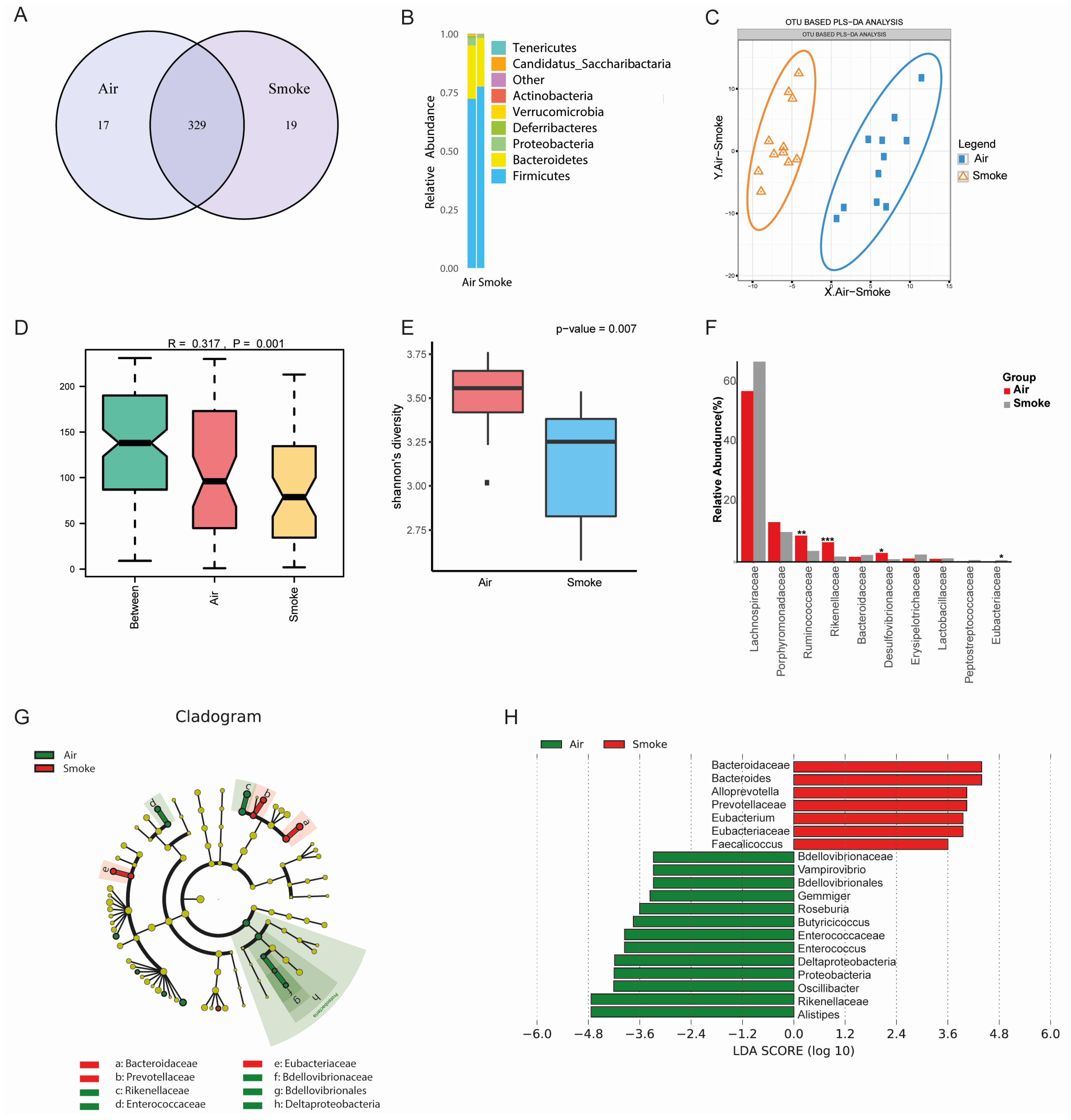
2.6. Altered Fecal Microbial Composition in Cigarette Smoke-Exposed Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cigarette Smoke Exposure
4.3. RNA Isolation and Gene Sequence
4.3.1. RNA Preparation
4.3.2. Non-Directional Sequencing of Polyadenylated (polyA) mRNA
4.4. Clinical Data
4.5. DNA Extraction of Fecal Samples, Library Construction, Gut Microbiota Sequencing
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. The Top 10 Causes of Death. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 14 December 2021).
