Abstract
Vascular endothelial growth factor 165 (VEGF165) is a prominent isoform of the VEGF-A protein that plays a crucial role in various angiogenesis-related diseases. It is homodimeric, and each of its monomers is composed of two domains connected by a flexible linker. DNA aptamers, which have emerged as potent therapeutic molecules for many proteins with high specificity and affinity, can also work for VEGF165. A DNA aptamer heterodimer composed of monomers of V7t1 and del5-1 connected by a flexible linker (V7t1:del5-1) exhibits a greater binding affinity with VEGF165 compared to either of the two monomers alone. Although the structure of the complex formed between the aptamer heterodimer and VEGF165 is unknown due to the highly flexible linkers, gaining structural information will still be valuable for future developments. Toward this end of accessing structural information, we adopt an ensemble docking approach here. We first obtain an ensemble of structures for both VEGF165 and the aptamer heterodimer by considering both small- and large-scale motions. We then proceed through an extraction process based on ensemble docking, molecular dynamics simulations, and binding free energy calculations to predict the structures of the VEGF165/V7t1:del5-1 complex. Through the same procedures, we reach a new aptamer heterodimer that bears a locked nucleic acid-modified counterpart of V7t1, namely RNV66:del5-1, which also binds well with VEGF165. We apply the same protocol to the monomeric units V7t1, RNV66, and del5-1 to target VEGF165. We observe that V7t1:del5-1 and RNV66:del5-1 show higher binding affinities with VEGF165 than any of the monomers, consistent with experiments that support the notion that aptamer heterodimers are more effective anti-VEGF165 aptamers than monomeric aptamers. Among the five different aptamers studied here, the newly designed RNV66:del5-1 shows the highest binding affinity with VEGF165. We expect that our ensemble docking approach can help in de novo designs of homo/heterodimeric anti-angiogenic drugs to target the homodimeric VEGF165.
1. Introduction
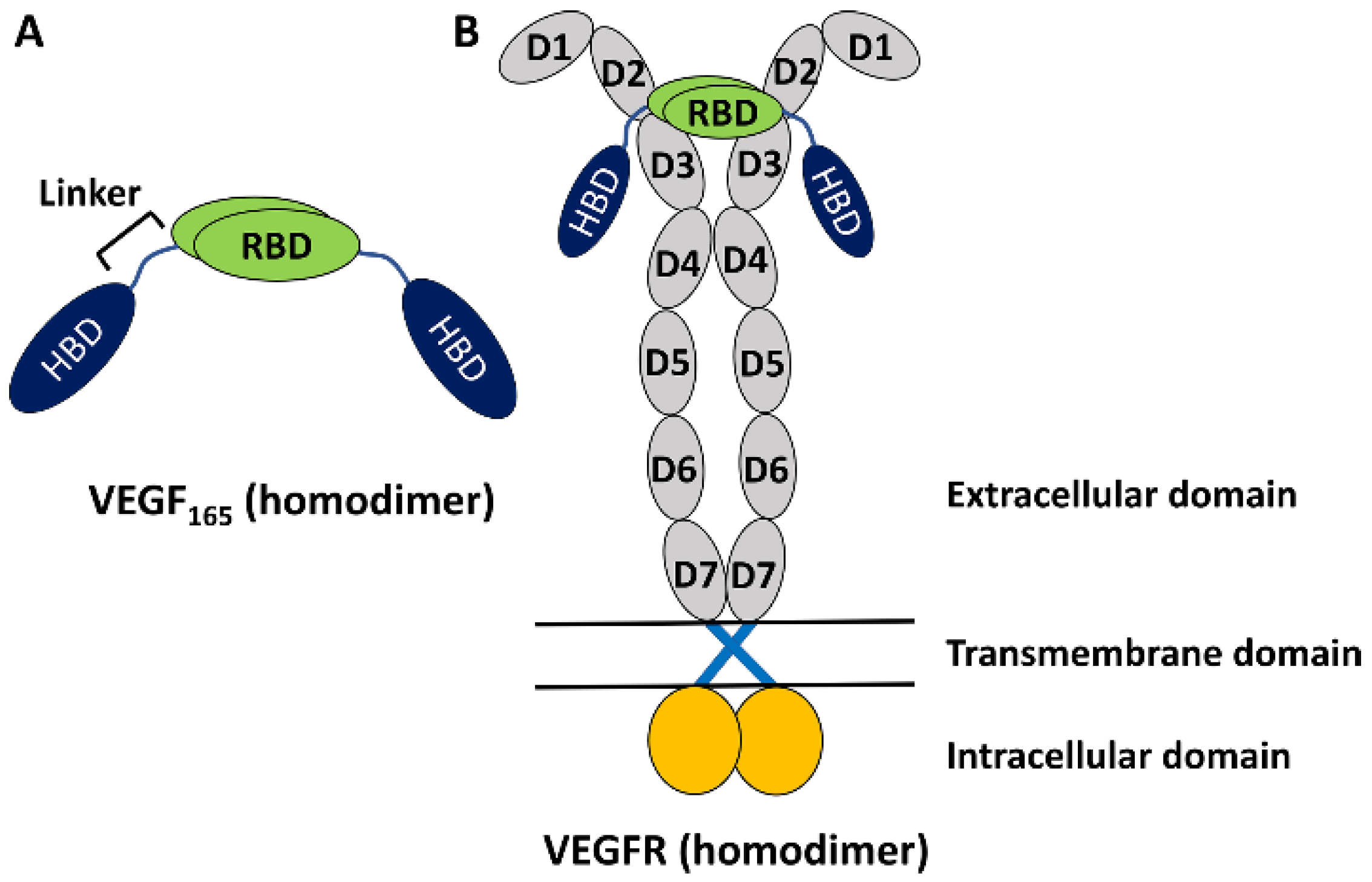
The vascular endothelial growth factor (VEGF) family of proteins plays key roles in regulating physiological vasculogenesis and angiogenesis, namely the process of new blood vessel formations [1,2,3]. At the same time, the VEGF family is also responsible for pathological angiogenesis in diseases such as tumor growth and neovascular age-related macular degeneration [2,3,4,5,6,7]. Therefore, VEGF’s family members are important targets for diagnosing these diseases [2,3]. The human VEGF family comprises five members [7], among which the most studied one is VEGF-A, which is often referred to simply as VEGF. It has several isoforms with different numbers of amino acid residues [7], but VEGF165 with 165 residues is the most abundant and is known to play an important role in pathological angiogenesis [7]. It is a homodimeric protein, with each monomer having a heparin-binding domain (HBD) and a receptor-binding domain (RBD) connected by a flexible linker (Figure 1A) [7,8]. The experimental structures of HBD and RBD are known [9,10], but due to the high flexibility of the linker region, the full structure of the homodimeric VEGF165 is experimentally unknown [8]. Functionally, VEGF binds with VEGF receptors (VEGFRs) and induces dimerization and activation of VEGFR. This triggers signal transduction pathways that are crucial for angiogenesis (Figure 1B) [11,12]. While there are a series of VEGFR variants, VEGFR-2 is known to play a main role in both physiological and pathological angiogenesis [13]. Up to now, significant research efforts have been directed toward discovering inhibitors targeting VEGF to prevent its binding to VEGFR [14]. Several VEGF inhibitors have currently been approved for anti-angiogenic treatment, such as Bevacizumab [15], Aflibercept [16], and Macugen [17].
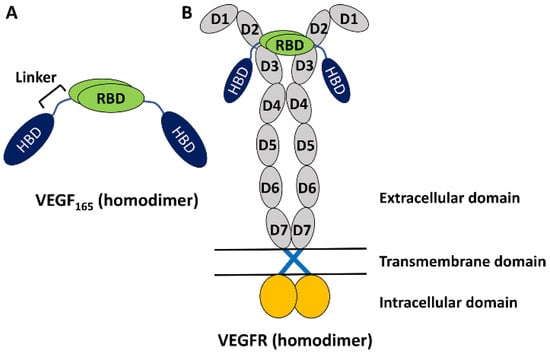
Figure 1.
(A) Schematic representation of VEGF165 as a homodimer with two monomers. Each monomer is composed of RBD and HBD, connected by a flexible linker. (B) VEGF165 binding with VEGFR toward its activation. VEGFR is also a homodimer and is composed of an extracellular domain with seven sub-domains in each monomer, a transmembrane domain lying in lipid, and an intracellular domain extruding into the cytoplasm.
Aptamers are short single-stranded DNA or RNA molecules used for the molecular recognition of targets with high affinity and specificity [18,19]. Not surprisingly, aptamers have also been used to inhibit the activity of VEGF. Indeed, Macugen is a Food and Drug Administration (FDA)-approved RNA aptamer used for treating age-related macular degeneration [17]. Several DNA aptamers, including VEa5, 2G19, and Vap7, have been further developed as anti-angiogenic agents against VEGF [20,21,22,23]. In addition, efforts have been made to enhance the binding affinity of aptamers to VEGF by designing G-quadruplex-forming aptamers [23,24,25] and aptamer dimers [20,23,26,27,28,29]. Naturally, it is important to find an aptamer with high binding affinity to VEGF. Experimentally, it has been shown that homodimers made for monomeric aptamers VEa5, del5-1, and 3R02 show higher affinities with VEGF165 than their respective monomers [26,29]. Another experimental study demonstrated the efficacy of an aptamer heterodimer composed of DNA aptamers V7t1 and del5-1 (V7t1:del5-1) [23]. In particular, V7t1:del5-1 showed higher affinity towards VEGF165 than its monomeric components, with del5-1 attaching to the HBD part and V7t1 attaching to the RBD part [23]. V7t1 is a 25-mer aptamer with a G-quadruplex structure and was derived from Vap7 [23]. On the other hand, del5-1 is a 50-mer aptamer derived from VEa5, and it encompasses three stem-loop regions [26]. In the heterodimer, V7t1 and del5-1 are connected using 10 thymine nucleotides as a linker [23]. The structure of the bound complex formed by the aptamer heterodimer and VEGF165 is not known experimentally, primarily due to the high flexibility of the linkers present in both components. Even still, understanding the interactions between the aptamer heterodimer and VEGF165 will be indispensable for developing new aptamer heterodimers with improved properties.
In the absence of experimental structures, in silico methods such as molecular docking and molecular dynamics (MD) simulations are useful tools [30,31,32]. The molecular docking approach facilitates the prediction of a ligand-binding site on the target molecule as well as the binding affinity and provides useful information on the position and orientation of the ligand [30]. Conversely, MD simulations capture the dynamic behavior of the ligand–target complex, providing useful information on stability, flexibility, and the associated conformational changes [31]. Additionally, end-point free energy methods [32,33,34] have been extensively utilized for calculating ligand–target binding free energies. They often use only the conformations in the free and bound states of the ligand and target and can be computationally less expensive than more rigorous alchemical- and pathway-based free energy methods [35,36,37]. Even though the end-point methods lack somewhat in accuracy compared to these rigorous methods, they can still offer higher accuracy than docking scoring functions [32].
With the above in mind, here we propose an ensemble docking approach for predicting an ensemble of complex structures of V7t1:del5-1 bound to VEGF165 by taking into account both small- and large-scale motions in both VEGF165 and the aptamer heterodimer. We first utilize anisotropic network model (ANM) analysis to gain insights into large-scale motions based on the lowest-frequency normal modes [38], followed by biased MD simulations to generate molecular structures considering these motions. In fact, identifying large-scale changes that typically involve low-frequency normal-mode motions in a complex system is a challenging task with atomistic MD. ANM can be a reasonable approach to generating large-scale collective motions occurring between the domains/monomers connected by a linker in both VEGF165 and the aptamer heterodimer. Through biased MD simulations, we obtain several conformations of VEGF165 and the aptamer heterodimer corresponding to their lowest-frequency normal modes. The obtained structures are then used for extraction based on ensemble docking, followed by unbiased MD simulations as well as binding free energy calculations based on molecular mechanics generalized Born surface area (MM/GBSA) [32], to predict the complex structures of the aptamer heterodimer bound to VEGF165.
We also designed a new aptamer heterodimer, RNV66:del5-1, against VEGF165 by replacing V7t1 with a locked nucleic acid (LNA) modified version of V7t1, namely RNV66. Although V7t1 effectively targets the RBD part of VEGF165, it exhibits polymorphism, resulting in multiple G-quadruplex structures [24]. In contrast, the 25-mer RNV66 generated by replacing guanine residues at positions 5, 21, and 24 of V7t1 with LNA-G residues has a single stable G-quadruplex structure [24]. Moreover, RNV66 by itself is an outstanding anti-VEGF aptamer and is known to inhibit cancer proliferation with higher binding affinity and nuclease resistance than V7t1 [25]. Using our ensemble docking protocol mentioned above, we also provide the complex structures of RNV66:del5-1 bound to VEGF165. For comparison, the three constituting monomeric units (V7t1, RNV66, and del5-1) are also included in the ensemble docking approach with VEGF165. All five VEGF165/aptamer complexes obtained are then analyzed in detail to explore binding poses, hydrogen bond (H-bond) interactions in VEGF165/aptamer complexes, and steric clashes between the aptamer and VEGFR-2 when the complex interacts with VEGFR-2. In addition, the stability of the G-quadruplex structure of RNV66 or V7t1 upon binding with VEGF165, either as a monomer or as part of a heterodimer, is also examined.
