
Ageing-Related Neurodegeneration and Cognitive Decline


Abstract
1. Introduction
2. Results
2.1. Subjects in the Fifth Decade
2.2. Subjects in the Sixth Decade
2.3. Subjects in the Seventh Decade
2.4. Subjects in the Eighth Decade
2.5. Subjects in the Ninth Decade
2.6. Subjects in the 10–11th Decade
2.7. Clinical Signs of Dementia
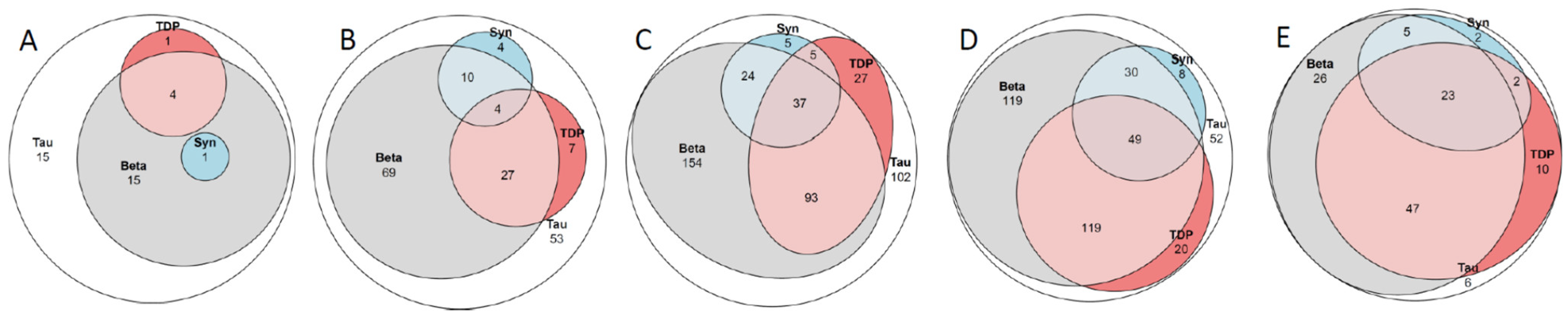
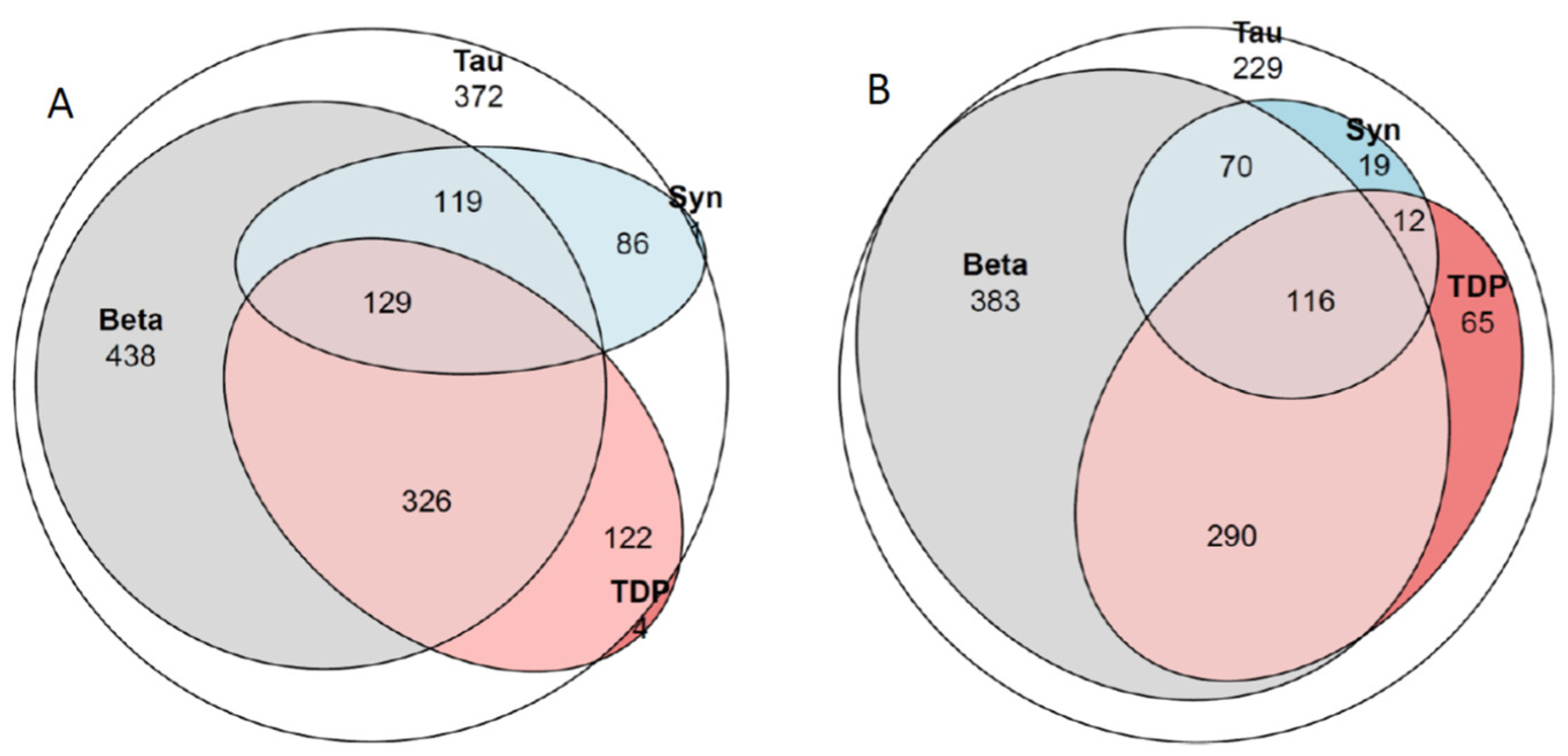
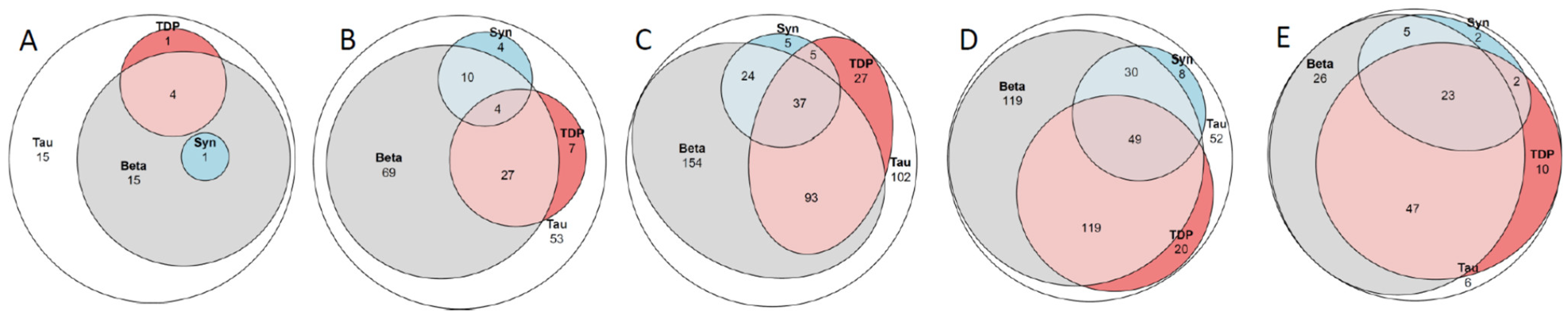
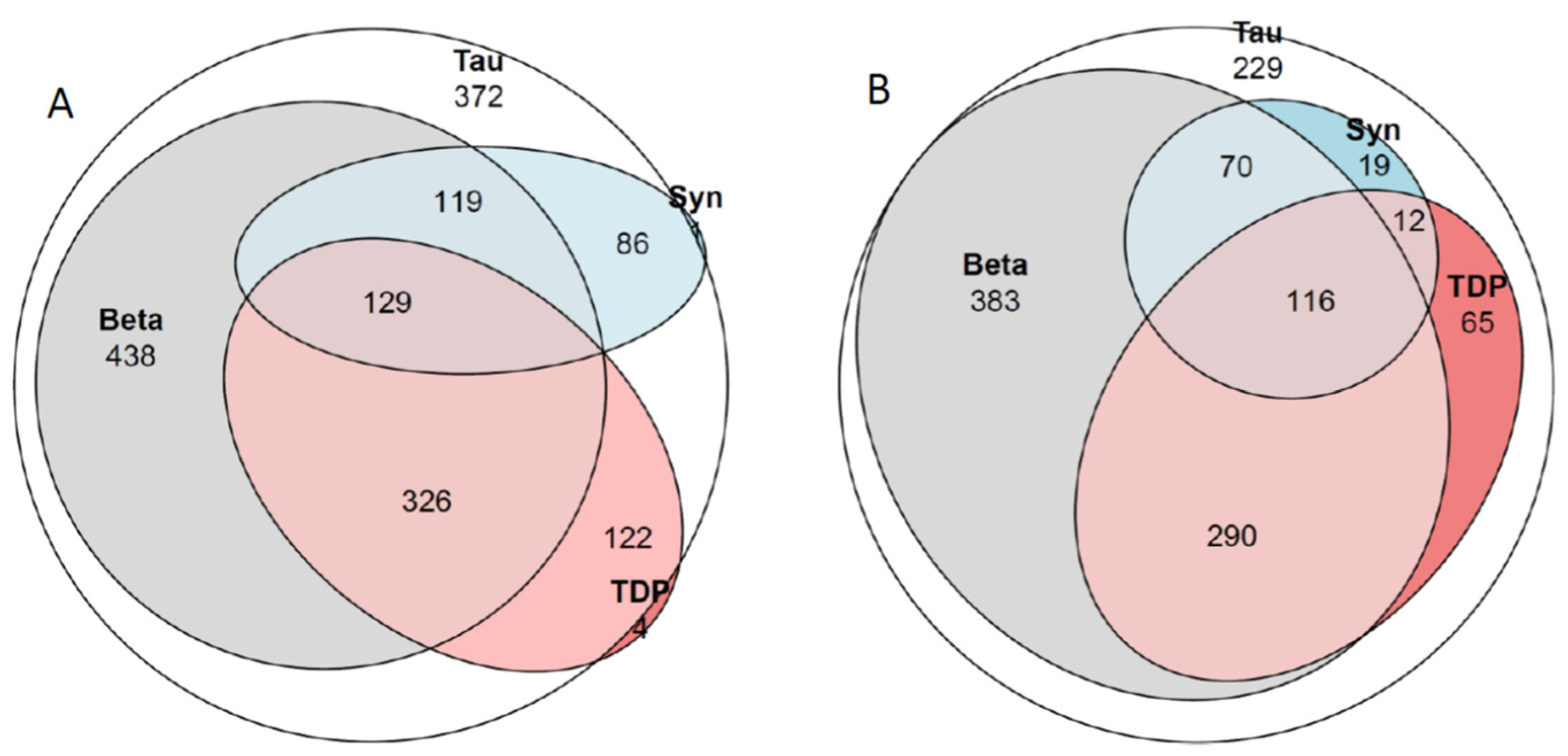
2.8. Concomitant Pathologies
Limbic-Predominant Age-Related TDP Encephalopathy (LATE)
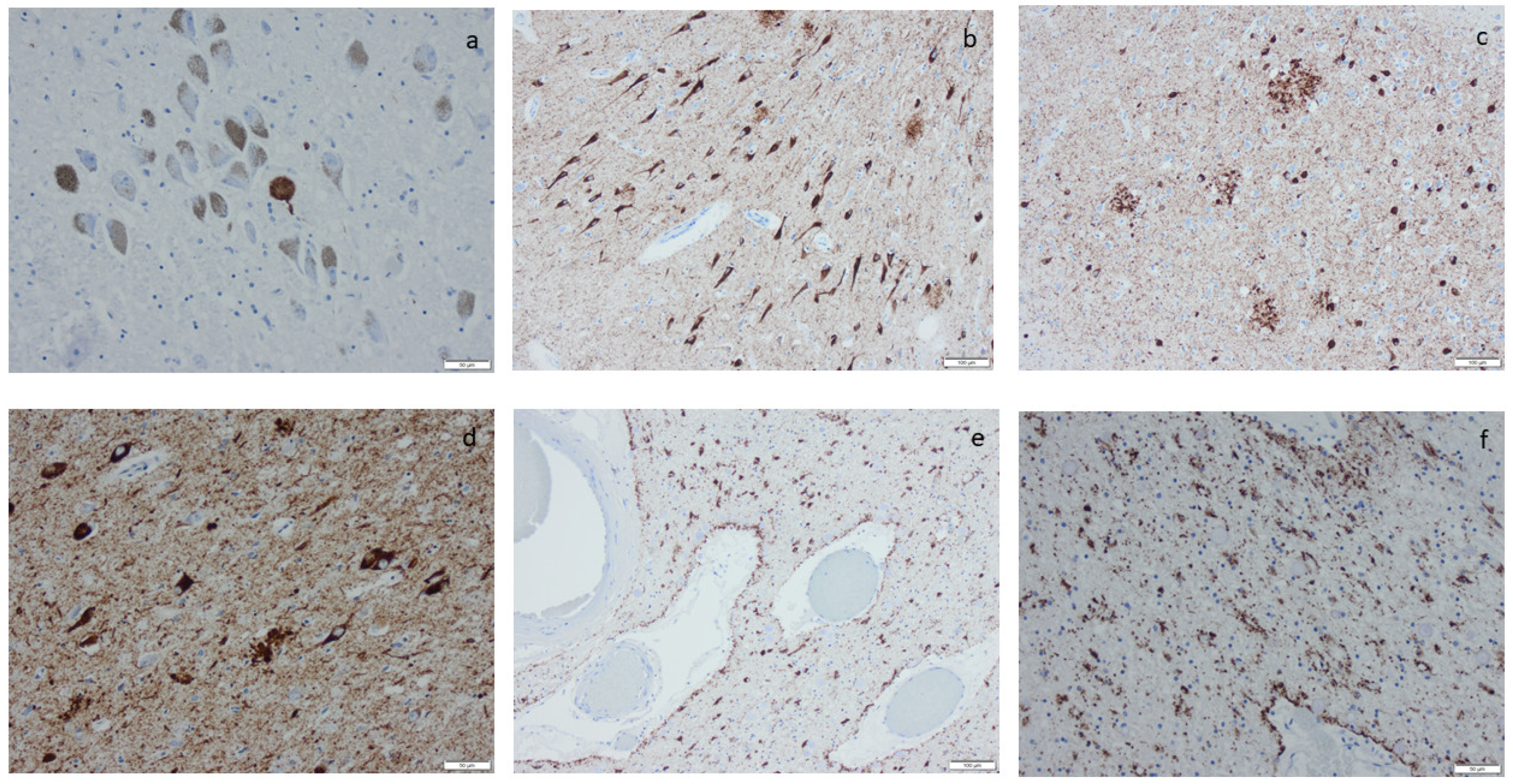
2.9. Age-Related Tau Astrogliopathy (ARTAG)
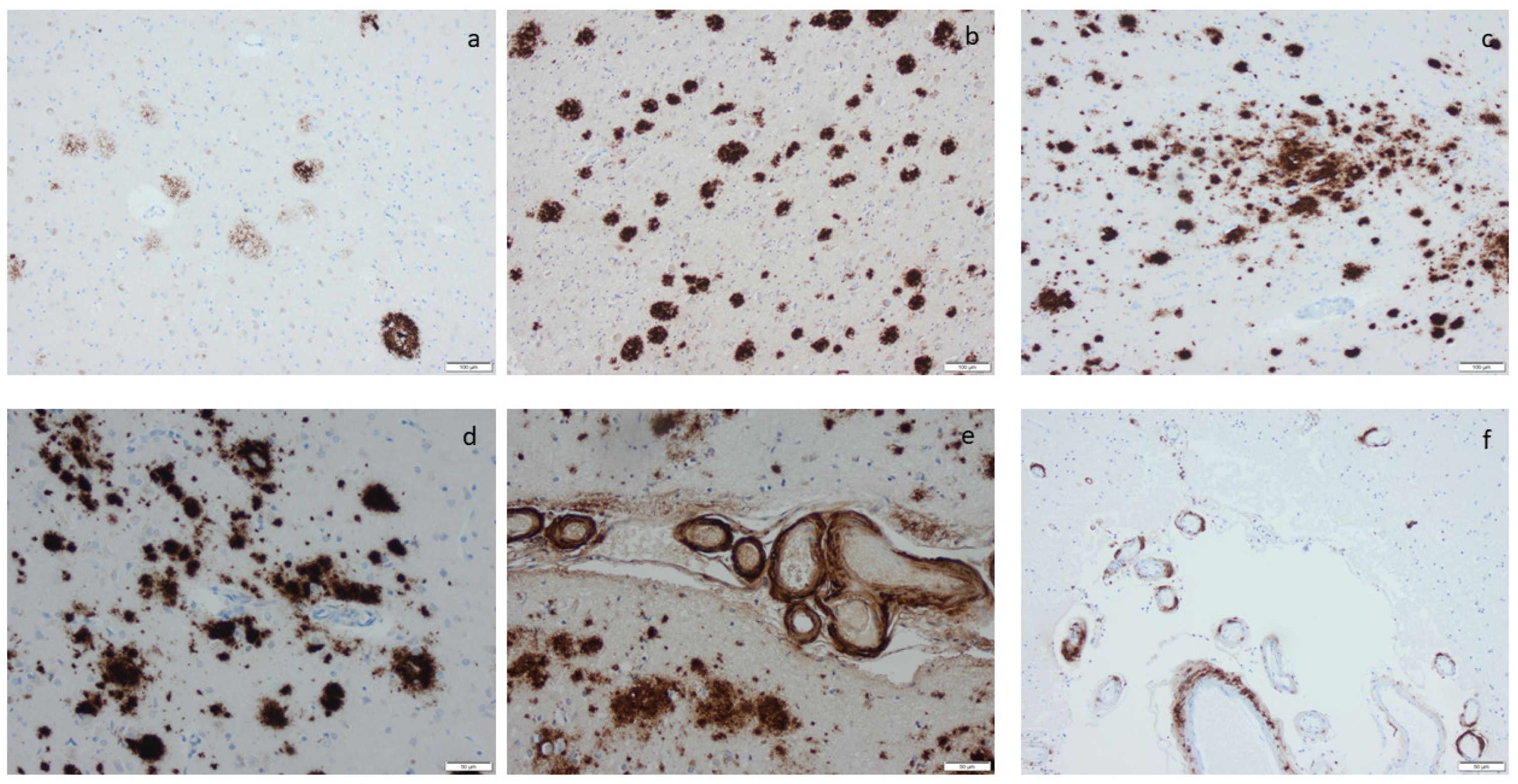
2.10. Cerebral Amyloid Angiopathy (CAA)
2.11. Vascular Neuropathologic Change (VNC)
3. Discussion
3.1. Autopsy Service and following Neuropathological Assessment
3.2. Clinical Signs of Cognitive Decline, Mild Cognitive Impairment, and Dementia
3.3. Hyperphosphorylated τ and β-Amyloid
3.4. Hyperphosphorylated τ in the Locus Coeruleus (LC)
3.5. ADNC and PART
3.6. Concomitant Pathologies
3.7. Vascular Neuropathologic Change (VNC)
3.8. Age-Related Tau Astrogliopathy (ARTAG)
3.9. Definite Diagnoses
3.10. Intervening with the Development and Progression of the Common Protein Alterations
4. Material and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, L.A.; Salmon, D.P.; Lowe, J.; Mirra, S.S.; Byrne, E.J.; et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 1996, 47, 1113–1124. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging. 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Alafuzoff, I.; Al-Sarraj, S.; Arzberger, T.; Bogdanovic, N.; Capellari, S.; Ferrer, I.; Gelpi, E.; Kövari, V.; Kretzschmar, H.; et al. Mixed Brain Pathologies in Dementia: The BrainNet Europe Consortium Experience. Dement. Geriatr. Cogn. Disord. 2008, 26, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Parisi, J.E.; Petrucelli, L.; Jack, C.R.; Petersen, R.C.; Dickson, D.W. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2013, 127, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Petrucelli, L.; Liesinger, A.M.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016, 131, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Lee, E.B.; Cykowski, M.D.; Alafuzoff, I.; Arfanakis, K.; Attems, J.; Brayne, C.; Corrada, M.M.; Dugger, B.N.; Flanagan, M.E.; et al. LATE-NC staging in routine neuropathologic diagnosis: An update. Acta Neuropathol. 2022, 145, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Ferrer, I.; Grinberg, L.T.; Alafuzoff, I.; Attems, J.; Budka, H.; Cairns, N.J.; Crary, J.F.; Duyckaerts, C.; Ghetti, B.; et al. Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol. 2015, 131, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Alafuzoff, I.; Libard, S. Mixed Brain Pathology Is the Most Common Cause of Cognitive Impairment in the Elderly. J. Alzheimer’s Dis. 2020, 78, 453–465. [Google Scholar] [CrossRef]