- Vutcovici, M.; Bitton, A.; Ernst, P.; Kezouh, A.; Suissa, S.; Brassard, P. Inflammatory bowel disease and risk of mortality in COPD. Eur. Respir. J. 2016, 47, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Raftery, A.L.; Tsantikos, E.; Harris, N.L.; Hibbs, M.L. Links Between Inflammatory Bowel Disease and Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11, 2144. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Hu, S.; Li, C.; Ma, H.; Wang, Q.; Meng, G.; Guo, T.; Zhang, J. Cigarette Smoke Induced Lung Barrier Dysfunction, EMT, and Tissue Remodeling: A Possible Link between COPD and Lung Cancer. BioMed Res. Int. 2019, 2019, 2025636. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, L.; Schultz, B.M.; Salazar, G.A.; Pardo-Roa, C.; Sebastián, V.P.; Álvarez-Lobos, M.M.; Bueno, S.M. Impact of Cigarette Smoking on the Gastrointestinal Tract Inflammation: Opposing Effects in Crohn’s Disease and Ulcerative Colitis. Front. Immunol. 2018, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pelgrim, C.E.; Marzal, L.N.P.; Korver, S.; van Ark, I.; Leusink-Muis, T.; van Helvoort, A.; Keshavarzian, A.; Kraneveld, A.D.; Garssen, J.; et al. Changes in intestinal homeostasis and immunity in a cigarette smoke- and LPS-induced murine model for COPD: The lung-gut axis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L266–L280. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, L.; Dong, S.; Ge, S.; Zhu, T. Immune regulation of the gut-brain axis and lung-brain axis involved in ischemic stroke. Neural Regen. Res. 2024, 19, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.D.; Olwenyi, O.A.; Bhyravbhatla, N.; Thurman, M.; Pandey, K.; Klug, E.A.; Johnston, M.; Dyavar, S.R.; Acharya, A.; Podany, A.T.; et al. Therapeutic implications of SARS-CoV-2 dysregulation of the gut-brain-lung axis. World J. Gastroenterol. 2021, 27, 4763–4783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Jiang, X.; Mao, L.; Xia, Y.; Fan, Y.; Li, N.; Jiang, Z.; Qin, X.; Jiang, Y.; et al. Disruption of the lung-gut-brain axis is responsible for cortex damage induced by pulmonary exposure to zinc oxide nanoparticles. Toxicology 2023, 485, 153390. [Google Scholar] [CrossRef]
- Wang, L.; Cai, Y.; Garssen, J.; Henricks, P.A.J.; Folkerts, G.; Braber, S. The Bidirectional Gut-Lung Axis in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2023, 207, 1145–1160. [Google Scholar] [CrossRef]
- Gokulan, K.; Joshi, M.; Khare, S.; Bartter, T. Lung microbiome, gut-lung axis and chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 2022, 28, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Wiertsema, S.P.; van Bergenhenegouwen, J.; Garssen, J.; Knippels, L.M.J. The Interplay between the Gut Microbiome and the Immune System in the Context of Infectious Diseases throughout Life and the Role of Nutrition in Optimizing Treatment Strategies. Nutrients 2021, 13, 886. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.A.; Garssen, J.; Witkamp, R.F. Pharma-nutrition interface: The gap is narrowing. Eur. J. Pharmacol. 2011, 651, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- De Smet, E.G.; Van Eeckhoutte, H.P.; Cobos, F.A.; Blomme, E.; Verhamme, F.M.; Provoost, S.; Verleden, S.E.; Venken, K.; Maes, T.; Joos, G.F.; et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. 