2. Results and Discussion
2.1. Designing Anti-VEGF165 Aptamers
In this paper, we study two aptamer heterodimers, RNV66:del5-1 and V7t1:del5-1, as well as three monomeric aptamers, RNV66, V7t1, and del5-1, for targeting VEGF165. Through computational means, we tried to incorporate small- and large-scale motions into the consideration of the protein–aptamer interaction [39] involving VEGF165.
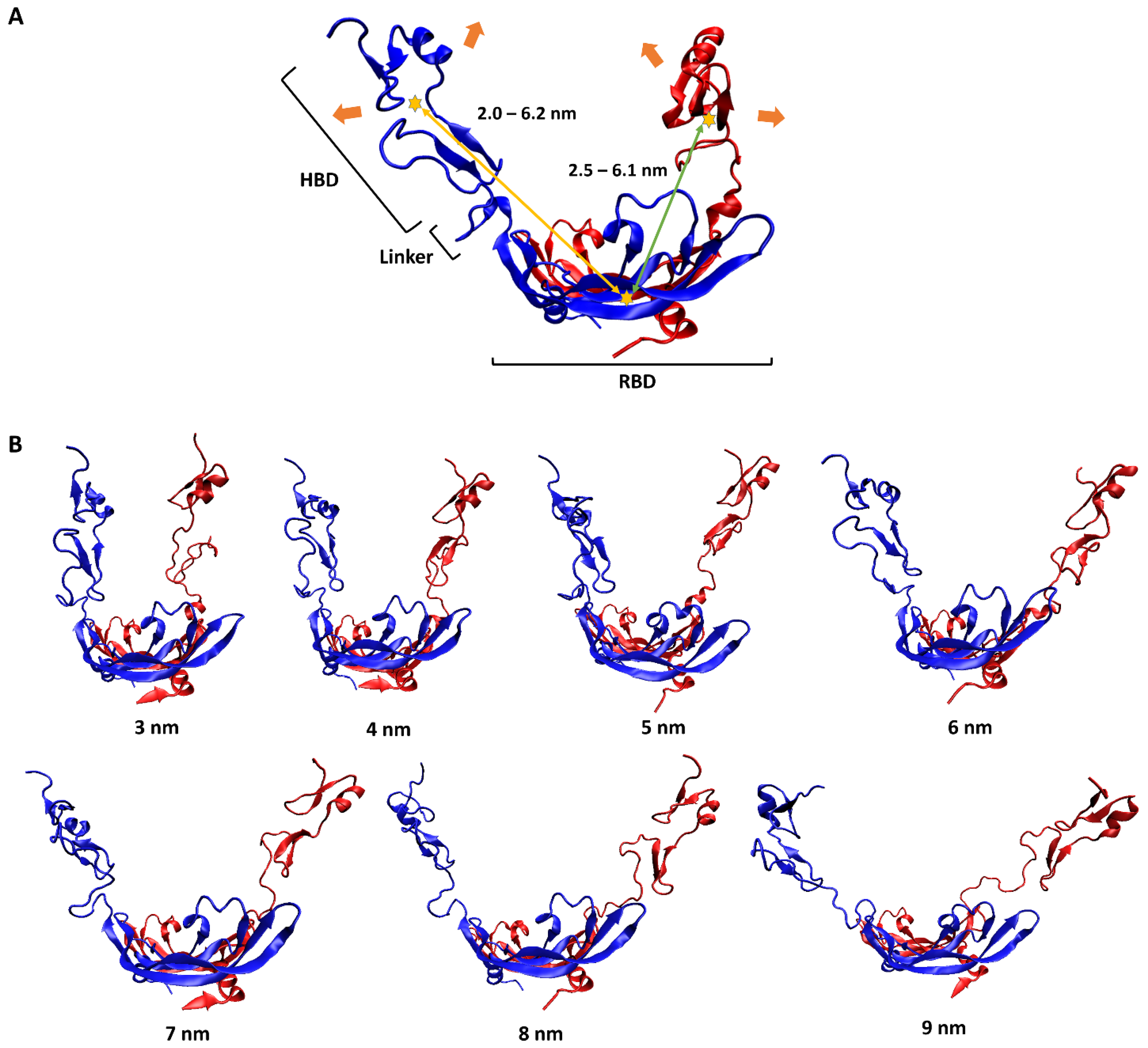
To have a pictorial sense of the large-scale motion, the lowest-frequency normal mode of VEGF165 was first predicted using ANM analysis on VEGF165. As mentioned above, this approach was selected because observing large-scale motions induced by flexible linkers solely through unbiased MD simulations is highly challenging due to the sampling difficulty. This motion could be characterized as a scissoring bending vibration between the two HBD units, with oscillations in the distance between the two (Figure 2A). To reflect this large-scale motion, we generated seven different VEGF165 conformations by using biased MD simulations, increasing the distance between the centers of mass (COMs) of the two HBD units from 3 nm to 9 nm in 1 nm intervals (Figure 2B). In addition, from each 10 ns long biased MD trajectory, VEGF165 snapshots were extracted at every 500 ps to account for small-scale motions of the protein. Accordingly, 20 VEGF165 structures were extracted from each biased MD simulation, resulting in a total of 140 (7 × 20) VEGF165 conformations. With these, we calculated all possible intra-monomer COM distances between RBD and HBD as well as the inter-monomer ones, and the distances ranged 2.0–6.2 nm and 2.5–6.1nm, respectively (Figure 2A).
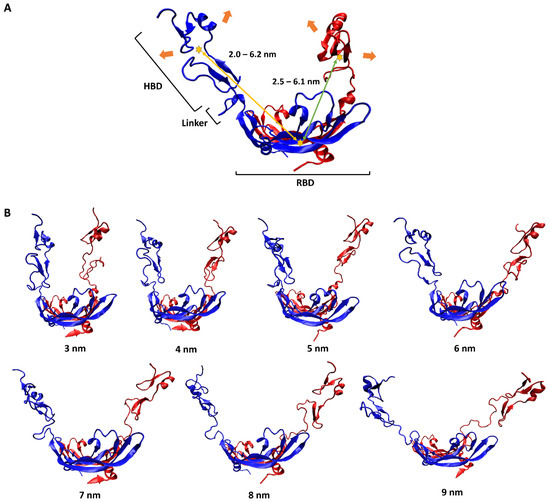
Figure 2.
(A) Schematic representation of VEGF165 motion. The two monomers of VEGF165 are shown in blue and red. The orange arrows indicate the lowest-frequency normal-mode motion obtained from ANM. The yellow stars represent the COMs of HBD and RBD, with the yellow and green arrows indicating, respectively, the intra-monomer and inter-monomer COM distances between HBD and RBD. (B) Biased MD simulation results for generating seven VEGF165 conformations, with HBD–HBD COM distances restrained at the designated distances.
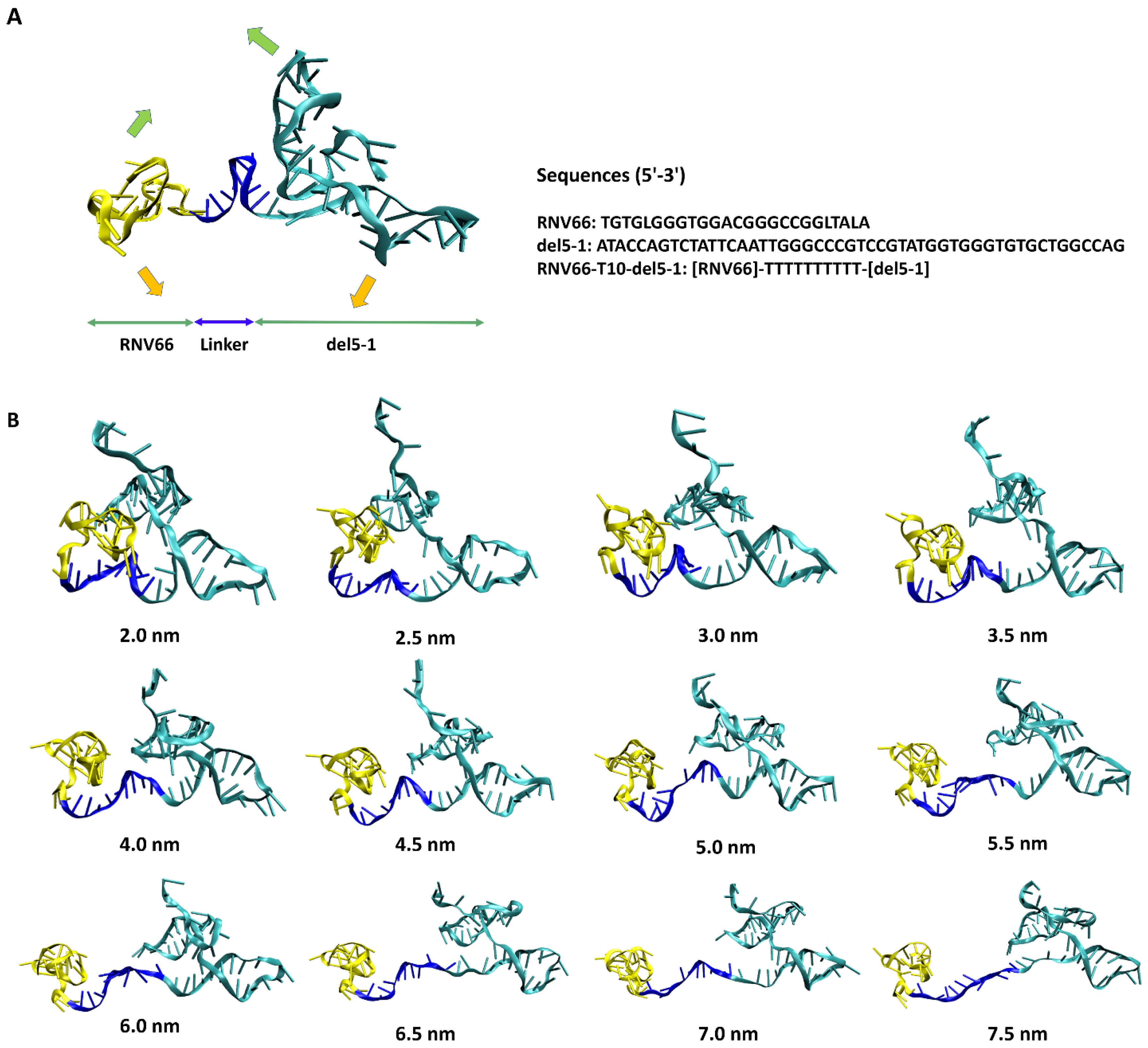
With these distance values in mind, we also performed ANM analyses on the equilibrated structures of RNV66:del5-1 and V7t1:del5-1 as obtained from the MD simulations. From these, wavy motions with which the two aptamer monomers became closer and farther from each other were commonly observed as the lowest-frequency normal mode in both heterodimers (Figure 3A). To better handle this large-scale motion, we performed biased MD simulations with restraints on the inter-monomer COM distances by increasing the restraining distances from 2.0 nm to 7.5 nm in steps of 0.5 nm intervals. For each COM distance, the biased simulations were continued up to 10 ns, leading to 12 distinct conformations for each heterodimer bearing large-scale motions (Figure 3B). Small-scale motions were then added by extracting snapshots at 500 ps intervals from each 10 ns biased MD simulation, leading to a total of 240 (12 × 20) structures obtained for each heterodimer. As RNV66 or V7t1 binds to RBD [23,24,25] while del5-1 binds to HBD [26], it was necessary to consider distance ranges between RNV66 and del5-1 and between V7t1 and del5-1 that could encompass the RBD-to-HBD distances. This was the reason we chose the heterodimer distance range of 2.0–7.5 nm. Because V7t1:del5-1 was effective with a linker made of 10 thymine nucleotides [23], we used the same linker for RNV66:del5-1 as well. For the cases of the 3 aptamer monomers, we similarly extracted snapshots at every 500 ps of a single 10 ns MD simulation of each without any restraint, resulting in 20 monomer structures for each monomer type.
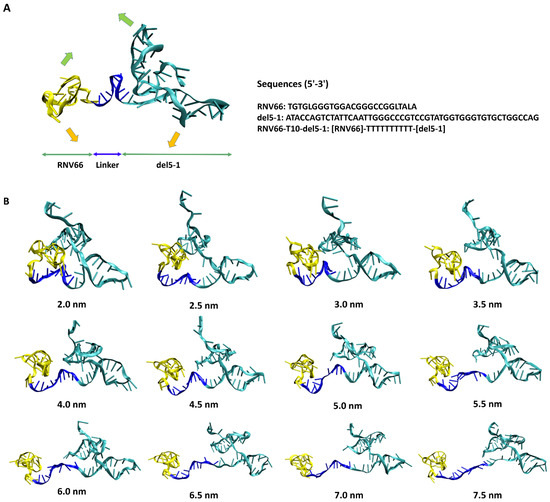
Figure 3.