- McAleese, K.E.; Colloby, S.J.; Thomas, A.J.; Al-Sarraj, S.; Ansorge, O.; Neal, J.; Roncaroli, F.; Love, S.; Francis, P.T.; Attems, J. Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia. Alzheimer’s Dement. 2021, 17, 1121–1133. [Google Scholar] [CrossRef]
- Skrobot, O.A.; Attems, J.; Esiri, M.; Hortobágyi, T.; Ironside, J.W.; Kalaria, R.N.; King, A.; Lammie, G.A.; Mann, D.; Neal, J.; et al. Vascular cognitive impairment neuropathology guidelines (VCING): The contribution of cerebrovascular pathology to cognitive impairment. Brain 2016, 139, 2957–2969. [Google Scholar] [CrossRef]
- Grossman, M.; Seeley, W.W.; Boxer, A.L.; Hillis, A.E.; Knopman, D.S.; Ljubenov, P.A.; Miller, B.; Piguet, O.; Rademakers, R.; Whitwell, J.L.; et al. Frontotemporal lobar degeneration. Nat. Rev. Dis. Primers. 2023, 9, 40. [Google Scholar] [CrossRef]
- Martínez-Hernández, A. The autopsy in crisis. Rev. Med. Chil. 2000, 128, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Tamsen, F.; Alafuzoff, I. When is a postmortem examination carried out? A retrospective analysis of all Swedish deaths 1999–2018. Virchows Arch. 2023, 482, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Friberg, N.; Ljungberg, O.; Berglund, E.; Berglund, D.; Ljungberg, R.; Alafuzoff, I.; Englund, E. Cause of death and significant disease found at autopsy. Virchows Arch. 2019, 475, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Cholerton, B.A.; Corrada, M.M.; Edland, S.D.; Flanagan, M.E.; Hemmy, L.S.; Kawas, C.H.; White, L.R. Concepts for brain aging: Resistance, resilience, reserve, and compensation. Alzheimer’s Res. Ther. 2019, 11, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Beardmore, R.; Hou, R.; Darekar, A.; Holmes, C.; Boche, D. The Locus Coeruleus in Aging and Alzheimer’s Dis-ease: A Postmortem and Brain Imaging Review. J. Alzheimers Dis. 2021, 83, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.K.; Del Tredici, K.; Thomas, T.L.; Braak, H.; Diamond, M.I. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.D. Alzheimer’s pathogenesis: Is there neuron-to-neuron propagation? Acta Neuropathol. 2011, 121, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Kaalund, S.S.; Passamonti, L.; Allinson, K.S.J.; Murley, A.G.; Robbins, T.W.; Spillantini, M.G.; Rowe, J.B. Locus coeruleus pathology in progressive su-pranuclear palsy, and its relation to disease severity. Acta Neuropathol. Commun. 2020, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, A.; Shaw, P.; Weintraub, S.; Bigio, E.; Mash, D.C.; Mesulam, M.M. Locus coeruleus neurofibrillary degeneration in ag-ing, mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging. 2007, 28, 327–335. [Google Scholar] [CrossRef]