2020, 13, 423–436. [Google Scholar] [CrossRef]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut-lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Brassard, P.; Vutcovici, M.; Ernst, P.; Patenaude, V.; Sewitch, M.; Suissa, S.; Bitton, A. Increased incidence of inflammatory bowel disease in Quebec residents with airway diseases. Eur. Respir. J. 2015, 45, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Papoutsopoulou, S.; Satsangi, J.; Campbell, B.J.; Probert, C.S. Review article: Impact of cigarette smoking on intestinal inflammation-direct and indirect mechanisms. Aliment. Pharmacol. Ther. 2020, 51, 1268–1285. [Google Scholar] [CrossRef]
- Obeidat, M.; Dvorkin-Gheva, A.; Li, X.; Bossé, Y.; Brandsma, C.-A.; Nickle, D.C.; Hansbro, P.M.; Faner, R.; Agusti, A.; Paré, P.D.; et al. The Overlap of Lung Tissue Transcriptome of Smoke Exposed Mice with Human Smoking and COPD. Sci. Rep. 2018, 8, 11881. [Google Scholar] [CrossRef]
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Cui, Z.-H.; Dong, Y.; Liang, X.-W.; Zhao, Y.-X.; Baranova, A.; Cao, H.; Wang, L. GPNMB contributes to a vicious circle for chronic obstructive pulmonary disease. Biosci. Rep. 2020, 40, BSR20194459. [Google Scholar] [CrossRef]
- Golovatch, P.; Mercer, B.A.; Lemaître, V.; Wallace, A.; Foronjy, R.F.; D’Armiento, J. Role for cathepsin K in emphysema in smoke-exposed guinea pigs. Exp. Lung Res. 2009, 35, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Dong, T.; Wang, S.; Jing, H.; Chen, J. Vitamin D3-vitamin D receptor axis suppresses pulmonary emphysema by maintaining alveolar macrophage homeostasis and function. EBioMedicine 2019, 45, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Yuan, X.; Song, L.-Z.; Roberts, L.; Zarinkamar, N.; Seryshev, A.; Zhang, Y.; Hilsenbeck, S.; Chang, S.-H.; Dong, C.; et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci. Transl. Med. 2012, 4, 117ra9. [Google Scholar] [CrossRef] [PubMed]
- Miao, T.; Xiao, W.; Du, L.; Mao, B.; Huang, W.; Chen, X.; Li, C.; Wang, Y.; Fu, J. High expression of SPP1 in patients with chronic obstructive pulmonary disease (COPD) is correlated with increased risk of lung cancer. FEBS Open Bio 2021, 11, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-X.; Su, Y.; Zhang, A.-L.; Xu, J.-W.; Fu, Q.; Yan, L. MiR-34c-5p plays a protective role in chronic obstructive pulmonary disease via targeting CCL22. Exp. Lung Res. 2019, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marjanović, N.; Bosnar, M.; Michielin, F.; Willé, D.R.; Anić-Milić, T.; Čulić, O.; Popović-Grle, S.; Bogdan, M.; Parnham, M.J.; Haber, V.E. Macrolide antibiotics broadly and distinctively inhibit cytokine and chemokine production by COPD sputum cells in vitro. Pharmacol. Res. 2011, 63, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Frankenberger, M.; Eder, C.; Hofer, T.P.J.; Heimbeck, I.; Skokann, K.; Kaßner, G.; Weber, N.; Möller, W.; Ziegler-Heitbrock, L. Chemokine expression by small sputum macrophages in COPD. Mol. Med. 2011, 17, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Almansa, R.; Socias, L.; Sanchez-Garcia, M.; Martín-Loeches, I.; del Olmo, M.; Andaluz-Ojeda, D.; Bobillo, F.; Rico, L.; Herrero, A.; Roig, V.; et al. Critical COPD respiratory illness is linked to increased transcriptomic activity of neutrophil proteases genes. BMC Res. Notes 2012, 5, 401. [Google Scholar] [CrossRef]