(A) RNV66:del5-1 together with its lowest-frequency normal-mode motion from ANM, illustrated with green and orange arrows. The sequence information is also given on the right. (B) Twelve RNV66:del5-1 structures obtained from biased MD simulations, with the inter-monomer COM distances restrained at the designated distances.
2.2. Ensemble Docking of Aptamers with VEGF165
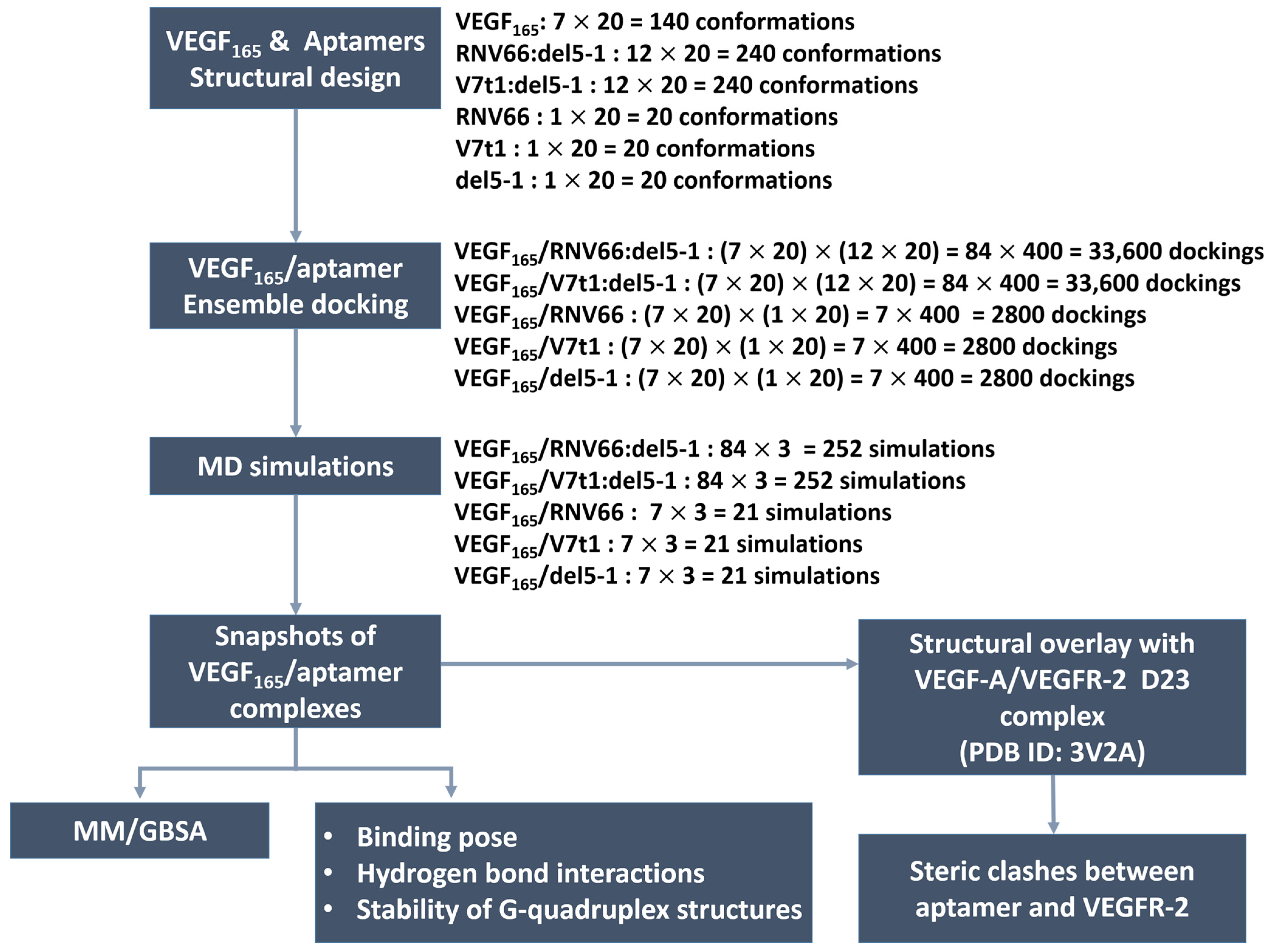
To generate the VEGF165/aptamer complexes, we utilized the HDOCK docking program [40,41] by utilizing all combinations of the conformations described in the previous section. In fact, HDOCK has been extensively adopted recently to analyze protein–RNA and protein–DNA interactions [42,43,44], and it has shown excellent performance [45] in the community-wide Critical Assessment of Prediction of Interactions (CAPRI) [46]. To further verify its applicability to our systems, we conducted a docking experiment between VEGF121 and domains 2 and 3 (D23) of VEGFR-2 with it. The docked structure (Figure S1 in Supporting Information (SI)) was a close match with the experimentally known VEGF-A/VEGFR-2 D23 structure (PDB ID: 3V2A) [47] after adding the missing residues in the crystal structure through homology modeling with SWISS-MODEL [48].
In total, 240 structures for each aptamer heterodimer and 20 for each aptamer monomer underwent docking with 140 VEGF165 structures. Consequently, a total of 33600 docked poses or structures (i.e., 140 × 240) for each aptamer heterodimer and 2800 poses (i.e., 140 × 20) for each aptamer monomer were obtained (Figure 4). Based on the obtained docking results, we extracted the initial structures required for subsequent MD simulations to calculate the binding free energies with MM/GBSA [32]. For each type of aptamer heterodimer, 400 docked structures (20 VEGF165 snapshots × 20 aptamer snapshots) were generated for each of the 84 variants (7 VEGF165 variants × 12 aptamer variants). Subsequently, the docked structures that did not meet the “attachment criteria,” i.e., RNV66 or V7t1 attaching to the RBD part and del5-1 attaching to the HBD part, were manually excluded. From the remaining docked structures for each type of aptamer heterodimer, the top-3 complexes with the highest docking scores were selected for each of the 84 variants. Similarly, for each type of monomeric aptamer, 400 docked structures (20 VEGF165 snapshots × 20 aptamer snapshots) were generated for each of the 7 variants (7 VEGF165 variants × 1 aptamer). Within the docked structures for each type of monomeric aptamer, those that did not satisfy the attachment criteria were eliminated first, and the top-three complexes with the highest docking scores were selected from the rest for each of the seven variants. Consequently, we obtained 252 complex structures each for VEGF165/RNV66:del5-1 and VEGF165/V7t1:del5-1 and 21 complex structures each for VEGF165/RNV66, VEGF165/V7t1, and VEGF165/del5-1 (Figure 4). Thus, a total of 567 VEGF165/aptamer structures were selected as initial structures for the subsequent MD simulations. The docking scores of these selected conformations are provided in SI (Tables S1 and S2).
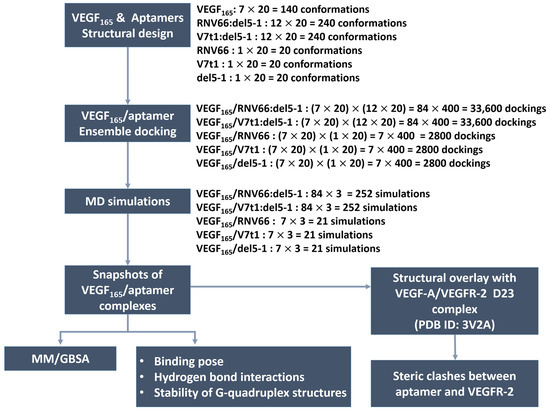
Figure 4.
Procedures of our ensemble docking.
2.3. Binding Affinity of Aptamers with VEGF165
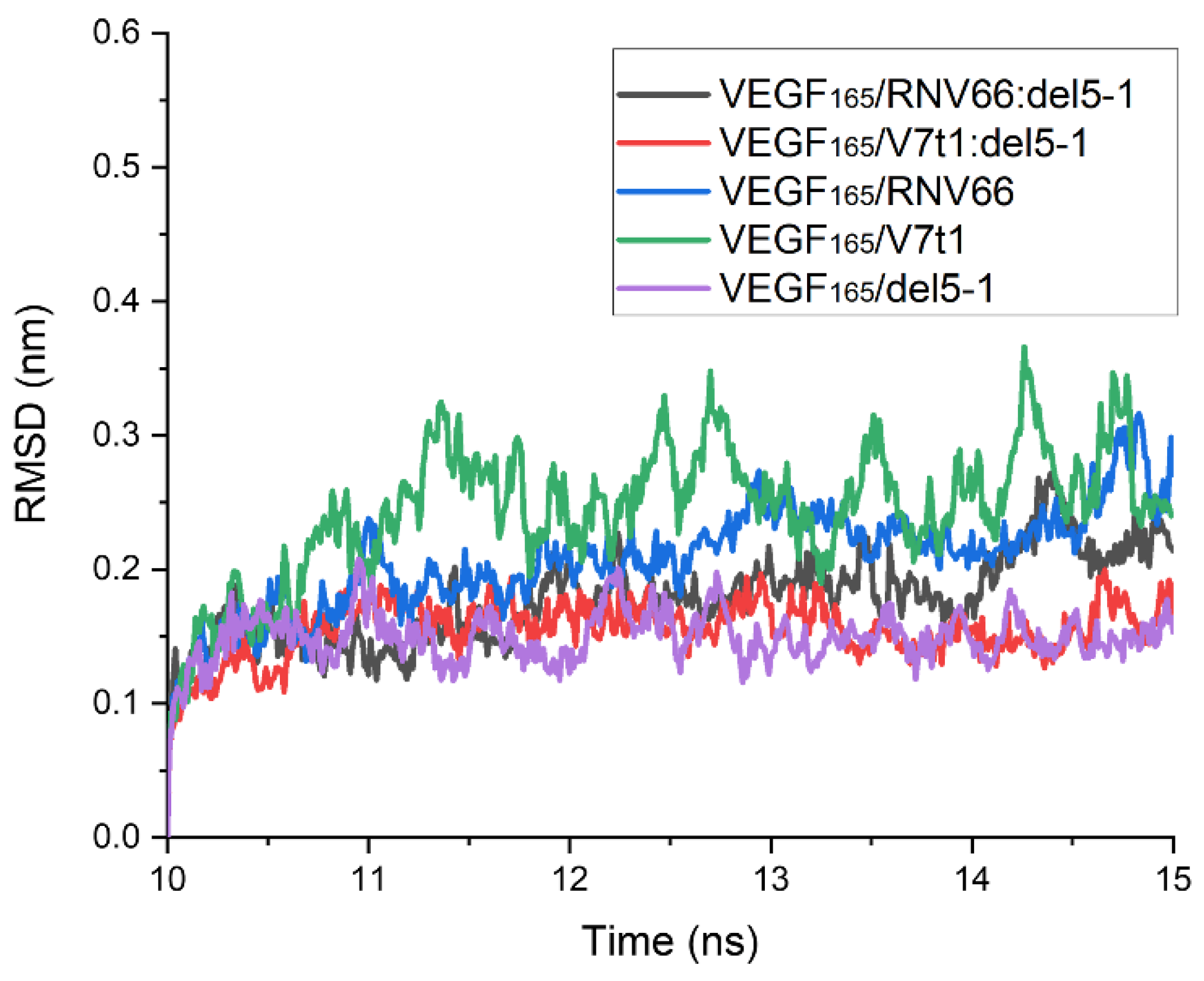
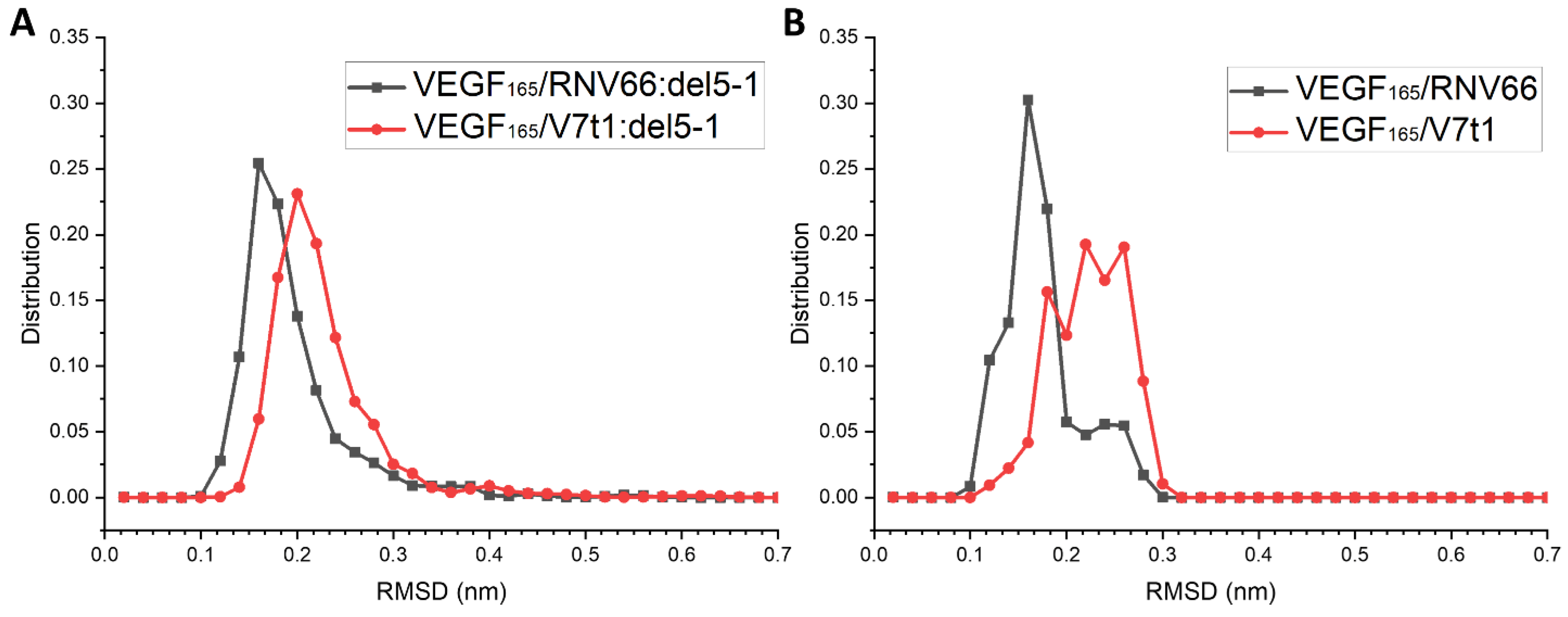
To obtain binding free energies using MM/GBSA, we first performed 15 ns MD simulations for each complex with the chosen structures. After analyzing the time course of the root-mean-square deviation (RMSD) for each complex from its 15 ns MD trajectory, we confirmed that all 567 structures were equilibrated within 10 ns. The time course of the RMSD for each complex was calculated with respect to its initial structure. However, upon observing the complex structures after the 15 ns period, we found that some complexes did not meet the attachment criteria mentioned above. Thus, these trajectories were excluded from the list, and there remained 491 trajectories in total for further analyses, as follows: 218 for VEGF165/RNV66:del5-1; 210 for VEGF165/V7t1:del5-1; 21 for VEGF165/RNV66; 21 for VEGF165/V7t1; and 21 for VEGF165/del5-1. For each MD trajectory, using the snapshots sampled at every 10 ps over its last 5 ns, we calculated the binding free energy using the single-trajectory approach of MM/GBSA [32]. The time courses of the RMSD values obtained from the MD trajectories with the lowest binding free energies for the five types of VEGF165/aptamer complexes are shown in Figure 5. In this case, the RMSD values were calculated using the complex structure at the 10 ns mark of each MD trajectory as the reference structure.