- Andrés-Benito, P.; Fernández-Dueñas, V.; Carmona, M.; Escobar, L.A.; Torrejón-Escribano, B.; Aso, E.; Ciruela, F.; Ferrer, I. Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol. Appl. Neurobiol. 2017, 43, 373–392. [Google Scholar] [CrossRef]
- Ghosh, A.; Torraville, S.E.; Mukherjee, B.; Walling, S.G.; Martin, G.M.; Harley, C.W.; Yuan, Q. An experimental model of Braak’s pretangle pro-posal for the origin of Alzheimer’s disease: The role of locus coeruleus in early symptom development. Alzheimers Res. Ther. 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Klimek, V.; Stockmeier, C.; Overholser, J.; Meltzer, H.Y.; Kalka, S.; Dilley, G.; Ordway, G.A. Reduced Levels of Norepinephrine Transporters in the Locus Coeruleus in Major Depression. J. Neurosci. 1997, 17, 8451–8458. [Google Scholar] [CrossRef] [PubMed]
- Hobson, J.A.; McCarley, R.W.; Wyzinski, P.W. Sleep Cycle Oscillation: Reciprocal Discharge by Two Brainstem Neuronal Groups. Science 1975, 189, 55–58. [Google Scholar] [CrossRef]
- Berridge, C.W.; Stellick, R.L.; Schmeichel, B.E. Wake-promoting actions of medial basal forebrain beta2 receptor stimulation. Behav. Neurosci. 2005, 119, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.; Thomas, A.J. Depression and dementia: Cause, consequence or coincidence? Maturitas 2014, 79, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Galgani, A.; Giorgi, F.S. Exploring the Role of Locus Coeruleus in Alzheimer’s Disease: A Com-prehensive Update on MRI Studies and Implications. Curr. Neurol. Neurosci. Rep. 2023, 23, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Bancher, C. Senile Dementia with Tangles (Tangle Predominant Form of Senile Dementia). Brain Pathol. 1998, 8, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Braak, H.; Brion, J.-P.; Buée, L.; Del Tredici, K.; Goedert, M.; Halliday, G.; Neumann, M.; Spillantini, M.G.; Tolnay, M.; et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015, 129, 749–756. [Google Scholar] [CrossRef]
- Ganguli, M.; Snitz, B.E.; Saxton, J.A.; Chang, C.-C.H.; Lee, C.-W.; Bilt, J.V.; Hughes, T.F.; Loewenstein, D.A.; Unverzagt, F.W.; Petersen, R.C. Outcomes of mild cognitive impairment by definition: A population study. Arch. Neurol. 2011, 68, 761–767. [Google Scholar] [CrossRef]
- Wharton, S.B.; Simpson, J.E.; Ince, P.G.; Richardson, C.D.; Merrick, R.; Matthews, F.E.; Brayne, C.; CFAS. Insights into the pathological basis of dementia from population-based neuropathology studies. Neuropathol. Appl. Neurobiol. 2023, 49, e12923. [Google Scholar] [CrossRef] [PubMed]