- Esteve-Codina, A.; Hofer, T.P.; Burggraf, D.; Heiss-Neumann, M.S.; Gesierich, W.; Boland, A.; Olaso, R.; Bihoreau, M.-T.; Deleuze, J.-F.; Moeller, W.; et al. Gender specific airway gene expression in COPD sub-phenotypes supports a role of mitochondria and of different types of leukocytes. Sci. Rep. 2021, 11, 12848. [Google Scholar] [CrossRef]
- Pelgrim, C.E.; Wang, L.; Marzal, L.N.P.; Korver, S.; van Ark, I.; Leusink-Muis, T.; Braber, S.; Folkerts, G.; Garssen, J.; van Helvoort, A.; et al. Increased exploration and hyperlocomotion in a cigarette smoke and LPS induced murine model of COPD: Linking pulmonary and systemic inflammation with the brain. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L251–L265. [Google Scholar] [CrossRef]
- Ochsenkühn, T.; Herrmann, K.; Schoenberg, S.O.; Reiser, M.F.; Göke, B.; Sackmann, M. Crohn disease of the small bowel proximal to the terminal ileum: Detection by MR-enteroclysis. Scand. J. Gastroenterol. 2004, 39, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, L.; Pardo-Roa, C.; Salazar, G.A.; Salazar-Echegarai, F.; Miranda, J.P.; Ramírez, G.; Chávez, J.L.; Kalergis, A.M.; Bueno, S.M.; Álvarez-Lobos, M. Mucosal Exposure to Cigarette Components Induces Intestinal Inflammation and Alters Antimicrobial Response in Mice. Front. Immunol. 2019, 10, 2289. [Google Scholar] [CrossRef] [PubMed]
- Castro-Dopico, T.; Colombel, J.-F.; Mehandru, S. Targeting B cells for inflammatory bowel disease treatment: Back to the future. Curr. Opin. Pharmacol. 2020, 55, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Wouters, E.F.M.; Creutzberg, E.C.; Schols, A.M.W.J. Systemic effects in COPD. Chest 2002, 121 (Suppl. 113S), 127S–130S. [Google Scholar] [CrossRef]
- Barnes, P.J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Furusho, J.K. Inflammatory bowel disease therapy: Blockade of cytokines and cytokine signaling pathways. Curr. Opin. Gastroenterol. 2018, 34, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Vanderslice, P.; Biediger, R.J.; Woodside, D.G.; Berens, K.L.; Holland, G.W.; Dixon, R.A. Development of cell adhesion molecule antagonists as therapeutics for asthma and COPD. Pulm. Pharmacol. Ther. 2004, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Xu, Y.; Zha, X.; Zeng, J.; Qiao, S.; Yang, S.; Zhang, H.; Tan, L.; Sun, L.; Pang, G.; et al. IL-21/IL-21R Signaling Aggravated Respiratory Inflammation Induced by Intracellular Bacteria through Regulation of CD4(+) T Cell Subset Responses. J. Immunol. 2021, 206, 1586–1596. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, X.; Zhu, J.; Yue, D.; Zhang, X.; Wang, X.; You, Y.; Wang, B.; Xu, Y.; Lu, C.; et al. IL-21/IL-21R signaling suppresses intestinal inflammation induced by DSS through regulation of Th responses in lamina propria in mice. Sci. Rep. 2016, 6, 31881. [Google Scholar] [CrossRef]
- Cornwell, W.D.; Kim, V.; Song, C.; Rogers, T.J. Pathogenesis of inflammation and repair in advanced COPD. Semin. Respir. Crit. Care Med. 2010, 31, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Huang, Y.; Zhong, X.; Tang, H. IL-21 is increased in peripheral blood of emphysema mice and promotes Th1/Tc1 cell generation in vitro. Inflammation 2014, 37, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Dai, Z.; Wang, Z.; Deng, Z.; Zhang, J.; Pu, J.; Cao, W.; Pan, T.; Zhou, Y.; Yang, Z.; et al. Gut microbiota dysbiosis contributes to the development of chronic obstructive pulmonary disease. Respir. Res. 2021, 22, 274. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, K.L.; Rehman, S.F.; Vaughan, A.; Lachner, N.; Budden, K.F.; Kim, R.Y.; Wood, D.L.A.; Gellatly, S.L.; Shukla, S.D.; Wood, L.G.; et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun. 2020, 11, 5886. [Google Scholar] [CrossRef]