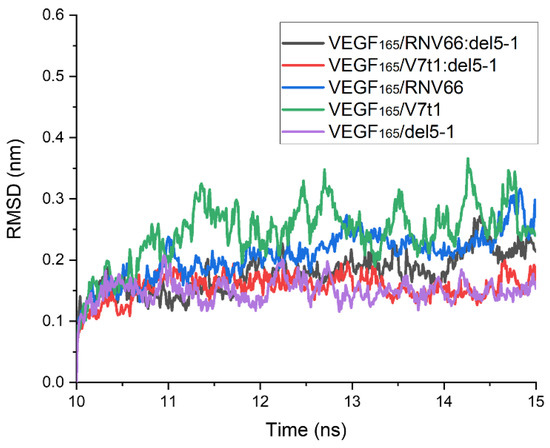
Figure 5.
Time courses of RMSD values for the five types of VEGF165/aptamer complexes. RMSD values are presented for the 10–15 ns part of the 15 ns long MD trajectory with the lowest binding free energy for each complex. The reference structures for measuring RMSDs were the ones at the 10 ns time point.
The average binding free energies for the five types of complexes are listed in Table 1, along with the number of trajectories adopted for generating the averages. The averages revealed that RNV66:del5-1 is the strongest binder and is much better than V7t1:del5-1. In general, heterodimers are better than monomers. Although any quantitative interpretation with specific numbers should be avoided due to the inherent limitation of MM/GBSA, these binding free energies are also in the same trend with the average docking scores also listed in Table 1 and are consistent with available experimental data that showed that V7t1:del5-1 exhibits a higher binding affinity to VEGF165 compared to either V7t1 or del5-1 [23]. Similarly, RNV66:del5-1 demonstrates a higher binding affinity than its monomeric counterparts, further supporting the notion that aptamer heterodimers are more effective anti-VEGF165 aptamers than monomers. Interestingly, from our results, RNV66:del5-1 appeared as a more potent binder than V7t1:del5-1, suggesting that an aptamer heterodimer designed with RNV66, which is known to be more effective in inhibiting than V7t1 [25], may potentially serve as a superior anti-VEGF165 inhibitor. To verify that we are not misguided by any outlying conformation, we selected the structures with the top-ten lowest binding free energies for each of the five types of VEGF165/aptamer complexes and re-calculated the averages based only on those top-ten contributors (Table S3). The results are still consistent with the ones observed with all structures, as discussed above.

Table 1.
Averaged binding free energies (ΔG) and docking scores for the five types of VEGF165/aptamer complexes.
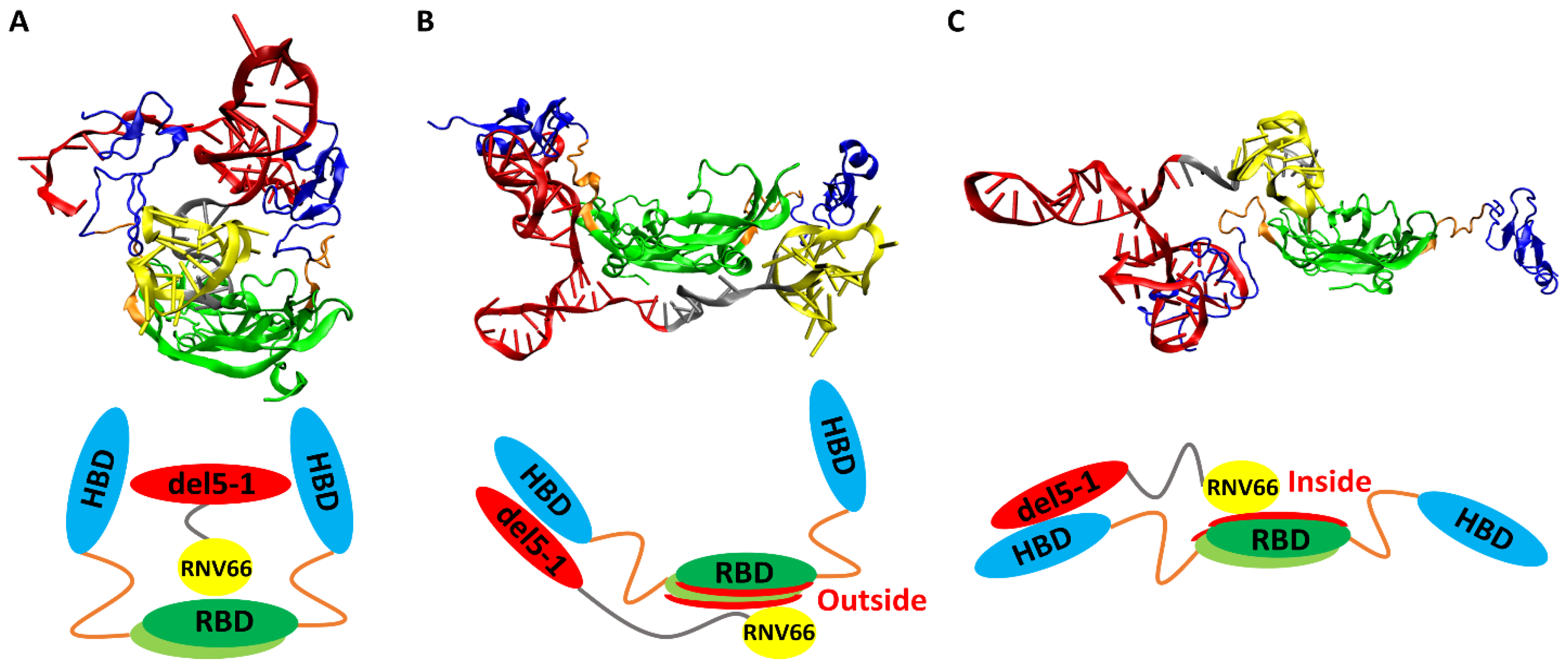
When we visually inspected the final snapshots of the 491 VEGF165/aptamer trajectories using VMD [49], we noticed some typical binding poses, and we classified them into three categories: sandwich, side, and hug poses. A sandwich pose refers to a configuration where the two HBD units surround the aptamer in the center. In other words, for the VEGF165/aptamer complex to exhibit a sandwich pose, both HBD units of VEGF165 must bind to a single aptamer domain (Figure 6A). Additionally, in the side pose, del5-1 binds to only one HBD of VEGF165, while RNV66 or V7t1 binds to the outside of VEGF165 (Figure 6B). In a hug pose, RNV66 or V7t1 is bound to the interior of VEGF165, while del5-1 binds to only one of the two HBD units, and the other unit is located far from del5-1 (Figure 6C). In the classification, to assess whether a specific aptamer has bound to a particular domain of VEGF165, we considered whether the distances between protein side-chain heavy atoms and DNA heavy atoms were within 0.45 nm [50]. More extensive pictorial representations than those in Figure 6 can be found in Figures S2 and S3.
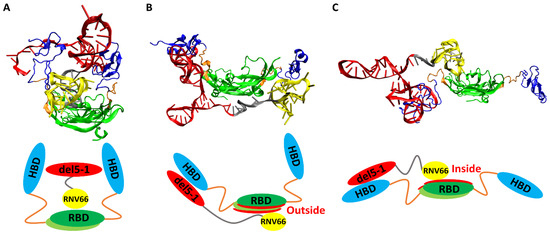
Figure 6.
Typical poses of VEGF165/aptamer complexes: (A) a sandwich pose of VEGF165/RNV66:del5-1, (B) a side pose of VEGF165/RNV66:del5-1, and (C) a hug pose of VEGF165/RNV66:del5-1. The structures obtained through VMD for each pose are presented alongside their schematic representations. The representative structures for each pose were arbitrarily chosen. For aptamers, RNV66 and V7t1 are shown in yellow, while del5-1 is in red, with the linker in silver. For VEGF165, RBD is in green, while HBD is in blue, with the linker in orange.
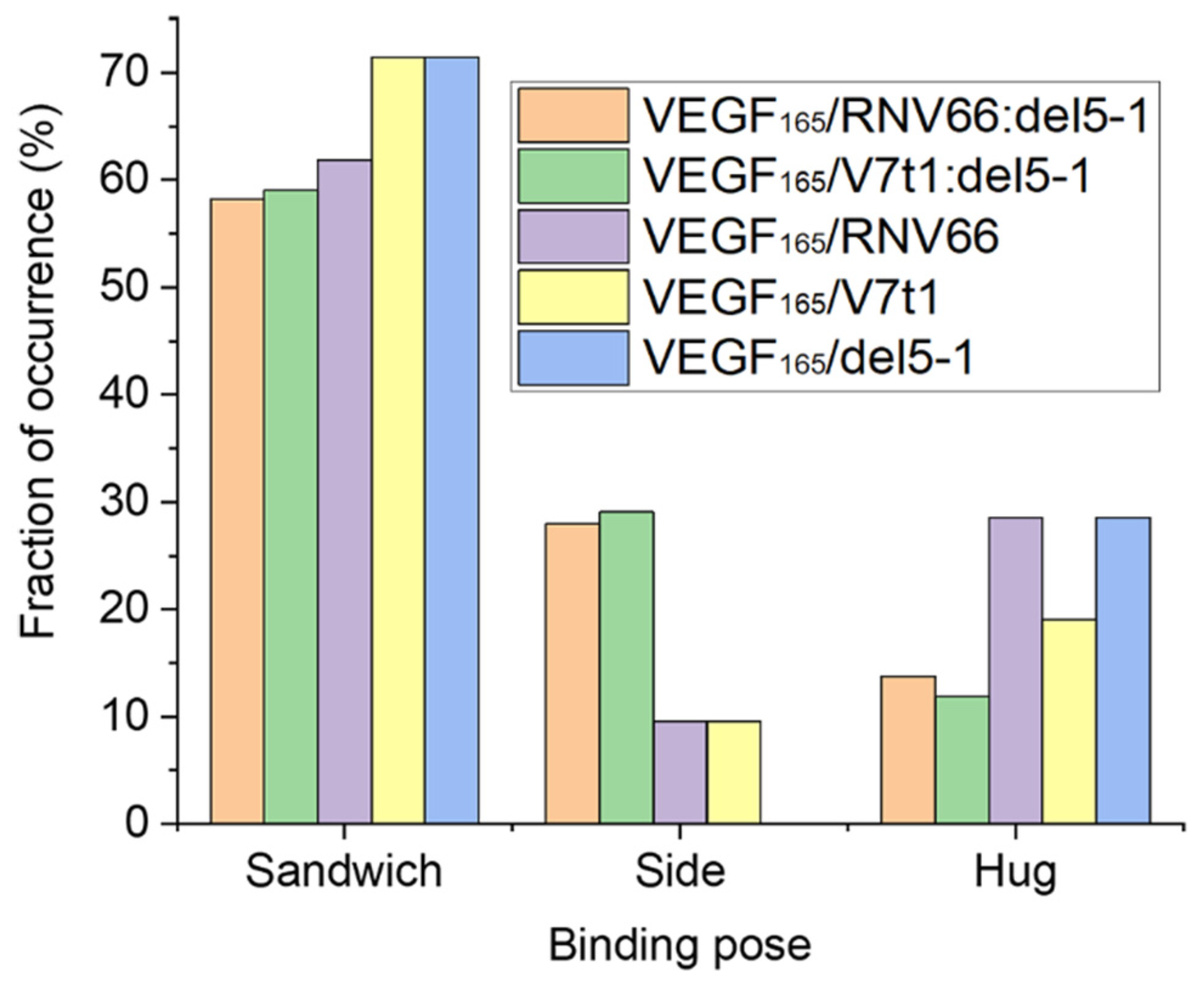
Upon computing the fraction of occurrences of these three binding poses, we observed that all the five aptamers we adopted preferred the sandwich pose when binding to VEGF165 (Figure 7). This indicates that the sandwich pose is the most stable structure for complexation. Interestingly, aptamer heterodimers displayed fewer sandwich and hug poses compared to monomeric aptamers. The binding poses of the top-ten complex structures for each type of VEGF165/aptamer complex can be found in Table S4.