- Gauthreaux, K.; A Bonnett, T.; Besser, L.M.; Brenowitz, W.D.; Teylan, M.; Mock, C.; Chen, Y.-C.; Chan, K.C.G.; Keene, C.D.; Zhou, X.-H.; et al. Concordance of Clinical Alzheimer Diagnosis and Neuropathological Features at Autopsy. J. Neuropathol. Exp. Neurol. 2020, 79, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Vitek, G.E.; Decourt, B.; Sabbagh, M.N. Lecanemab (BAN2401): An anti–beta-amyloid monoclonal antibody for the treatment of Alzheimer disease. Expert. Opin. Investig. Drugs 2023, 32, 89–94. [Google Scholar] [CrossRef] [PubMed]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017, 27, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F.H.; Williams, B.; Ritz, E. Essential hypertension. Lancet 2007, 370, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Ogrodowczyk, M.; Dettlaff, K.; Jelinska, A. Beta-Blockers: Current State of Knowledge and Perspectives. Mini Rev. Med. Chem. 2016, 16, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Rabin, J.S.; Schultz, A.P.; Hedden, T.; Viswanathan, A.; Marshall, G.A.; Kilpatrick, E.; Klein, H.; Buckley, R.F.; Yang, H.-S.; Properzi, M.; et al. Interactive Associations of Vascular Risk and β-Amyloid Burden With Cognitive Decline in Clinically Normal Elderly Individuals: Findings From the Harvard Aging Brain Study. JAMA Neurol. 2018, 75, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Trudeau, K.; Behl, Y.; Dhar, S.; Chronopoulos, A. New Insights into Hyperglycemia-induced Molecular Changes in Microvascular Cells. J. Dent. Res. 2009, 89, 116–127. [Google Scholar] [CrossRef] [PubMed]
- van Arendonk, J.; Neitzel, J.; Steketee, R.M.E.; van Assema, D.M.E.; Vrooman, H.A.; Segbers, M.; Ikram, M.A.; Vernooij, M.W. Diabetes and hypertension are related to amy-loid-beta burden in the population-based Rotterdam Study. Brain 2023, 146, 337–348. [Google Scholar] [CrossRef]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Brunnström, H.; Englund, E. Clinicopathological Concordance in Dementia Diagnostics. Am. J. Geriatr. Psychiatry 2009, 17, 664–670. [Google Scholar] [CrossRef] [PubMed]
- McAleese, K.E.; Walker, L.; Erskine, D.; Johnson, M.; Koss, D.; Thomas, A.J.; Attems, J. Concomitant LATE-NC in Alzheimer’s disease is not associated with increased tau or amyloid-β pathological burden. Neuropathol. Appl. Neurobiol. 2020, 46, 722–734. [Google Scholar] [CrossRef]
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.B.; Fitzpatrick, A.L.; Lopez, O.; Jackson, S.; Lyketsos, C.; Jagust, W.; Ives, D.; DeKosky, S.T.; Kuller, L.H. Dementia and Alzheimer’s disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J. Am. Geriatr. Soc. 2005, 53, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Plessis, D.D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.A.; Lee, V.M.Y. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Petrou, M.; Dwamena, B.A.; Foerster, B.R.; MacEachern, M.P.; Bohnen, N.I.; Müller, M.L.; Albin, R.L.; Frey, K.A. Amyloid deposition in Parkinson’s disease and cognitive impairment: A systematic review. Mov. Disord. 2015, 30, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Madonna, R.; Balistreri, C.R.; Geng, Y.-J.; De Caterina, R. Diabetic microangiopathy: Pathogenetic insights and novel therapeutic approaches. Vasc. Pharmacol. 2017, 90, 1–7. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Marković, M.; Milošević, J.; Wang, W.; Cao, Y. Passive Immunotherapies Targeting Amyloid-β in Alzheimer’s Disease: A Quantitative Systems Pharmacology Perspective. Mol. Pharmacol. 2023, 105, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Cummings, J.; Doody, R.; Kramer, L.; Salloway, S.; Selkoe, D.J.; Sims, J.; Sperling, R.A.; Vellas, B. The Future of Anti-Amyloid Trials. J. Prev. Alzheimers Dis. 2020, 7, 146–151. [Google Scholar] [CrossRef]
- Killinger, B.A.; Madaj, Z.; Sikora, J.W.; Rey, N.; Haas, A.J.; Vepa, Y.; Lindqvist, D.; Chen, H.; Thomas, P.M.; Brundin, P.; et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5280. [Google Scholar] [CrossRef]
- Lu, H.-T.; Shen, Q.-Y.; Xie, D.; Zhao, Q.-Z.; Xu, Y.-M. Lack of association between appendectomy and Parkinson’s disease: A systematic review and meta-analysis. Aging Clin. Exp. Res. 2019, 32, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Leino, M.; Popova, S.N.; Alafuzoff, I. Transactive DNA Binding Protein 43 Rather Than Other Misfolded Proteins in the Brain is Associated with Islet Amyloid Polypeptide in Pancreas in Aged Subjects with Diabetes Mellitus. J. Alzheimer’s Dis. 2017, 59, 43–56. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Ince, P.G.; Arzberger, T.; Al-Sarraj, S.; Bell, J.; Bodi, I.; Bogdanovic, N.; Bugiani, O.; Ferrer, I.; Gelpi, E.; et al. Staging/typing of Lewy body related α-synuclein pathology: A study of the BrainNet Europe Consortium. Acta Neuropathol. 2009, 117, 635–652. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Thal, D.R.; Arzberger, T.; Bogdanovic, N.; Al-Sarraj, S.; Bodi, I.; Boluda, S.; Bugiani, O.; Duyckaerts, C.; Gelpi, E.; et al. Assessment of beta-amyloid deposits in human brain: A study of the BrainNet Europe Consortium. Acta Neuropathol. 2009, 117, 309–320. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Argyrophilic grains: Characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci. Lett. 1987, 76, 124–127. [Google Scholar] [CrossRef]
- Rösler, T.W.; Marvian, A.T.; Brendel, M.; Nykänen, N.-P.; Höllerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-repeat tauopathies. Prog. Neurobiol. 2019, 180, 101644. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
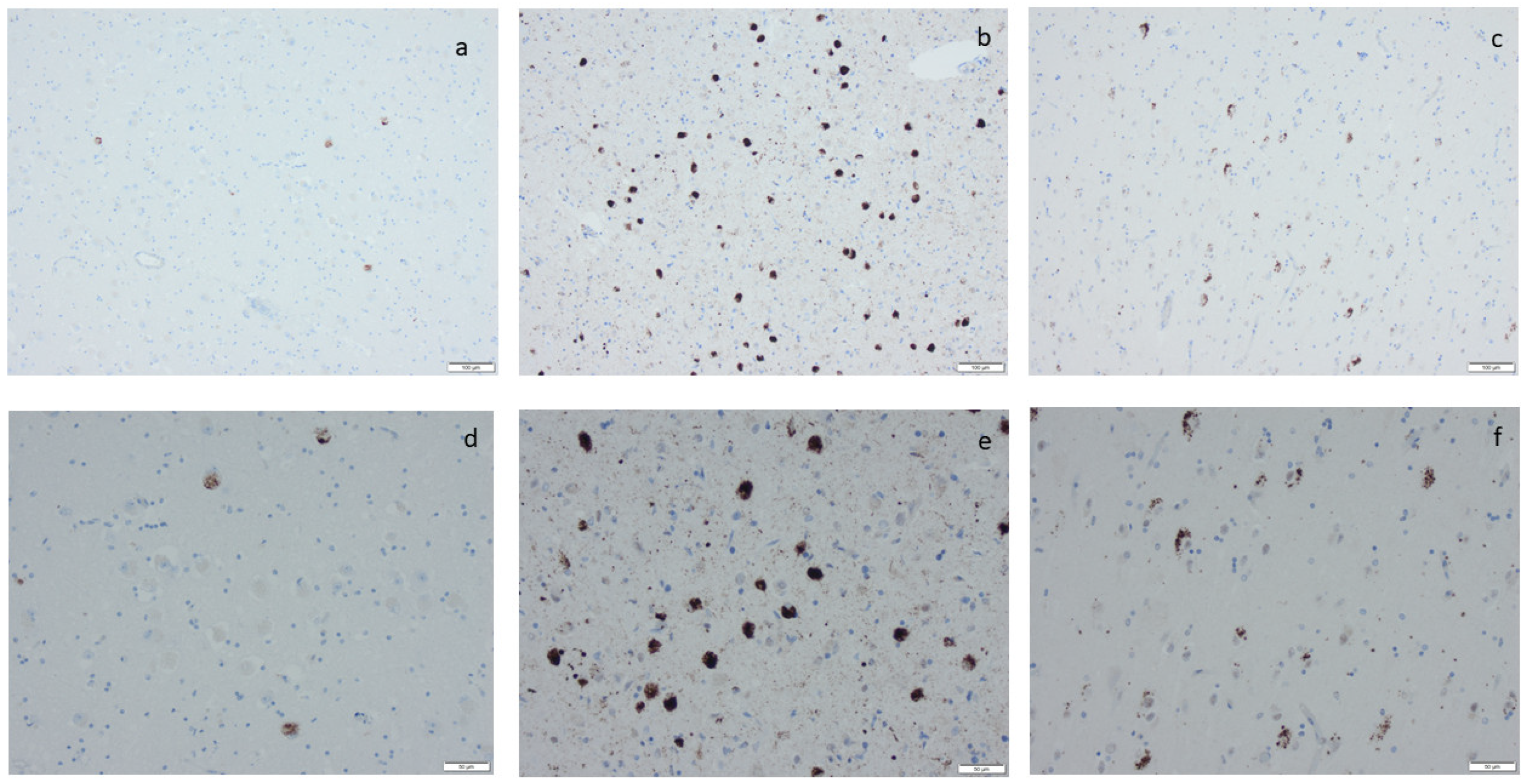{kind=link}
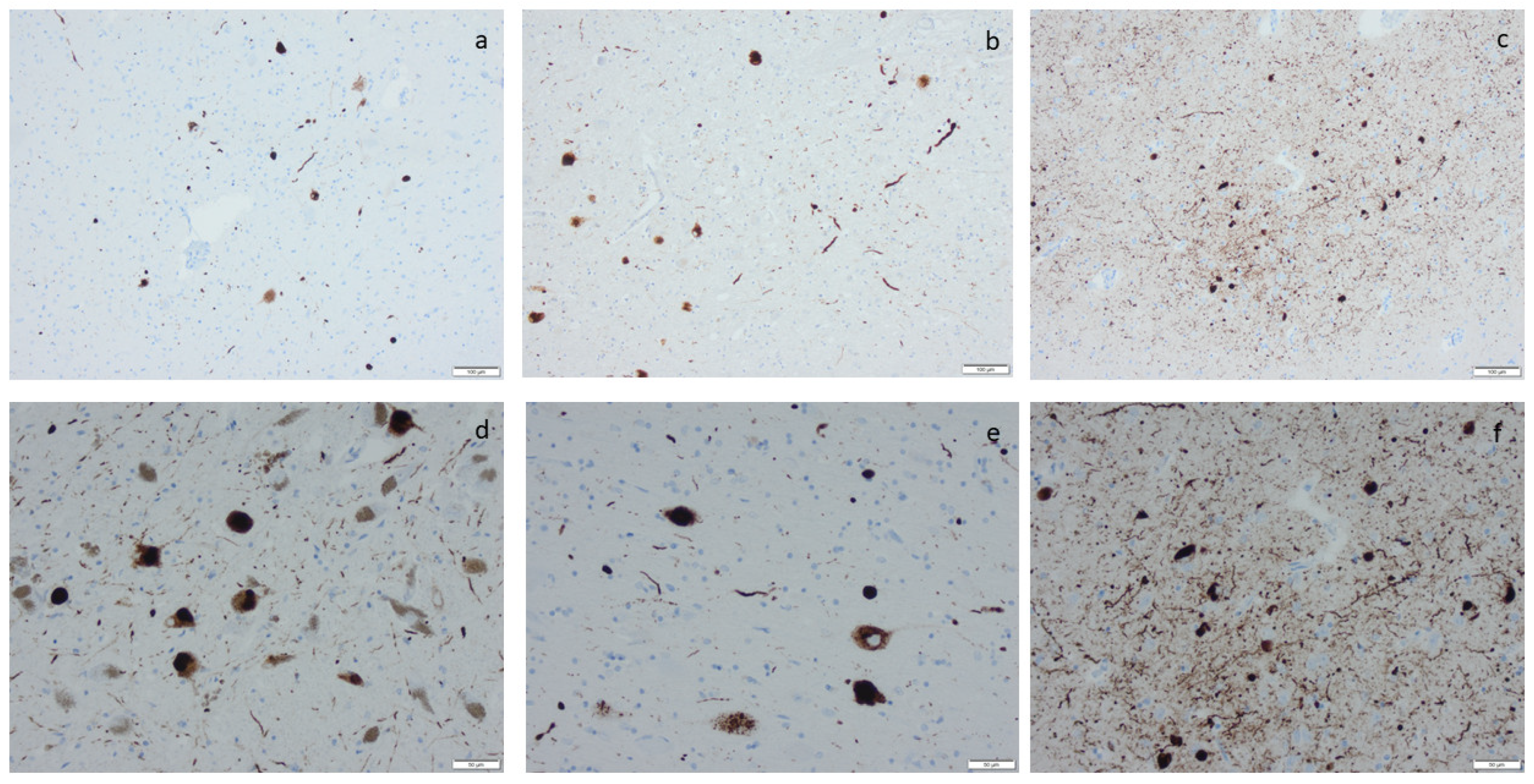{kind=link}