- Chiu, Y.-C.; Lee, S.-W.; Liu, C.-W.; Lin, R.C.-J.; Huang, Y.-C.; Lan, T.-Y.; Wu, L.S.-H. Comprehensive profiling of the gut microbiota in patients with chronic obstructive pulmonary disease of varying severity. PLoS ONE 2021, 16, e0249944. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wan, X.; Wu, X.; Zhang, C.; Liu, J.; Hou, S. Eubacterium rectale contributes to colorectal cancer initiation via promoting colitis. Gut Pathog. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.T.; Marsland, B.J. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol. 2019, 12, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liao, Y.; Yang, R.; Zhu, Z.; Zhang, L.; Wu, Z.; Sun, X. An engineered probiotic secreting Sj16 ameliorates colitis via Ruminococcaceae/butyrate/retinoic acid axis. Bioeng. Transl. Med. 2021, 6, e10219. [Google Scholar] [CrossRef] [PubMed]
- Rowan, F.; Docherty, N.G.; Murphy, M.; Murphy, B.; Coffey, J.C.; O’Connell, P.R. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis. Colon. Rectum 2010, 53, 1530–1536. [Google Scholar] [CrossRef]
- Lo Presti, A.; Zorzi, F.; Del Chierico, F.; Altomare, A.; Cocca, S.; Avola, A.; De Biasio, F.; Russo, A.; Cella, E.; Reddel, S.; et al. Fecal and Mucosal Microbiota Profiling in Irritable Bowel Syndrome and Inflammatory Bowel Disease. Front. Microbiol. 2019, 10, 1655. [Google Scholar] [CrossRef]
- Wang, L.; Pelgrim, C.E.; Swart, D.H.; Krenning, G.; van der Graaf, A.C.; Kraneveld, A.D.; Leusink-Muis, T.; van Ark, I.; Garssen, J.; Folkerts, G.; et al. SUL-151 Decreases Airway Neutrophilia as a Prophylactic and Therapeutic Treatment in Mice after Cigarette Smoke Exposure. Int. J. Mol. Sci. 2021, 22, 4991. [Google Scholar] [CrossRef] [PubMed]
- Roda, M.A.; Xu, X.; Abdalla, T.H.; Sadik, M.; Szul, T.; Bratcher, P.E.; Viera, L.; Solomon, G.M.; Wells, J.M.; McNicholas, C.M.; et al. Proline-Glycine-Proline Peptides Are Critical in the Development of Smoke-induced Emphysema. Am. J. Respir. Cell Mol. Biol. 2019, 61, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, A.; Betson, T.; Gama, M.V.; McAdam, K. Variation in tobacco and mainstream smoke toxicant yields from selected commercial cigarette products. Regul. Toxicol. Pharmacol. 2015, 71, 409–427. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Koelink, P.J.; Garssen, J.; Folkerts, G.; Henricks, P.A.J.; Braber, S. Gut Microbiome and Transcriptomic Changes in Cigarette Smoke-Exposed Mice Compared to COPD and CD Patient Datasets. Int. J. Mol. Sci. 2024, 25, 4058. https://doi.org/10.3390/ijms25074058
Wang L, Koelink PJ, Garssen J, Folkerts G, Henricks PAJ, Braber S. Gut Microbiome and Transcriptomic Changes in Cigarette Smoke-Exposed Mice Compared to COPD and CD Patient Datasets. International Journal of Molecular Sciences. 2024; 25(7):4058. https://doi.org/10.3390/ijms25074058
Chicago/Turabian StyleWang, Lei, Pim J. Koelink, Johan Garssen, Gert Folkerts, Paul A. J. Henricks, and Saskia Braber. 2024. "Gut Microbiome and Transcriptomic Changes in Cigarette Smoke-Exposed Mice Compared to COPD and CD Patient Datasets" International Journal of Molecular Sciences 25, no. 7: 4058. https://doi.org/10.3390/ijms25074058