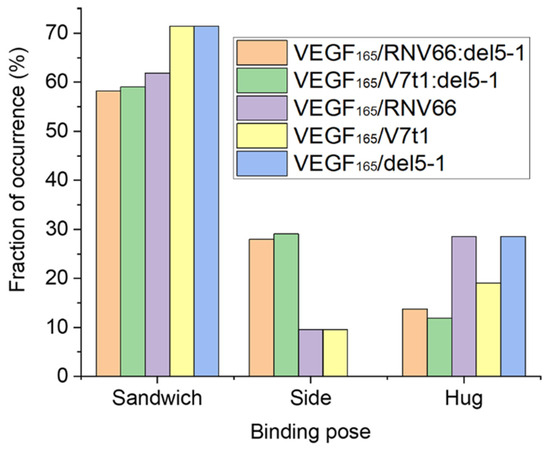
Figure 7.
Fraction of occurrences of the sandwich, the side, and the hug poses for each type of VEGF165/aptamer complexes.
2.4. Hydrogen Bonds between Aptamer and Key Residues of VEGF165
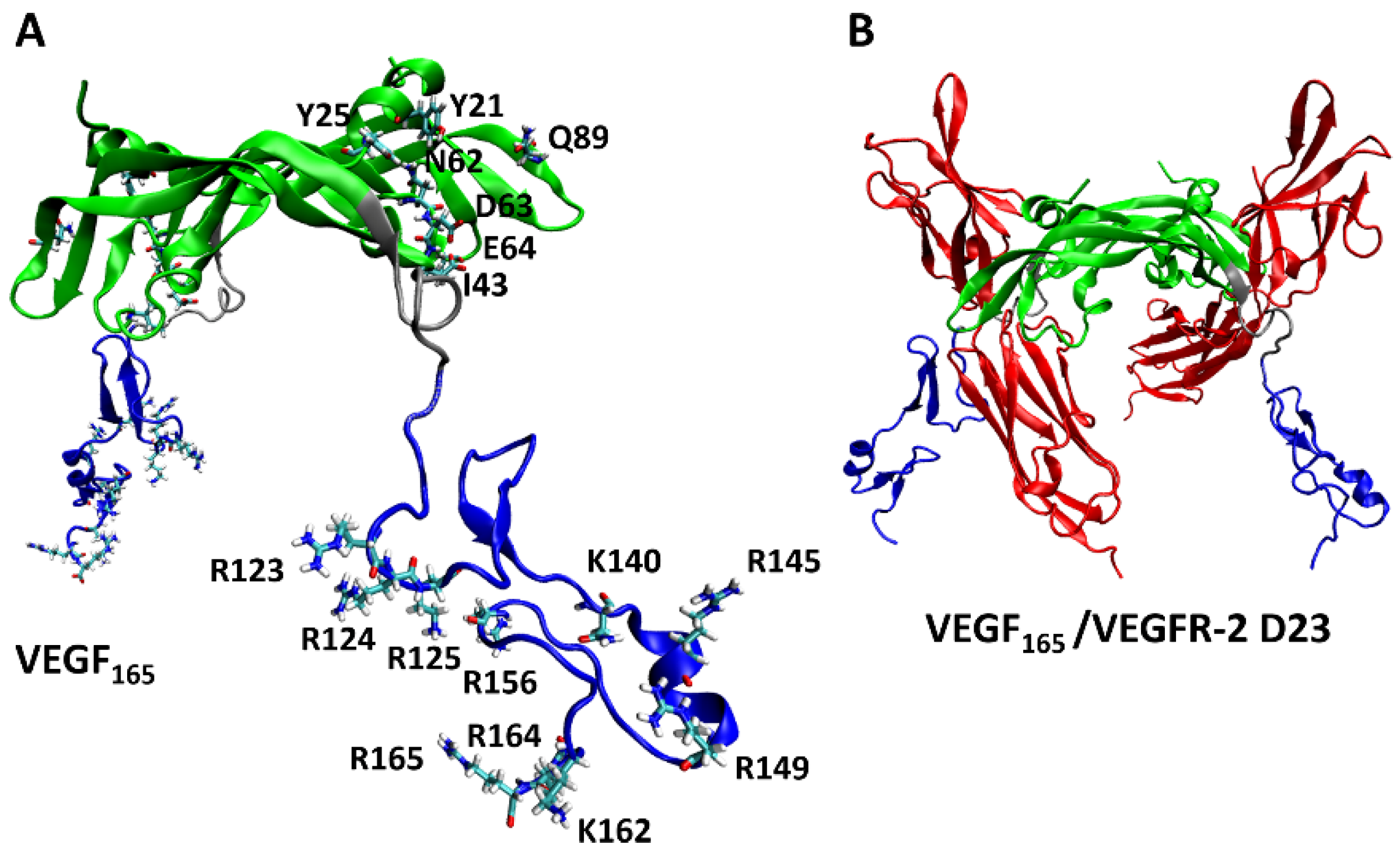
From experiments, the key VEGF165 residues involved in its binding with heparin [9] and VEGFR-2 [47] are known. For comparison, we investigated to identify which of these key VEGF165 residues are involved in binding with the five aptamers through H-bonds. We focused specifically on examining the formation of H-bonds with the seven residues (Y21, Y25, I43, N62, D63, E64, and Q89) in the RBD part, which were identified as the key residues for binding with VEGFR-2 [47], as well as the ten residues (R123, R124, K125, K140, R145, R149, R156, K162, R164, and R165) in the HBD part, known to interact with heparin (Figure 8) [9].
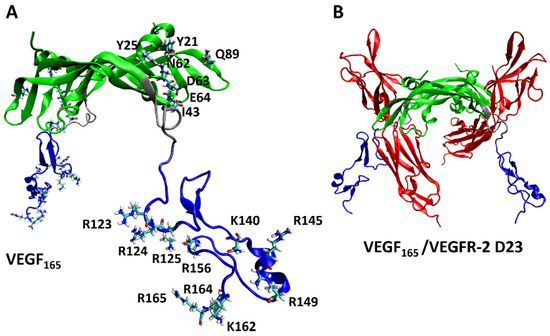
Figure 8.
(A) Key residues of VEGF165 in binding with VEGFR-2 and heparin, with the RBD/HBD/linker unit depicted in green/blue/silver. (B) Schematic representation of the complex formed by VEGF165 together with VEGFR-2 D23. VEGFR-2 D23 is highlighted in red.
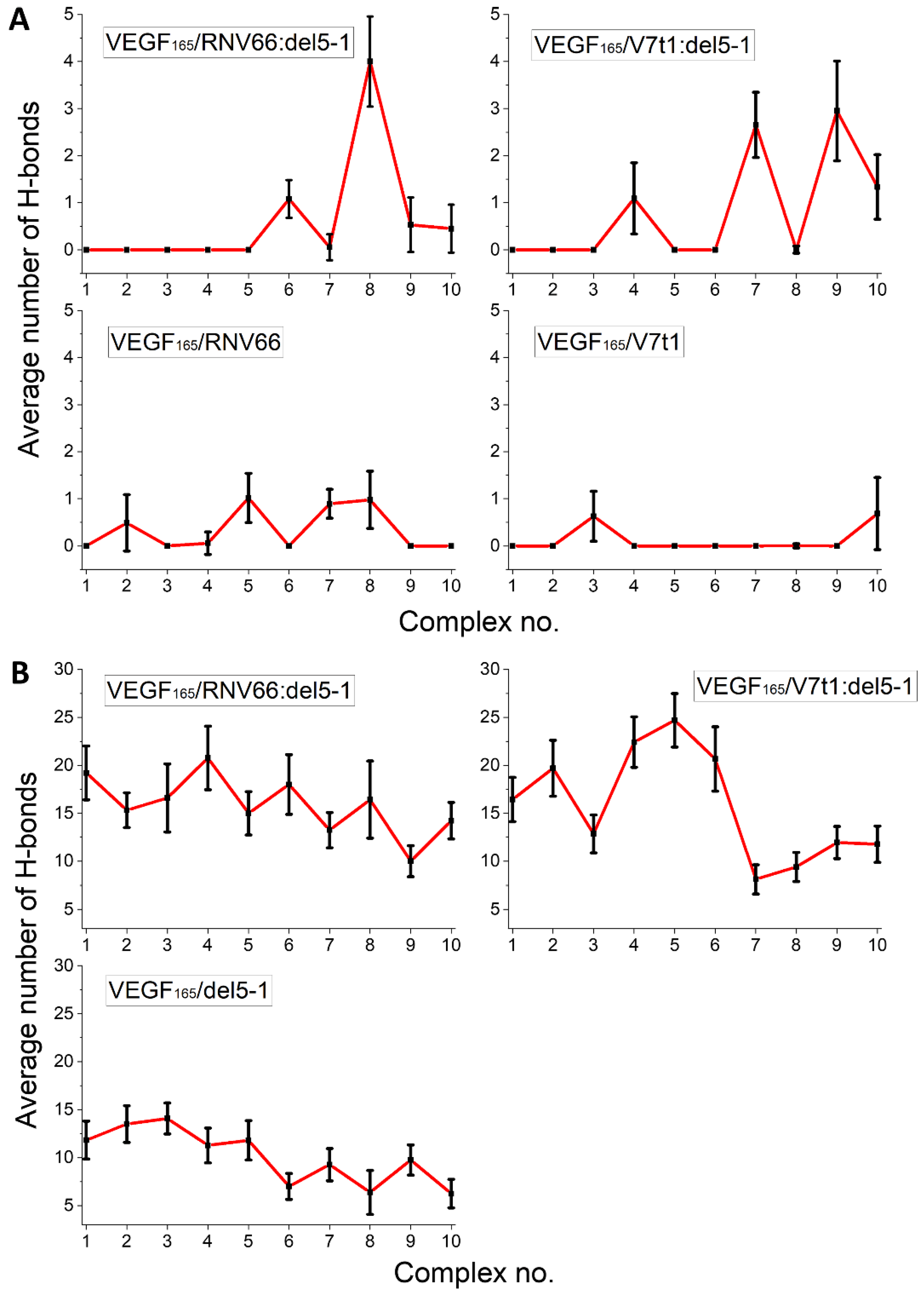
We adopted the MD trajectories corresponding to the top-ten complex structures with the lowest binding free energies, mentioned in an earlier section, by taking the snapshots at every 10 ps during the last 5 ns of each trajectory and counting the number of H-bonds in each snapshot. The criterion for a H-bond was a maximum donor–acceptor distance of 3.5 Å and a maximum H-donor–acceptor angle of 30 deg [51,52]. We then calculated the time average of the number of H-bonds over the 5 ns period for each trajectory. The results are shown in Figure 9 separately for the RBD residues and the HBD residues. In general, HBD is more prone to forming H-bonds than RBD, and heterodimers tend to form more H-bonds. In a sense, this is not surprising, as we already observed that RNV66:del5-1 and V7t1:del5-1 are more effective binders than their constituting monomers. Out of the 17 key VEGF165 residues, R123, R145, R149, R156, K162, R164, and N62 were the most involved in forming H-bonds, frequently with the five aptamers.