| 36,034 deceased subjects in the morgue during 15 years | ||
| 4389 (12% of the deceased) autopsies performed | ||
| 1630 (37% of the autopsied and 5% of the diseased) neuropathological assessments carried out | ||
| 1610 of the subjects age at death ≥41 years | ||
| 1576 subjects displayed hyperphosphorylated τ in the locus coeruleus or with the distribution as seen in ADNC/PART | 34 subjects 14 lacked neuropathological changes 3 subjects displayed only amyloid β-protein in the brain 17 displayed primary tauopathies | |
| 1421 subjects displayed hyperphosphorylated τ in the locus coeruleus or with the distribution as seen in ADNC/PART | 155 subjects displayed hyperphosphorylated τ in locus coeruleus or in Braak stage I–II and concomitant significant other neuropathologic change 113 subjects with α-synuclein Braak stage ≥ 4 [10] 26 subjects with TDP43 pathology Josephs stage ≥ 3 [13] 21 subjects with frontotemporal lobar degeneration | |
| 1184 subjects with hyperphosphorylated τ in Braak stages I–VI | 237 subjects with hyperphosphorylated τ limited to the locus coeruleus | |
| No HPτ | with HPτ/LC | with PART | with ADNC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Concomitant NC | L | I | L | I | H | ∑ | % of ∑ | ||
| All cases | 29 | 247 | 340 | 51 | 426 | 215 | 9 | 1317 | |
| No concomitant NC | 14 | 197 | 211 | 15 | 250 | 54 | 1 | 742 | 56 |
| +LATE-NC a/b | 22/1 | 45/3 | 14/2 | 74/11 | 80/17 | 3/2 | 274 | 21 | |
| +LATE-NC + CAA | 7 | 2 | 9 | <1 | |||||
| +LBD-NC a/b | 10/7 | 14/22 | 2/1 | 21/34 | 6/18 | 1/0 | 136 | 10 | |
| +LBD + CAA | 0/3 | 1 | 4 | <1 | |||||
| +MSA + CAA | 1 | 1 | <1 | ||||||
| +LATE-NC + LBD-NC | 3 | 14 | 6 | 27 | 35 | 2 | 87 | 7 | |
| +Argyrophilic grains (Ag) | 2 | 1 | 2 | 1 | 1 | 7 | <1 | ||
| +Ag + LATE-NC | 1 | 2 | 3 | <1 | |||||
| +Ag + LBD-NC a/b | 1/0 | 1 | 2 | <1 | |||||
| +Ag + LATE-NC + LBD-NC | 1 | 1 | 2 | <1 | |||||
| +Ag + PSP | 1 | 1 | 2 | <1 | |||||
| +PSP/CBD | 5 | 1 | 6 | <1 | |||||
| +PSP/CBD + LATE-NC | 1 | 3 | 1 | 5 | <1 | ||||
| +CBD + AgD + LATE-NC | 1 | 1 | <1 | ||||||
| +FTLD | 3 | 2 | 3 | 8 | <1 | ||||
| +ARTAG | 7 | 33 | 108 | 29 | 139 | 111 | 7 | 435 | 33 |
| +VNC | 11 | 130 | 181 | 25 | 280 | 149 | 7 | 783 | 59 |
| +severe VNC | 1 | 4 | 2 | 24 | 14 | 45 | 3 | ||
| No HPτ | with HPτ/LC | with PART | with ADNC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Concomitant NC | L | I | H | L | I | H | ∑ | % of ∑ | ||
| All cases | 5 | 4 | 20 | 9 | 1 | 17 | 135 | 102 | 293 | |
| No concomitant NC | 34 | 12 | 46 | 16 | ||||||
| +LATE-NC a/b | 0/2 | 1/0 | 0/1 | 36/14 | 31/16 | 101 | 34 | |||
| +LATE-NC + CAA a/b | 1/0 | 1/0 | 2 | <1 | ||||||
| +LBD-NC a/b | 0/1 | 0/3 | 0/3 | 0/4 | 2/11 | 1/8 | 33 | 11 | ||
| +LBD-NC + CAA a/b | 0/1 | 1 | <1 | |||||||
| +MSA | 1 | 1 | <1 | |||||||
