
Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study




Abstract
1. Introduction
2. Results and Discussion
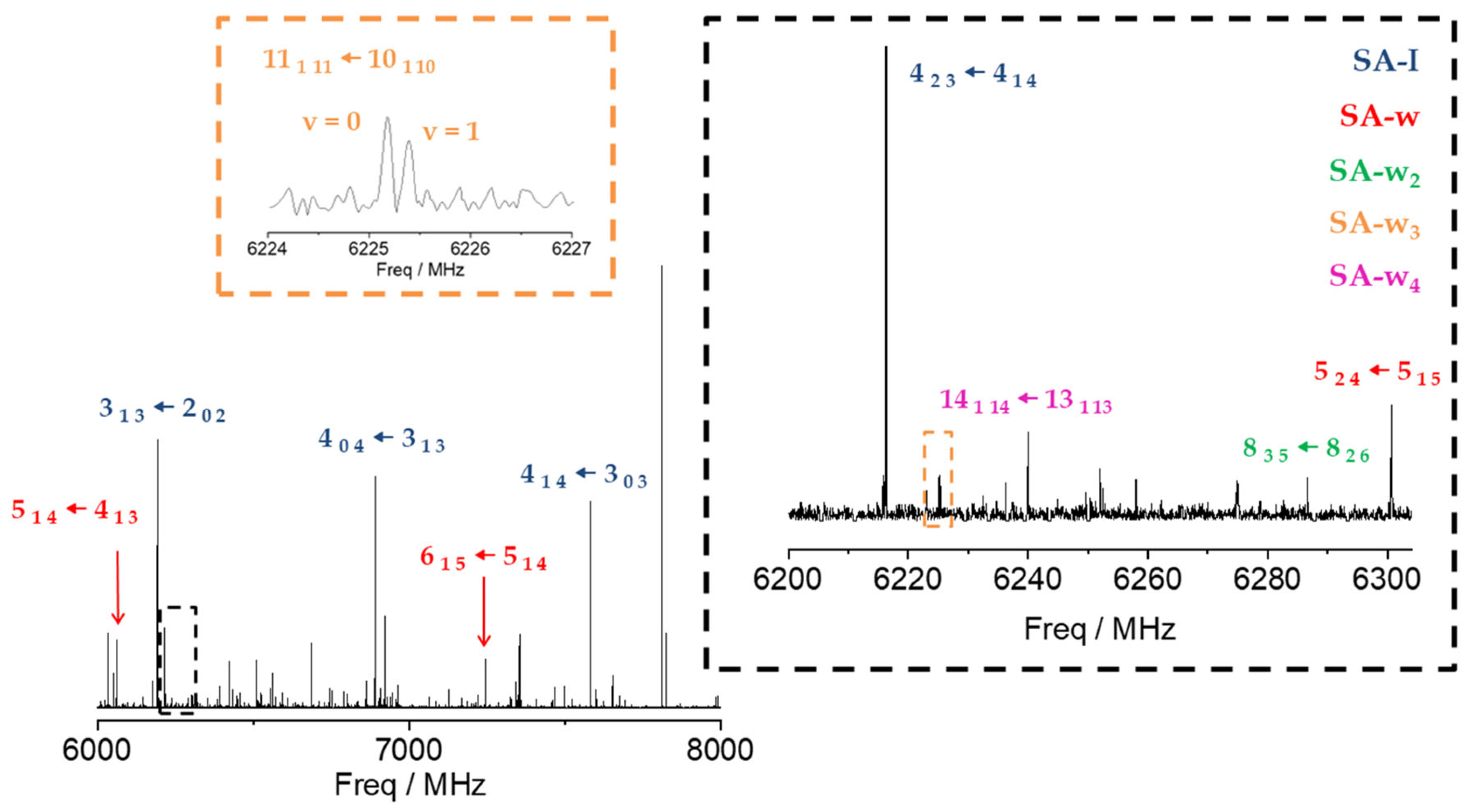
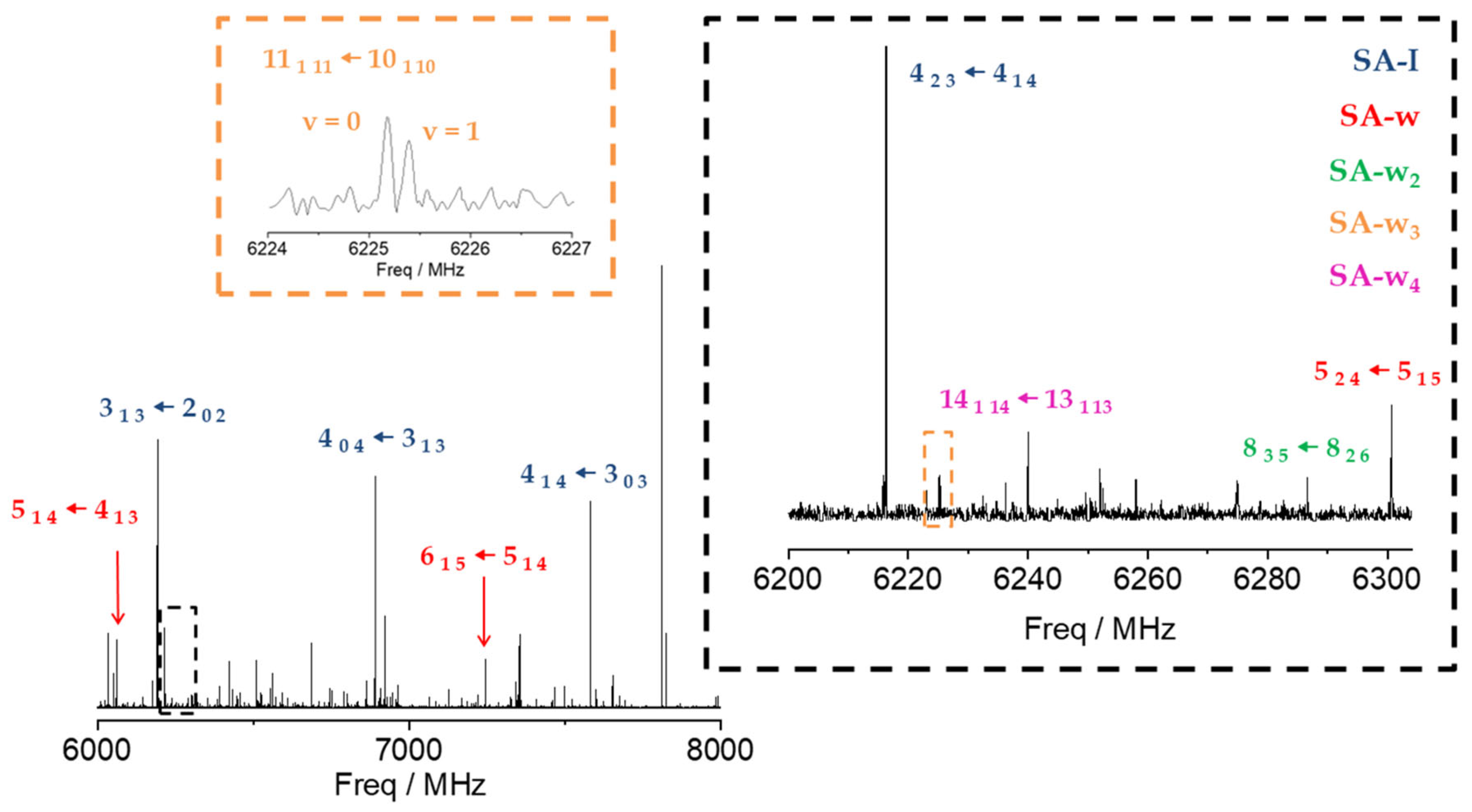
2.1. Rotational Spectrum
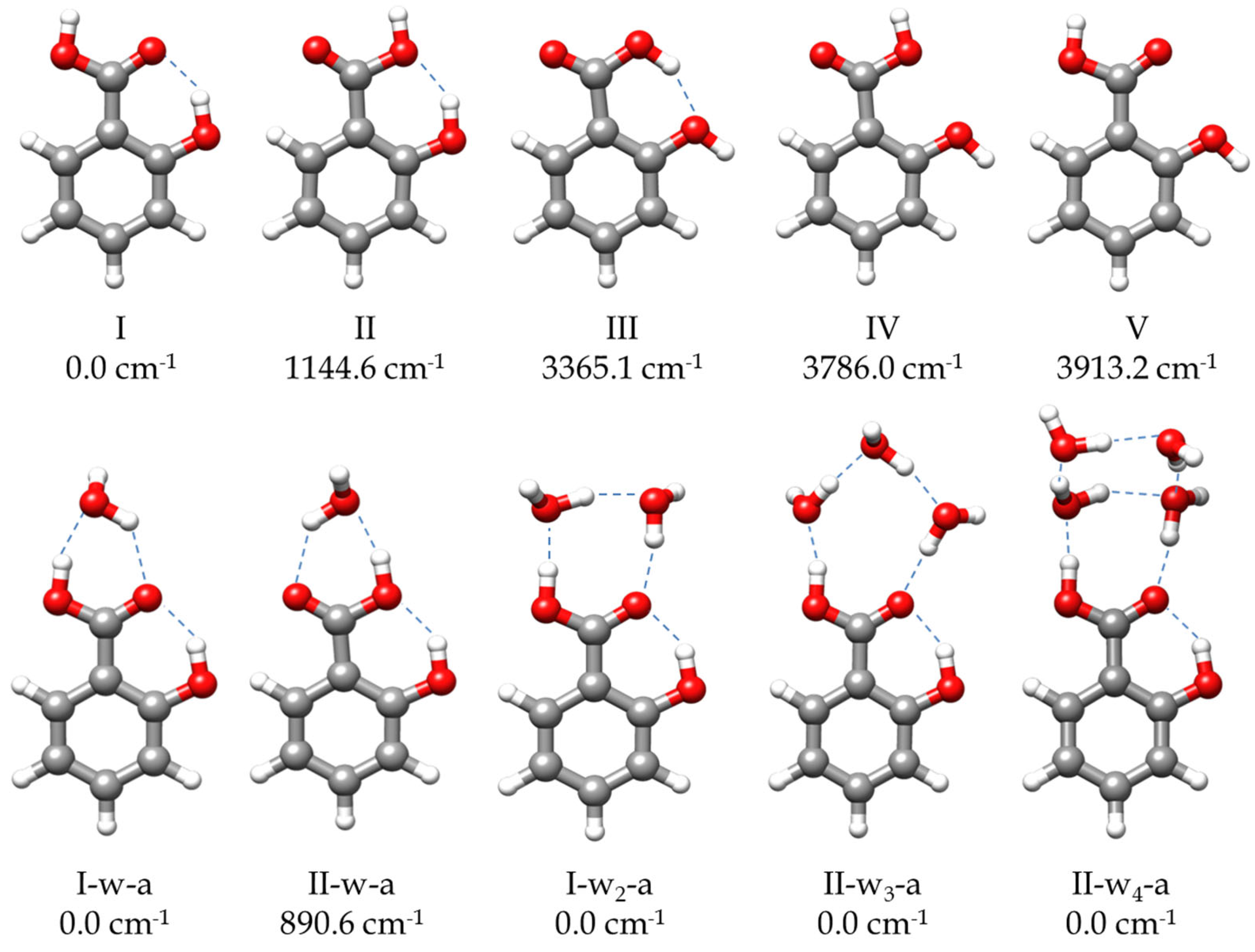
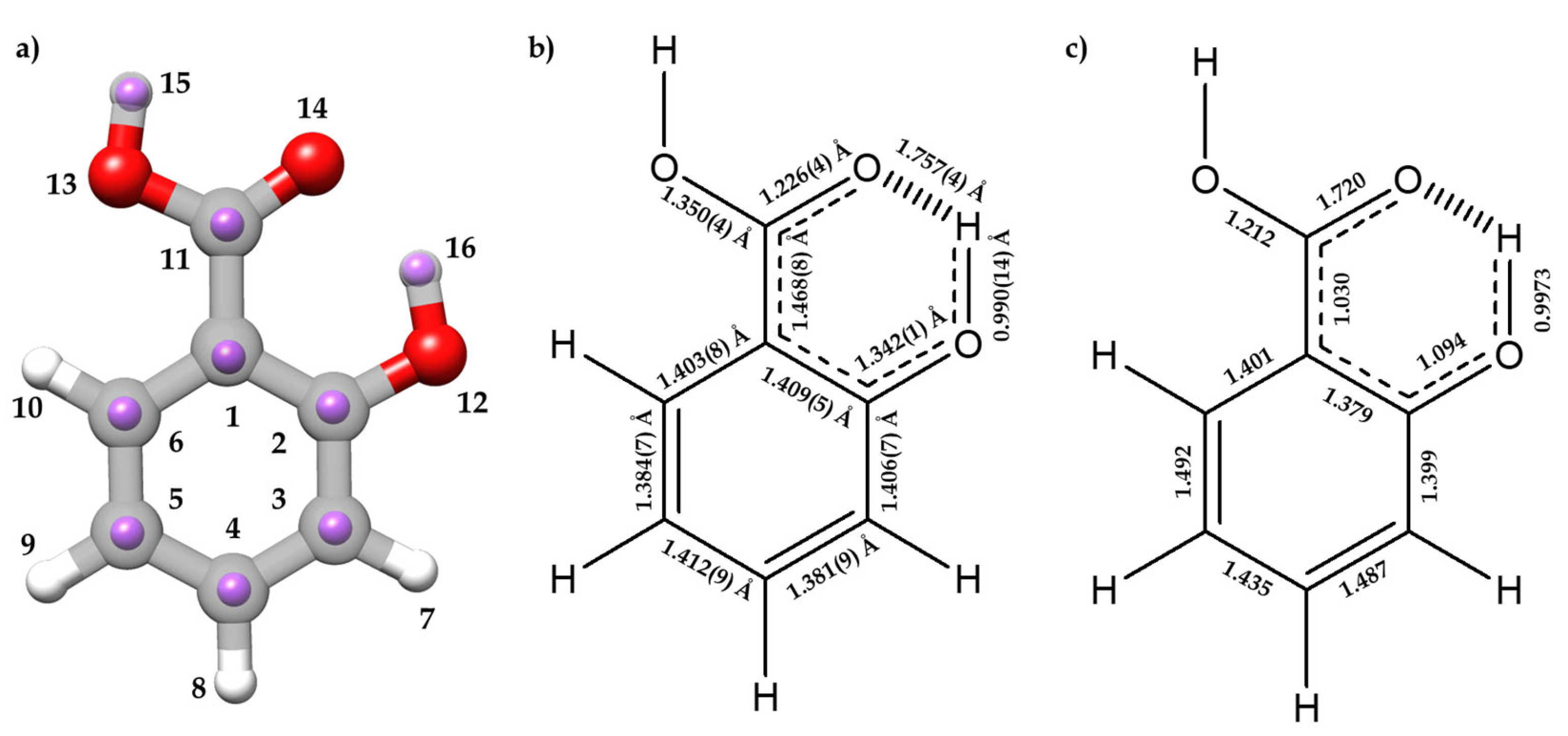
2.2. Molecular Structure
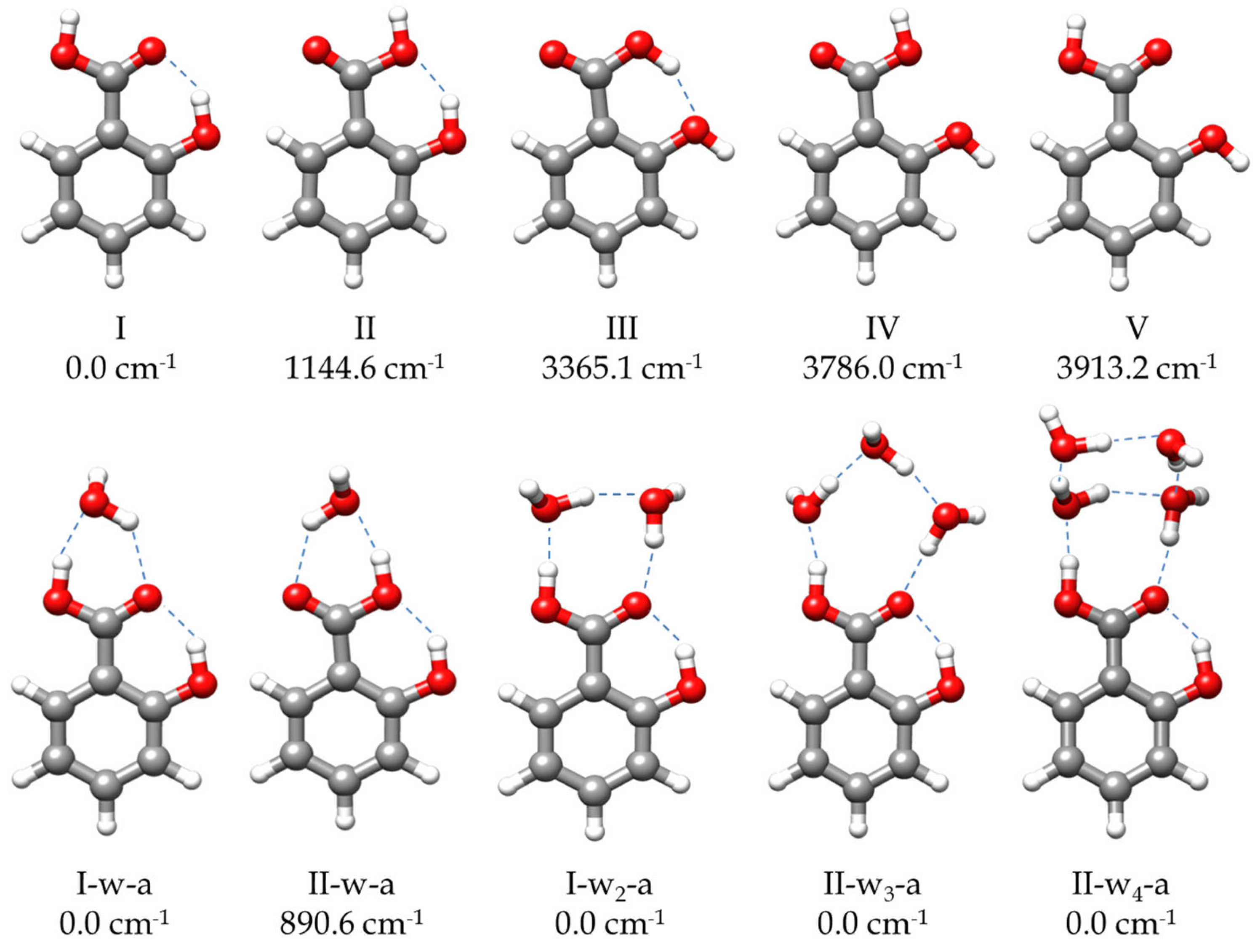
2.2.1. Monomer (SA-I)
2.2.2. Monohydrated Complex (SA-w)
2.2.3. Dihydrated Complex (SA-w2)
2.2.4. Trihydrated Complex (SA-w3)