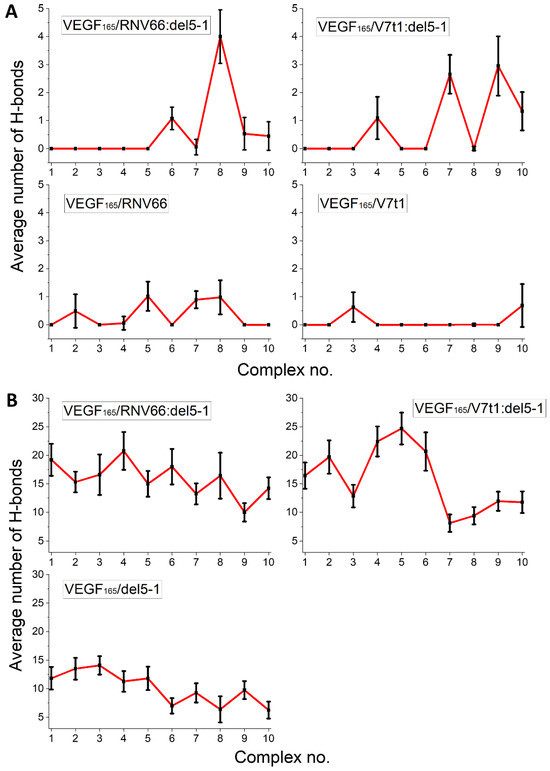
Figure 9.
Average number of H-bonds formed between the aptamers and the key residues of VEGF165 in (A) the RBD and (B) the HBD parts. Horizontal axes denote the top-ten complex structures for the given complex type.
2.5. Hydrogen Bonds between Aptamer and the Other Residues of VEGF165
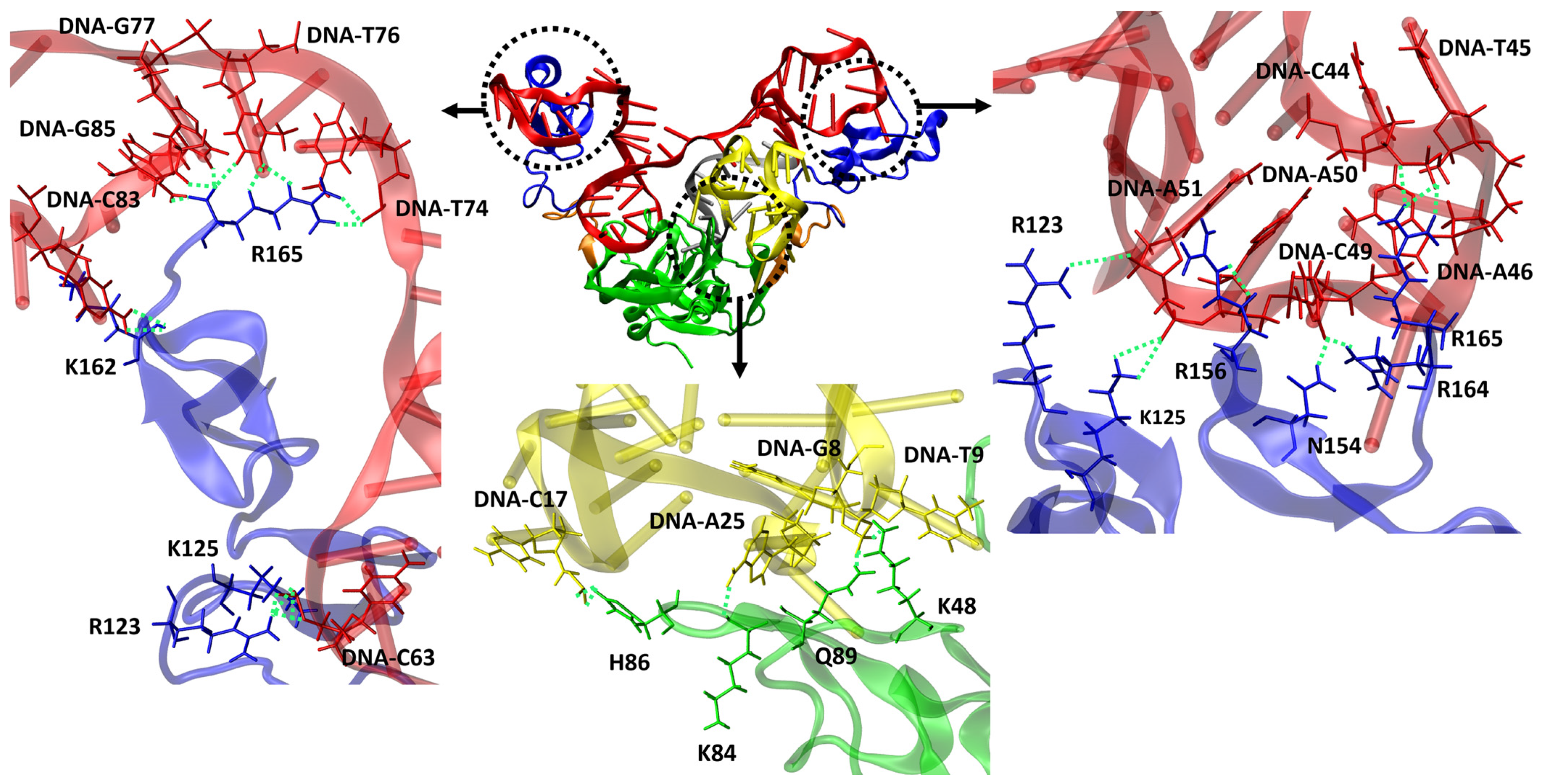
We also conducted an analysis of H-bond formations involving the other residues of VEGF165 to understand why heterodimer aptamers exhibit higher binding affinity compared to monomeric aptamers. This analysis specifically focused on the top-ten structures based on the binding free energy for each VEGF165/aptamer complex. For RNV66 and V7t1, the interactions predominantly involved VEGF165 residues D35, Q37, R56, and H99 when forming H-bonds with the RBD part. For del5-1, significant involvements were observed with R123, R124, K125, K136, S138, K140, Y142, D143, S144, R145, R149, N154, R156, R159, R164, and R165 in the HBD part. In contrast, the two heterodimer aptamers (RNV66:del5-1 and V7t1:del5-1) formed H-bonds not only with D35, Q37, R56, and H99 but also with K48, K84, H86, and Q89 in the RBD part (Figure 10). In the HBD part, in addition to the important VEGF165 residues mentioned for del5-1, interactions with K147, Q150, and K163 were also observed. These aspects illustrate that the higher binding affinities of the heterodimer aptamers result from their ability to bind to both the RBD and the HBD units of VEGF165, providing a distinct advantage in binding. We also speculate that homodimer aptamers will likely bind only to either RBD or HBD, and they will have lower binding affinities with VEGF165 than the heterodimers.
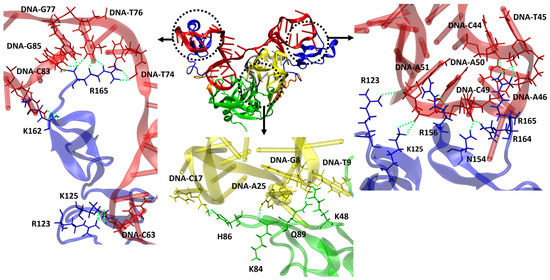
Figure 10.
Representation of VEGF165/RNV66:del5-1 complex and H-bonds formed between aptamer and VEGF165. RNV66 is shown in yellow, del5-1 in red, the linker between RNV66 and del5-1 in silver, the RBD units in green, the HBD units in blue, and the linker between RBD and HBD in orange. The upper panels represent the important residues for forming H-bonds between the HBD part and del5-1, while the lower panel depicts the crucial residues for forming H-bonds between the RBD part and RNV66. H-bonds are depicted as green dashed lines.
2.6. Steric Clashes between Aptamer and VEGFR-2
We believe that an aptamer can act as an effective inhibitor of VEGFR-2 activation because it can block the VEGF165-to-VEGFR-2 interaction itself. To fulfill this purpose, there should be a considerable steric clash between VEGFR-2 and the VEGF165/aptamer complex. To estimate this aspect, we employed the PISA server [53] to calculate the total area of the sterically clashing regions between VEGFR-2 and the aptamer based on the final snapshots from the MD trajectories of the top-ten complex structures with the lowest binding free energies. A detailed description is provided later in the Materials and Methods section. Here again, aptamer heterodimers generally exhibited significantly larger areas of steric clashes with VEGFR-2, further supporting the superior effectiveness of the heterodimers (Table 2).
Table 2.
Total area of steric clashes in nm2 between aptamer and VEGFR-2 for the top-10 complex structures of each VEGF165/aptamer complex type.
2.7. Stability of G-Quadruplex Structures
RNV66 and V7t1 are known to possess G-quadruplex structures [24]. We attempted to see whether the G-quadruplex structures of RNV66 and V7t1 were maintained during their binding to VEGF165, either as a monomeric aptamer or as part of a heterodimer. We adopted the trajectories of the VEGF165/aptamer complexes containing either RNV66 or V7t1, for a total of 470 trajectories each 15 ns long, as explained in Section 2.3. In the case of RNV66, to assess the stability of a G-quadruplex structure, we calculated the RMSD using the twelve residues that constitute the G-quadruplex region with respect to its NMR structure (PDB ID: 2M53) [24]. For V7t1, we adopted the corresponding twelve residues for the RMSD calculation, and the reference was the energy-minimized structure of V7t1, namely the energy minimization result that started from the NMR structure of RNV66 after replacing LNA-G residues with DNA-G residues. The results are shown in Figure 11, in which it can be observed that the binding of RNV66 and V7t1 to VEGF165 either as a monomeric aptamer or as part of a heterodimer did not significantly alter their G-quadruplex structures. Interestingly, RNV66 preserved the G-quadruplex structure better than V7t1, and the extent of the preservation was larger in the monomer case (Figure 11B). This aspect is consistent with the fact that the LNA modifications in RNV66 enhance the stability of the G-quadruplex structure [24,54] and further supports our prediction that RNV66:del5-1 will likely work better than V7t1:del5-1.
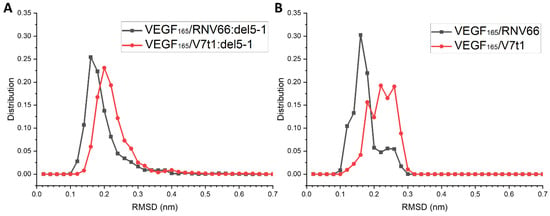
Figure 11.
RMSD distributions of the G-quadruplex structures in RNV66 and V7t1 within (A) the complexes with heterodimeric aptamers and (B) the complexes with monomeric aptamers.
3. Materials and Methods
3.1. Modeling DNA Aptamers
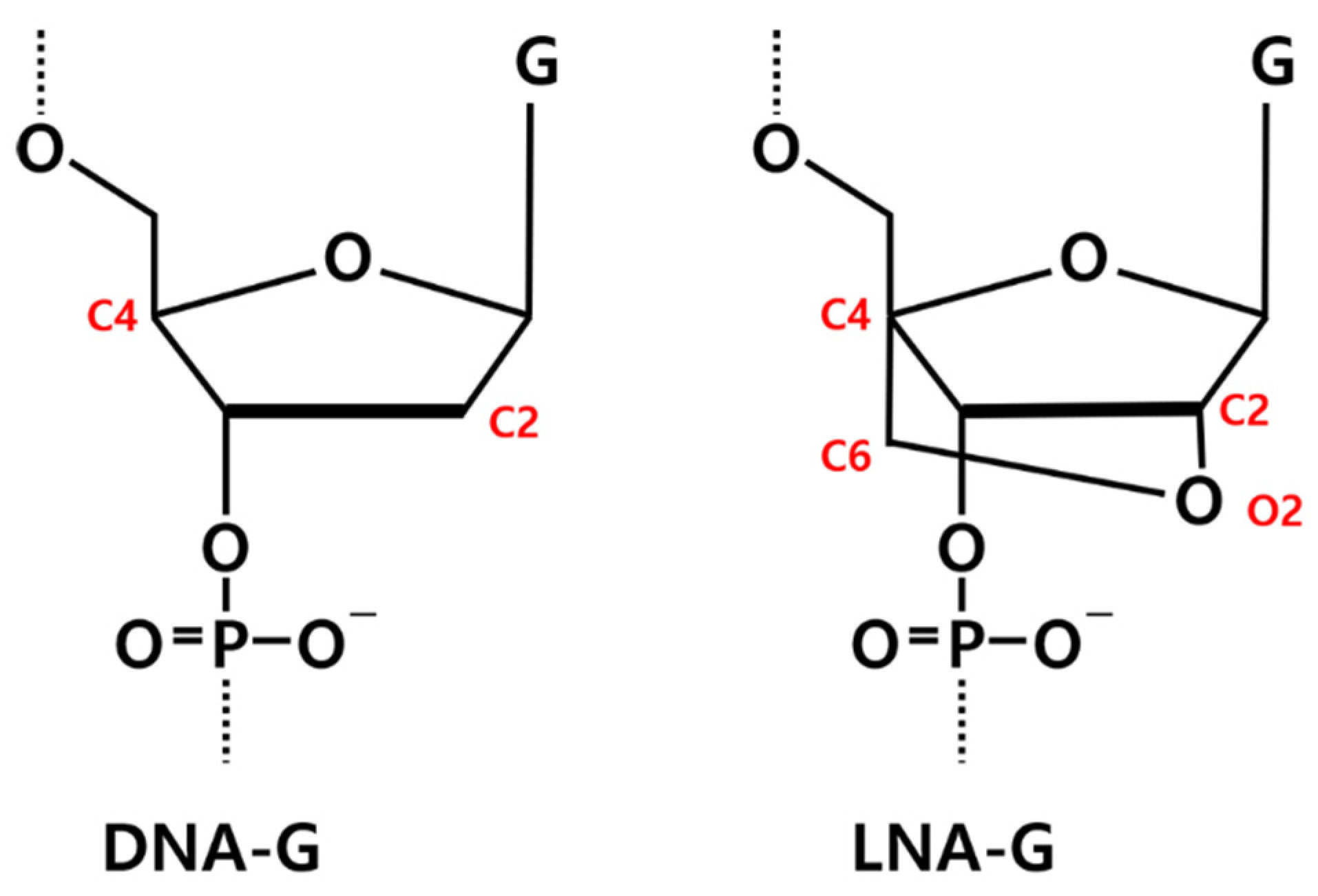
The initial structure of RNV66 was taken from its NMR structure deposited in the Protein Data Bank (PDB ID: 2M53) [24]. Because the experimental structure of V7t1 (5′-TGTGGGGGTGGACGGGCCGGGTAGA-3′) was not available, we generated it from the structure of RNV66 (5′-TGTGLGGGTGGACGGGCCGGLTALA-3′) by replacing its LNA-G residues at positions 5, 21, and 24 with DNA-G residues. This was achieved by removing O2 and C6 atoms, followed by adding single hydrogen atoms to C2 and C4 (Figure 12). Two K+ ions were placed between the three G-quartet planes to help maintain the G-quadruplex structures (Figure S4).