| +LATE-NC + LBD-NC | 1 | 6 | 2 | 5 | 31 | 34 | 79 | 27 | ||
| +Argyrophilic grains (Ag) | 1 | 1 | <1 | |||||||
| +Ag +LATE-NC+ CAA | 1 | 1 | 2 | <1 | ||||||
| +PSP/CBD | 4 | 4 | 1 | |||||||
| +PSP + LATE-NC | 1 | 1 | <1 | |||||||
| +PSP + LBD-NC | 0/1 | 1 | <1 | |||||||
| +FTLD- | 2 | 8 | 6 | 5 | 21 | 7 | ||||
| +ARTAG | 2 | 1 | 14 | 6 | 8 | 76 | 48 | 155 | 52 | |
| +VNC | 3 | 14 | 7 | 10 | 91 | 82 | 207 | 71 | ||
| +severe VNC | 1 | 12 | 4 | 17 | 6 | |||||
| ALL n (%) | PART n (%) | ADNC n (%) | Statistics | |
|---|---|---|---|---|
| All | 1184 | 362 (31) | 822 (69) | |
| Male/female | 652/532 | 212/150 | 440/382 | Ns |
| Age range | 48–102 | 48–102 | 50–102 | |
| Mean age at death ± standard error | 77.8 ± 0.3 | 75.8 ± 0.5 | 78.7 ± 0.3 | MWU < 0.001 |
| Eith dementia | 248 (21) | 11 (3) | 237 (29) | FE < 0.001 |
| Brain weight in grams | 1351 ± 5 | 1373 ± 9 | 1341 ± 6 | MWU < 0.002 |
| With HPτ, Braak stages I–II [2] | 661 (56) | 301 (83) | 361 (44) | FE < 0.001 |
| With HPτ, Braak stages III–IV [2] | 410 (35) | 60 (17) | 350 (43) | FE < 0.001 |
| With HPτ, Braak stages V–VI [2] | 112 (10) | 1 (<1) | 111 (14) | FE < 0.001 |
| With ARTAG | 487 (41) | 130 (36) | 357 (43) | FE 0.018 |
| With argyrophilic grains | 10 (1) | 7 (2) | 3 (<1) | FE 0.012 |
| With LATE-NC (Josephs ≥1) [13] | 482 (41) | 93 (26) | 389 (47) | FE < 0.001 |
| With severe LATE-NC (Josephs ≥3) [13] | 83 (7) | 2 (<1) | 81 (10) | FE < 0.001 |
| With LBD-NC [10] | 216 (18) | 36 (10) | 180 (22) | FE < 0.001 |
| With severe LBD-NC (Braak stage ≥4) [10] | 136 (12) | 11 (3) | 125 (15) | FE < 0.001 |
| With cerebral amyloid angiopathy (CAA) | 388 (33) | 37 (10) | 351 (43) | FE < 0.001 |
| Hippocampal sclerosis (HS) | 32 (3) | 8 (2) | 24 (3) | |
| With V-NC | 759 (64) | 191 (53) | 568 (69) | FE < 0.001 |
| With severe V-NC | 55 (5) | 5 (1) | 50 (6) | FE < 0.001 |
| Age at Death in Years | 50–59 | 60–69 | 70–79 | 80–89 | 90–102 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lesions | PART | ADNC | PART | ADNC | PART | ADNC | PART | ADNC | PART | ADNC |
| 36 | 179 | 447 | 400 | 121 | ||||||
| Number | 16 | 20 | 71 | 108 | 155 | 292 | 94 | 306 | 25 | 96 |
| Male/female | 10/6 | 11/9 | 48/23 | 64/44 | 90/65 | 152/150 | 54/40 | 160/146 | 9/16 | 53/43 |
| Results given in percent and difference when comparing ADNC with PART cases Fischer’s exact test | ||||||||||
| With dementia | 10 | 80.012 | 5 | 24<0.001 | 1 | 41<0.001 | 12 | 320.049 | ||
| +HPτ, Braak stages I–II [2] | 100 | 750.053 | 92 | 750.006 | 87 | 54<0.001 | 76 | 28<0.001 | 56 | 250.007 |
| +HPτ, Braak stages III–IV [2] | 15 | 9 | 18 | 13 | 33<0.001 | 25 | 58<0.001 | 44 | 56 | |
| +HPτ, Braak stages V–VI [2] | 10 | 70.023 | <1 | 13<0.001 | 15<0.001 | 190.023 | ||||
| +ARTAG | 6 | 10 | 25 | 25 | 34 | 39 | 46 | 48 | 64 | 71 |
| +Argyrophilic grain | 1 | 1 | 0 | 3 | <1 | 4 | 1 | |||
| +LATE-NC (Josephs ≥1) [13] | 6 | 20 | 16 | 300.033 | 25 | 42<0.001 | 29 | 54<0.001 | 60 | 69 |
| +LATE-NC (Josephs ≥3) [13] | 3 | <1 | 70.002 | 1 | 14<0.001 | 170.041 | ||||
| +LBD-NC (Braak stage ≥1) [10] | 5 | 9 | 16 | 8 | 20<0.001 | 14 | 250.024 | 20 | 27 | |
| +LBD-NC (Braak stage ≥4) [10] | 1 | 6 | 3 | 13<0.001 | 4 | 20<0.001 | 8 | 22 | ||
| +CAA | 250.053 | 6 | 32<0.001 | 10 | 40<0.001 | 13 | 48<0.001 | 20 | 510.006 | |
| +V-NC | 44 | 70 | 42 | 52 | 51 | 650.004 | 61 | 760.006 | 75 | 79 |
| +Severe V-NC | 5 | 2 | 1 | 5 | 3 | 8 | 9 | |||
| All | PART | ADNC | Dementia | Non-Dementia | |
|---|---|---|---|---|---|
| Number | 1184 | 362 | 822 | 248 | 936 |
| Hyperphosphorylated (HP) τ with age | 0.36 2 | 0.28 2 | 0.33 2 | −0.16 1 | 0.35 2 |
| Amyloid β-protein (Aβ) with age | 0.22 2 | 0.22 2 | −0.06 ns | 0.16 2 | |
| TDP43 with age | 0.26 2 | 0.21 2 | 0.25 2 | 0.14 ns | 0.25 2 |
| α Synuclein (αS) with age | 0.14 2 | 0.15 2 | 0.12 2 | 0.13 ns | 0.11 2 |
| Aβ with HPτ | 0.62 2 | 0.66 2 | 0.59 2 | 0.43 2 | |
| TDP43 with HPτ | 0.50 2 | 0.29 2 | 0.51 2 | 0.32 2 | 0.41 2 |
| αS with HPτ | 0.33 2 | 0.23 2 | 0.32 2 | 0.06 ns | 0.26 2 |
| TDP43 with Aβ | 0.39 2 | 0.43 2 | 0.32 2 | 0.26 2 | |
| αS with Aβ | 0.28 2 | 0.29 2 | 0.11 ns | 0.18 2 | |
| TDP43 with αS | 0.21 2 | 0.12 1 | 0.20 2 | 0.10 ns | 0.14 2 |
| Altered Protein | Staging Criteria | Stages | Based on the Visualization of the Altered Protein in |
|---|---|---|---|
| Hyperphosphorylated τ | Braak stage [2] | I to VI | The hippocampus reaching towards occipital cortex |
| Amyloid β protein | Thal phase [4] | 1 to 5 | The neocortex reaching towards cerebellum |
| α synuclein | Braak stage [10] | 1 to 6 | The medulla, nucleus vagus, reaching towards the parietal/frontal cortices |
| Transactive DNA-binding protein 43 | Josephs phase [13] | 1 to 5 | The amygdala reaching towards frontal cortex |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alafuzoff, I.; Libard, S. Ageing-Related Neurodegeneration and Cognitive Decline. Int. J. Mol. Sci. 2024, 25, 4065. https://doi.org/10.3390/ijms25074065
Alafuzoff I, Libard S. Ageing-Related Neurodegeneration and Cognitive Decline. International Journal of Molecular Sciences. 2024; 25(7):4065. https://doi.org/10.3390/ijms25074065
Chicago/Turabian StyleAlafuzoff, Irina, and Sylwia Libard. 2024. "Ageing-Related Neurodegeneration and Cognitive Decline" International Journal of Molecular Sciences 25, no. 7: 4065. https://doi.org/10.3390/ijms25074065
APA StyleAlafuzoff, I., & Libard, S. (2024). Ageing-Related Neurodegeneration and Cognitive Decline. International Journal of Molecular Sciences, 25(7), 4065. https://doi.org/10.3390/ijms25074065