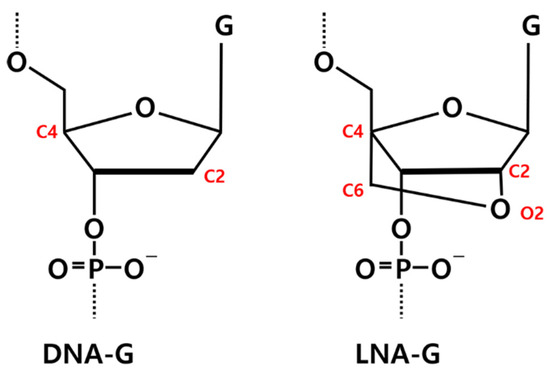
Figure 12.
Chemical structures of DNA-guanine (DNA-G) and LNA-guanine (LNA-G). Atoms involved in the LNA bridging are highlighted in red.
In the case of del5-1, due to the absence of an available three-dimensional (3D) structure, it was generated from its sequence information (5′- ATACCAGTCTATTCAATTGGGCCCGTCCGTATGGTGGGTGTGCTGGCCAG-3′) using various nucleic acid folding programs. The energetically most stable secondary DNA structure of del5-1 that can be written in a two-dimensional (2D) form was created with its sequence information using the RNAstructure web server [55]. In fact, the same 2D structure was obtained when we adopted the Mfold web server [56], further validating the structure. However, because predicting a 3D DNA structure based on 2D DNA structure information is not practically feasible, we instead generated a 3D RNA structure first based on the 2D DNA structure information after the apparent nucleobase replacements [57] using the RNAComposer web server [58]. The 3D DNA structure of del5-1 was then obtained from the 3D RNA structure by inversely replacing uracil with thymine. This indirect computational method, including the generation of a 3D DNA structure from a 3D RNA structure, while it is approximate and should be used with care, has been utilized in various earlier studies [59,60]. To generate the V7t1:del5-1 structure, V7t1 and del5-1 were linked by using ten thymine nucleotides. The RNV66:del5-1 heterodimer was generated in a similar manner. Structures, including the RNV66 obtained from the Protein Data Bank and those modeled through various steps, such as V7t1, del5-1, RNV66:del5-1, and V7t1:del5-1, underwent further refinement through energy minimization using GROMACS 2022 [61]. The minimization process was halted when the maximum force convergence reached below 1000 kJ mol−1 nm−1. Subsequently, 2 ns MD simulations under NPT conditions were conducted for equilibration purposes.
3.2. Modeling VEGF165
For the protein side, the full experimental structure of the homodimeric VEGF165 is not available. However, the X-ray crystal structure of the homodimeric RBD (PDB ID: 2VPF) [10] and the NMR structure of the monomeric HBD (PDB ID: 1VGH) [9] are available, and we manually linked HBD to each RBD monomer using the sequence information [8,39]. The interdomain linker sequence was RPKKDRARQENP, where RPKKD and ARQENP corresponded to the X-ray crystal structure of RBD (PDB ID: 2VPF) and the NMR structure of HBD (PDB ID: 1VGH), respectively. The missing residue R110 was generated using Avogadro [62]. Employing VMD [49], the individual components were manually linked to form the complete structure of the homodimeric VEGF165. Following this, the structure was refined through 500 steps of energy minimization and 2 ns of MD simulation under NPT conditions. We stress that the structure after 2 ns MD should not be considered a representative equilibrium structure. Because VEGF165 with the linker will inevitably be very flexible at room temperature, it will exist by forming a diverse structural ensemble. This is actually the most important reason for using the ensemble docking method.
3.3. Anisotropic Network Model (ANM) Analysis
The lowest-frequency normal modes of the aptamer heterodimers and VEGF165 were analyzed using the ANM web server [63]. ANM is a simple normal-mode analysis method at the residue level, showing the collective motions of large molecules [38]. While we utilized ANM to identify the lowest-frequency normal modes of VEGF165 and aptamer heterodimers (Figure 2 and Figure 3), we conducted additional 500 ns MD simulations for them to confirm that the motions from ANM appear consistently. The rationale behind this additional endeavor is the fact that both VEGF165 and aptamers are quite flexible without any uniquely defined representative equilibrium structures, and how ANM will behave without a well-defined equilibrium structure may be rather unclear. Upon applying ANM to the resulting structures of these long MD simulations, we confirmed that, like the initial structures we employed, the lowest-frequency normal modes were scissoring motions for VEGF165 and wavy motions for the aptamer heterodimers.
3.4. Docking with HDOCK
All docking calculations were carried out using HDOCK [40,41], with which a target was immobilized and only the ligand was allowed to wander in translational and rotational space with a fixed step size. An interval of 15 deg was used for the rotational sampling, and an interval of 0.12 nm was used for the fast Fourier transform (FFT)-based translational sampling [40]. A shape-based pairwise scoring function was employed to score the binding modes obtained through the sampling. The top-ten translations with the best shape complementarity were re-scored and optimized for each rotation by an iterative knowledge-based scoring function. The best-scored translation was then kept for each rotation [40].
3.5. MD Simulations
In the case of unbiased MD simulations, systems were solvated with TIP3P water [64] with a minimum distance of 10 Å between the solute and the edge of the box. Namely, for the initial structures, VEGF165 was placed in a simulation box with dimensions of 9.9 × 11.2 × 11.0 nm3; RNV66:del5-1 and V7t1:del5-1 were placed in a box with dimensions of 10.9 × 8.7 × 9.5 nm3; RNV66 and V7t1 were placed in a box with dimensions of 5.5 × 6.3 × 5.9 nm3; and del5-1 was placed in a box with dimensions of 6.9 × 7.9 × 7.8 nm3. For charge neutralization, counter ions were added. The DNA aptamers were described using the AMBER OL15 force field [65], with the missing parameters for the LNA-G residues in RNV66 taken from an earlier work [54]. The force field parameters for VEGF165 were taken from AMBER ff19SB [66]. The ions were modeled using the parameters proposed by Joung and Cheatham [67]. The topology files for the simulations were generated using AmberTools22 [68]. For each system, energy minimization using the steepest descent method [69] was first taken, followed by equilibration for 2 ns under NPT conditions. The minimization convergence was declared when the maximum force became smaller than 1000 kJ mol−1 nm−1.
For biased MD simulations, the conformations after 2 ns of initial equilibration were solvated in TIP3P water with a minimum distance of 60 Å between the solute and the edge of the box. With this, the simulation box sizes were the following: for VEGF165, 20.6 × 20.1 × 20.3 nm3; for RNV66:del5-1, 18.2 × 17.7 × 16.9 nm3; and for V7t1:del5-1, 19.2 × 17.0 × 19.7 nm3. To generate discrete conformations of VEGF165 and aptamer heterodimers along the lowest-frequency normal modes, biasing potentials with a force constant of 1000 kJ mol−1 nm−2 were applied along the distance between COMs. At this stage, the umbrella sampling tool [70] was employed as a practical tool for conducting biased MD simulations. The duration of any simulation was 10 ns after undergoing energy minimization and equilibration. For each monomeric aptamer that did not require a biased simulation, to be fair, 10 ns of MD simulations were performed without adding any bias. These processes were performed for all the complexes extracted based on the docking scores for further MD simulations, and production MD simulations were performed for 15 ns under NPT conditions, following energy minimization and 100 ps of equilibration under NVT conditions.
All simulations were performed with periodic boundary conditions using GROMACS 2022 [61]. The SHAKE algorithm was used to constrain the bonds involving hydrogen atoms, allowing a time step of 2 fs [71]. The short-range Lennard-Jones interactions were truncated at a cutoff distance of 1.2 nm, and long-range dispersion corrections were applied for energy and pressure. The long-range electrostatic interactions were treated with the Particle Mesh Ewald (PME) approach [72] with a real-space cutoff of 1.2 nm. The temperature was maintained at 300 K using a velocity-rescale thermostat [73] with a relaxation time of 1 ps. The pressure was isotropically coupled at 1 bar employing a c-rescale barostat [74] with a coupling constant of 1 ps and a compressibility of 4.5 × 10−5 bar−1.
3.6. MM/GBSA Binding Free Energy
The MM/GBSA method [32] is an end-point free energy calculation method that maintains a good balance between computational efficiency and accuracy [75]. In particular, it has been widely used for the re-scoring of docked poses in structure-based drug design by calculating binding free energies from MD simulations of the docked poses [76]. In the MM/GBSA method, the binding free energy of a ligand–target complex is calculated as the sum of ΔEMM, ΔGsol, and −TΔS, where they represent the changes in the gas-phase molecular mechanics (MM) energy, the solvation free energy, and the conformational entropy upon ligand–target binding, respectively [32]. ΔEMM is calculated based on the force field used for the simulations, while the polar and nonpolar components of ΔGsol are calculated, respectively, using the generalized Born (GB) model and the solvent-accessible surface area (SASA)-based approach [77,78]. The −TΔS term is usually calculated either by a normal-mode analysis or by a quasi-harmonic analysis [79], which are computationally burdensome and tend to bear a large margin of errors [80,81,82]. Therefore, MM/GBSA binding free energies are often calculated by ignoring the −TΔS term [25,32,80,81,82], especially when the relative binding free energies of closely related ligands with the same target are needed, as in our case [32]. We adopted the single-trajectory MM/GBSA approach [32], which ignores ligand and target conformational changes upon binding, leading to a significant reduction in noise in the binding free energy calculation [82]. All MM/GBSA calculations were performed using AmberTools22 [68].
3.7. Total Area of Steric Clashes between Aptamer and VEGFR-2
To estimate the total area of steric clashes between aptamer and VEGFR-2 when the VEGF165/aptamer complex binds to VEGFR-2, we first overlaid the final snapshot of the VEGF165/aptamer complex obtained after 15 ns of MD simulations onto the X-ray crystal structure of the VEGF-A/VEGFR-2 D23 complex (PDB ID: 3V2A). The structural overlay was performed by aligning the RBD part of the VEGF165/aptamer complex with that of the VEGF-A/VEGFR-2 D23 complex, which resulted in significant steric clashes between the aptamer and VEGFR-2. The buried surface area between the aptamer and VEGFR-2 was then calculated using the PISA server [53]. This value was equivalent to the total area of steric clashes between the aptamer and VEGFR-2.
4. Conclusions
The aptamer heterodimer V7t1:del5-1 has been shown experimentally to be a superior inhibitor of VEGF165 compared to its monomeric counterparts, V7t1 and del5-1 [23], but the flexible linker present in both VEGF165 and the aptamer heterodimer is preventing the experimental structure determination of the complex formed between the aptamer heterodimer and VEGF165. To overcome this limitation, an ensemble docking approach, considering both small- and large-scale motions in both VEGF165 and the aptamer heterodimer, is proposed here to obtain trustworthy structures of the aptamer heterodimer complexed with VEGF165. This ensemble docking approach was used to find the complex structures of V7t1:del5-1 and the newly designed RNV66:del5-1, as well as their monomeric counterparts (V7t1, RNV66, and del5-1) with VEGF165. The binding free energy analysis found that RNV66:del5-1 has the highest binding affinity with VEGF165 among the five types of aptamers studied, suggesting RNV66:del5-1 as a promising new aptamer heterodimer against VEGF165. Additionally, aptamer heterodimers show higher binding affinities to VEGF165 compared to their monomeric counterparts, consistent with the experimental results. This adds reliability to our ensemble docking approach for generating the structures of VEGF165/aptamer complexes. It was found that all five types of aptamers predominantly prefer a sandwich pose while forming complexes with VEGF165. In the sandwich pose, the aptamer is sandwiched between the two HBD units of VEGF165. Side and hug poses were also observed in the structures of VEGF165/aptamer complexes. Compared to monomeric aptamers, aptamer heterodimers have more H-bonds with the key residues of VEGF165 involved in the heparin and VEGFR-2 binding events, as well as a larger area of steric clashes with VEGFR-2 when the VEGF165/aptamer complex interacts with VEGFR-2, suggesting aptamer heterodimers as the most effective anti-VEGF165 aptamers over monomeric aptamers. Based on our simulation results, we found that the G-quadruplex structure of RNV66 or V7t1 is not significantly damaged upon binding with VEGF165, either as a monomeric aptamer or as part of an aptamer heterodimer. Moreover, the G-quadruplex structure of RNV66 was found to be more stable than that of V7t1 in the complexes of both monomeric and heterodimeric aptamers with VEGF165. The structural stability of RNV66 over V7t1 found in our simulations is consistent with the findings from previous experimental and molecular simulation studies that demonstrated that the presence of LNA residues in RNV66 stabilizes its G-quadruplex structure [24,54]. We believe that our ensemble docking approach, which incorporates both small- and large-scale motions, will be valuable in the development of new homo/heterodimeric therapeutic drugs against the homodimeric VEGF165.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25074066/s1.
Author Contributions
Conceptualization, Y.J.G. and M.K.; methodology, Y.J.G. and M.K.; validation, M.K. and Y.M.R.; investigation, Y.J.G. and M.K.; resources, Y.M.R.; data curation, Y.J.G.; writing—original draft preparation, Y.J.G.; writing—review and editing, M.K. and Y.M.R.; visualization, Y.J.G.; supervision, M.K. and Y.M.R.; project administration, Y.M.R.; funding acquisition, Y.M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Creative Materials Discovery Program (Grant 2018M3D1A1058813) and the Mid-Career Researcher Program (Grant 2021R1A2C2094153) through the National Research Foundation, funded by the Ministry of Science and ICT of Korea.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The initially designed structures of the DNA aptamers and VEGF165, along with the structures utilized in MD simulations, docking, MM/GBSA calculations, and associated files, i.e., input, parameters, topology files, and scripts for data analyses, are accessible to the public on GitHub (https://github.com/GoYeonju/Ensemble-docking (accessed on 24 November 2023)). The MD simulations, analyses, and MM/GBSA calculations were performed using GROMACS 2022 and AmberTools22, and all docking procedures were performed using HDOCK.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular Endothelial Growth Factor A in Intraocular Vascular Disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef]
- Ng, E.W.M.; Adamis, A.P. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can. J. Ophthalmol. 2005, 40, 352–368. [Google Scholar] [CrossRef]
- Bhisitkul, R.B. Vascular endothelial growth factor biology: Clinical implications for ocular treatments. Br. J. Ophthalmol. 2006, 90, 1542–1547. [Google Scholar] [CrossRef]
- Uciechowska-Kaczmarzyk, U.; Babik, S.; Zsila, F.; Bojarski, K.K.; Beke-Somfai, T.; Samsonov, S.A. Molecular dynamics-based model of VEGF-A and its heparin interactions. J. Mol. Graph. Model. 2018, 82, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, W.J.; Champe, M.A.; Christinger, H.W.; Keyt, B.A.; Starovasnik, M.A. Solution structure of the heparin-binding domain of vascular endothelial growth factor. Structure 1998, 6, 637–648. [Google Scholar] [CrossRef]
- Muller, Y.A.; Christinger, H.W.; Keyt, B.A.; de Vos, A.M. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: Multiple copy flexibility and receptor binding. Structure 1997, 5, 1325–1338. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.J.; Kalathingal, M.; Rhee, Y.M. Elucidating activation and deactivation dynamics of VEGFR-2 transmembrane domain with coarse-grained molecular dynamics simulations. PLoS ONE 2023, 18, e0281781. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Ahmad, A.; Nawaz, M.I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J. Cell. Biochem. 2022, 123, 1938–1965. [Google Scholar] [CrossRef] [PubMed]
- Muhsin, M.; Graham, J.; Kirkpatrick, P. Bevacizumab. Nat. Rev. Drug Discov. 2004, 3, 995–996. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W.; Grippon, S.; Kirkpatrick, P. Aflibercept. Nat. Rev. Drug Discov. 2012, 11, 269–270. [Google Scholar] [CrossRef]
- Kalathingal, M.; Rhee, Y.M. Molecular mechanism of binding between a therapeutic RNA aptamer and its protein target VEGF: A molecular dynamics study. J. Comput. Chem. 2023, 44, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Fukaya, T.; Abe, K.; Savory, N.; Tsukakoshi, K.; Yoshida, W.; Ferri, S.; Sode, K.; Ikebukuro, K. Improvement of the VEGF binding ability of DNA aptamers through in silico maturation and multimerization strategy. J. Biotechnol. 2015, 212, 99–105. [Google Scholar] [CrossRef]
- Hasegawa, H.; Sode, K.; Ikebukuro, K. Selection of DNA aptamers against VEGF165 using a protein competitor and the aptamer blotting method. Biotechnol. Lett. 2008, 30, 829–834. [Google Scholar] [CrossRef]
- Ikebukuro, K.; Hasegawa, H.; Sode, K. Selection and characterization of DNA aptamers against VEGF165 with aptamer blotting method and its application. In Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 2007; pp. 399–400. [Google Scholar]
- Nonaka, Y.; Sode, K.; Ikebukuro, K. Screening and Improvement of an Anti-VEGF DNA Aptamer. Molecules 2010, 15, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Marusic, M.; Veedu, R.N.; Wengel, J.; Plavec, J. G-rich VEGF aptamer with locked and unlocked nucleic acid modifications exhibits a unique G-quadruplex fold. Nucleic Acids Res. 2013, 41, 9524–9536. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Poongavanam, V.; Kanwar, J.R.; Roy, K.; Hillman, K.M.; Prasad, N.; Leth-Larsen, R.; Petersen, M.; Marusic, M.; Plavec, J.; et al. Targeting VEGF with LNA-stabilized G-rich oligonucleotide for efficient breast cancer inhibition. Chem. Commun. 2015, 51, 9499–9502. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Taira, K.I.; Sode, K.; Ikebukuro, K. Improvement of aptamer affinity by dimerization. Sensors 2008, 8, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Manochehry, S.; McConnell, E.M.; Li, Y. Unraveling Determinants of Affinity Enhancement in Dimeric Aptamers for a Dimeric Protein. Sci. Rep. 2019, 9, 17824. [Google Scholar] [CrossRef]
- Manochehry, S.; Gu, J.; McConnell, E.M.; Salena, B.J.; Li, Y. In Vitro Selection of New DNA Aptamers for Human Vascular Endothelial Growth Factor 165. Chembiochem 2020, 21, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, Y.; Yoshida, W.; Abe, K.; Ferri, S.; Schulze, H.; Bachmann, T.T.; Ikebukuro, K. Affinity improvement of a VEGF aptamer by in silico maturation for a sensitive VEGF-detection system. Anal. Chem. 2013, 85, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T.J. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Sham, Y.Y.; Chu, Z.T.; Tao, H.; Warshel, A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins 2000, 39, 393–407. [Google Scholar] [CrossRef]
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.G.; Polasa, A.; Agrawal, S.; Kumar, T.K.S.; Moradi, M. Binding affinity estimation from restrained umbrella sampling simulations. Nat. Comput. Sci. 2023, 3, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Gindin, T.; Zhang, B.S.; Wang, L.L.; Abel, R.; Murret, C.S.; Xu, F.; Bao, A.; Lu, N.J.; Zhou, T.Q.; et al. Free Energy Perturbation Calculation of Relative Binding Free Energy between Broadly Neutralizing Antibodies and the gp120 Glycoprotein of HIV-1. J. Mol. Biol. 2017, 429, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.W.; Chen, P.C.; Wang, J.; Sun, Y.C. Computation of relative binding free energy for an inhibitor and its analogs binding with Erk kinase using thermodynamic integration MD simulation. J. Comput. Aided Mol. Des. 2012, 26, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Park, J.W.; Go, Y.J.; Kim, W.J.; Rhee, Y.M. Considering both small and large scale motions of vascular endothelial growth factor (VEGF) is crucial for reliably predicting its binding affinities to DNA aptamers. RSC Adv. 2021, 11, 9315–9326. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.M.; Zhang, D.; Zhou, P.; Li, B.T.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Yan, Y.M.; Tao, H.Y.; He, J.H.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Lee, S.Y.; Birkholz, N.; Fineran, P.C.; Park, H.H. Molecular basis of anti-CRISPR operon repression by Aca10. Nucleic Acids Res. 2022, 50, 8919–8928. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Liu, C.; Yu, J.; Han, J.; Du, J.; Liang, S.; Wang, W.; Liu, Q.; Lian, R.; Zhu, T.; et al. PHF14 enhances DNA methylation of SMAD7 gene to promote TGF-β-driven lung adenocarcinoma metastasis. Cell Discov. 2023, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Mohammad, T.; Malik, A.; Hassan, M.I.; Domashevskiy, A.V. Iron response elements (IREs)-mRNA of Alzheimer’s amyloid precursor protein binding to iron regulatory protein (IRP1): A combined molecular docking and spectroscopic approach. Sci. Rep. 2023, 13, 5073. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wen, Z.; Wang, X.; Huang, S.Y. Addressing recent docking challenges: A hybrid strategy to integrate template-based and free protein-protein docking. Proteins 2017, 85, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Henrick, K.; Moult, J.; Eyck, L.T.; Sternberg, M.J.; Vajda, S.; Vakser, I.; Wodak, S.J. CAPRI: A Critical Assessment of PRedicted Interactions. Proteins 2003, 52, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Brozzo, M.S.; Bjelic, S.; Kisko, K.; Schleier, T.; Leppanen, V.M.; Alitalo, K.; Winkler, F.K.; Ballmer-Hofer, K. Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood 2012, 119, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Huang, C.-C.; Liu, Y.-C.; Oyang, Y.-J.; Huang, C.-K. DNA-binding residues and binding mode prediction with binding-mechanism concerned models. BMC Genom. 2009, 10, S23. [Google Scholar] [CrossRef]
- Wohlert, M.; Benselfelt, T.; Wågberg, L.; Furó, I.; Berglund, L.A.; Wohlert, J. Cellulose and the role of hydrogen bonds: Not in charge of everything. Cellulose 2022, 29, 1–23. [Google Scholar] [CrossRef]
- Kalathingal, M.; Sumikama, T.; Mori, T.; Oiki, S.; Saito, S. Structure and dynamics of solvent molecules inside the polytheonamide B channel in different environments: A molecular dynamics study. Phys. Chem. Chem. Phys. 2018, 20, 3334–3348. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Deb, I.; Sarzynska, J.; Lahiri, A. LNA-induced dynamic stability in a therapeutic aptamer: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 41, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Bellaousov, S.; Reuter, J.S.; Seetin, M.G.; Mathews, D.H. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013, 41, W471–W474. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Xiao, Y. 3dDNA: A Computational Method of Building DNA 3D Structures. Molecules 2022, 27, 5936. [Google Scholar] [CrossRef] [PubMed]
- Antczak, M.; Popenda, M.; Zok, T.; Sarzynska, J.; Ratajczak, T.; Tomczyk, K.; Adamiak, R.W.; Szachniuk, M. New functionality of RNAComposer: An application to shape the axis of miR160 precursor structure. Acta. Biochim. Pol. 2016, 63, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef]
- Sabri, M.Z.; Hamid, A.A.A.; Hitam, S.M.S.; Rahim, M.Z.A. The assessment of three dimensional modelling design for single strand DNA aptamers for computational chemistry application. Biophys. Chem. 2020, 267, 106492. [Google Scholar] [CrossRef]
- Bauer, P.; Hess, B.; Lindahl, E. GROMACS 2022 Source Code (Version 2022). Available online: https://zenodo.org/records/6103835 (accessed on 22 February 2022).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Eyal, E.; Lum, G.; Bahar, I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbova, M.; Sponer, J.; Otyepka, M.; Jurecka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.Z.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.S.; Cheatham, T.E., 3rd. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes: The Art of Scientific Computing, 3rd ed.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Torrie, G.M.; Valleau, J.P. Non-Physical Sampling Distributions in Monte-Carlo Free-Energy Estimation—Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints—Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Virtanen, S.I.; Niinivehmas, S.P.; Pentikäinen, O.T. Case-specific performance of MM-PBSA, MM-GBSA, and SIE in virtual screening. J. Mol. Graph. Model. 2015, 62, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.R.W.; Cardozo, M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J. Chem. Inf. Model. 2008, 48, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Honig, B. Calculation of the Total Electrostatic Energy of a Macromolecular System—Solvation Energies, Binding-Energies, and Conformational-Analysis. Proteins 1988, 4, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Hou, T.J.; Xu, X.J. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput. Aid Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.H.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, A.; Edvinsson, L.; Johansson, A.; Andersson, C.D.; Andersson, I.E.; Raubacher, F.; Linusson, A. Postprocessing of docked protein–ligand complexes using implicit solvation models. J. Chem. Inf. Model. 2011, 51, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631–1639. [Google Scholar] [CrossRef]
- Hou, T.J.; Wang, J.M.; Li, Y.Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).