2.2.5. Tetrahydrated Complex (SA-w4)
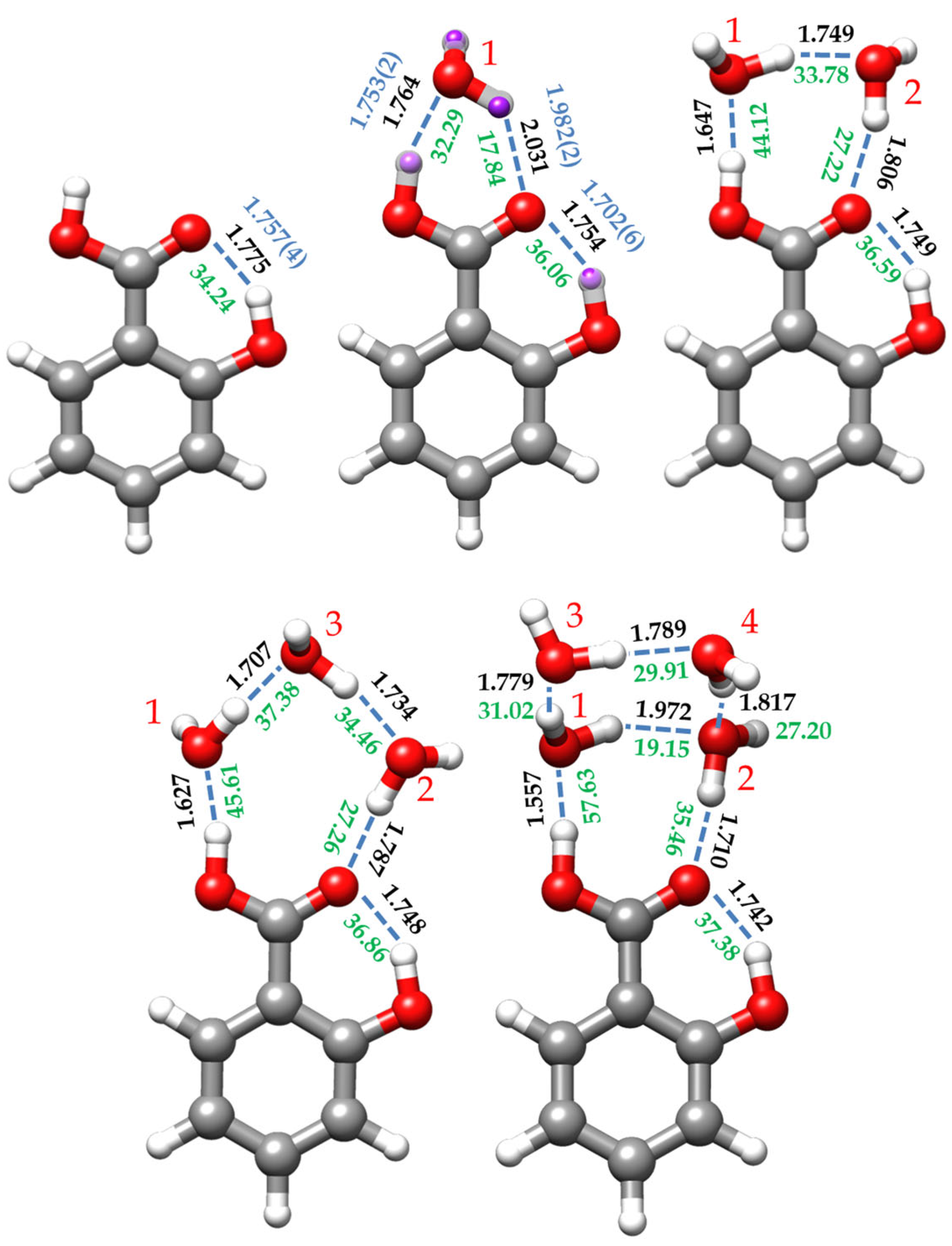
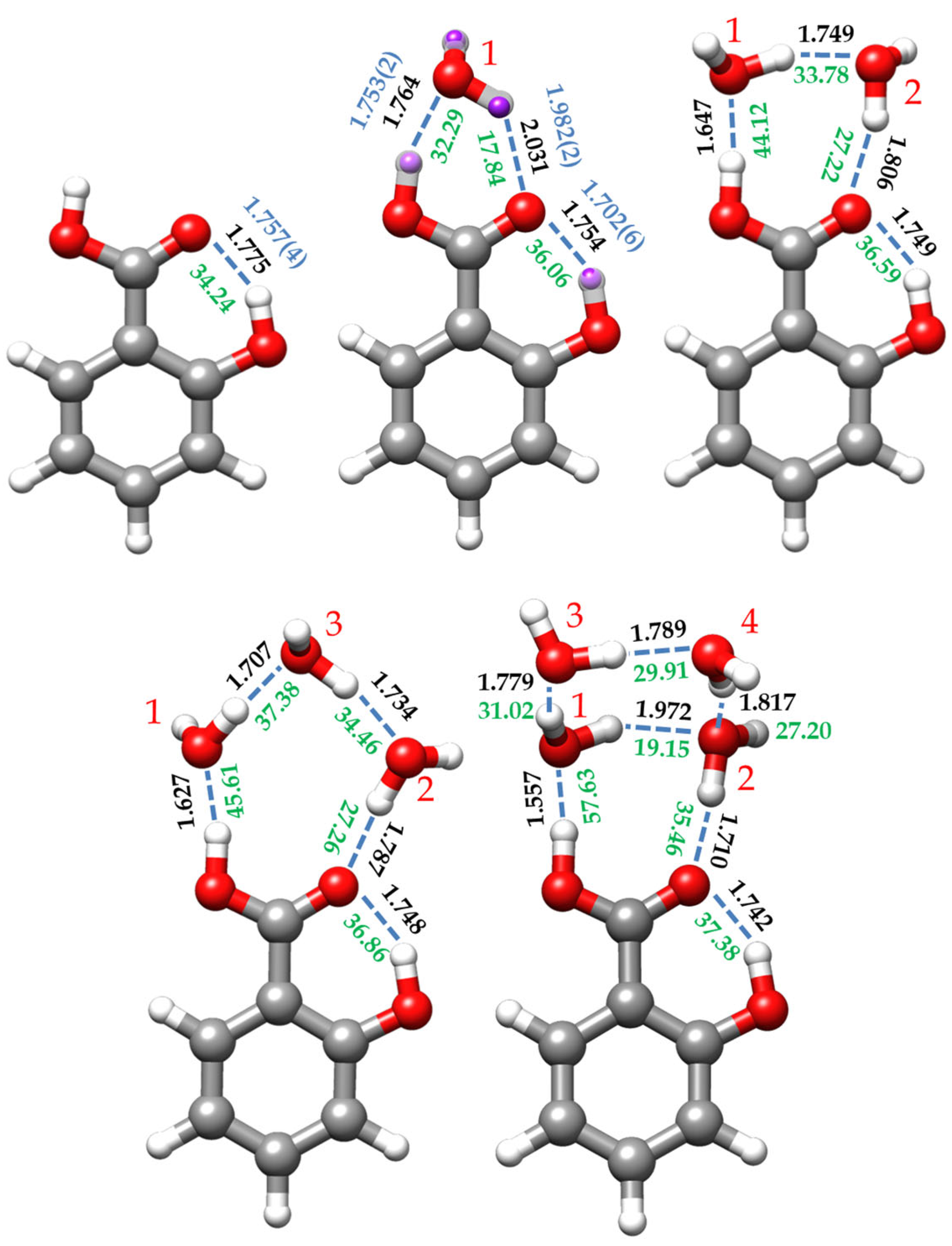
2.3. Cooperative Effects and Dissociation Energies
3. Materials and Methods
3.1. Experimental Details
3.2. Theoretical Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayat, S.; Ahmad, A. Salicylic Acid: A Plant Hormone; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Grosser, T.; Smyth, E.; FitzGerald, G.A. Anti-inflammatory, Antipyretic, and Analgesic Agents; Pharmacotherapy of Gout. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 12th ed.; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2015. [Google Scholar]
- Palleros, D.R. Experimental Organic Chemistry; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Bosund, I.; Erichsen, I.; Molin, N. The Bacteriostatic Action of Benzoic and Salicylic Acids. Physiol. Plant. 1960, 13, 800–811. [Google Scholar] [CrossRef]
- Madan, R.K.; Levitt, J. A review of toxicity from topical salicylic acid preparations. J. Am. Acad. Dermatol. 2014, 70, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, F.B.; Figus, M.; Attygalle, A.B. Ortho effect in electron ionization mass spectrometry of N-acylanilines bearing a proximal halo substituent. J. Am. Soc. Mass Spectrom. 2008, 19, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Wisiorowski, M.; Howard, S.T.; Stolarczyk, L.Z. Angular-group-induced bond alternation. I. Origin of the effect from Ab Initio calculations. Tetrahedron 1997, 53, 13027–13036. [Google Scholar] [CrossRef]
- Howard, S.T.; Krygowski, T.M.; Ciesielski, A.; Wisiorowski, M. Angular group-induced bond alternation II. The magnitude and the nature of the effect and its application to polynuclear benzenoid systems. Tetrahedron 1998, 54, 3533–3548. [Google Scholar] [CrossRef]
- Yahagi, T.; Fujii, A.; Ebata, T.; Mikami, N. Infrared Spectroscopy of the OH Stretching Vibrations of Jet-Cooled Salicylic Acid and Its Dimer in S0 and S1. J. Phys. Chem. A 2001, 105, 10673–10680. [Google Scholar] [CrossRef]
- Evangelisti, L.; Tang, S.; Velino, B.; Caminati, W. Microwave spectrum of salicylic acid. J. Mol. Struct. 2009, 921, 285–288. [Google Scholar] [CrossRef]
- Aarset, K.; Page, E.M.; Rice, D.A. Molecular Structures of Benzoic Acid and 2-Hydroxybenzoic Acid, Obtained by Gas-Phase Electron Diffraction and Theoretical Calculations. J. Phys. Chem. A 2006, 110, 9014–9019. [Google Scholar] [CrossRef]
- Bisht, P.B.; Petek, H.; Yoshihara, K.; Nagashima, U. Excited state enol-keto tautomerization in salicylic acid: A supersonic free jet study. J. Chem. Phys. 1995, 103, 5290–5307. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Intramolecular Hydrogen Bonding in the S1(ππ*) Excited State of Anthranilic Acid and Salicylic Acid: TDDFT Calculation of Excited-State Geometries and Infrared Spectra. J. Phys. Chem. A 2004, 108, 10917–10922. [Google Scholar] [CrossRef]
- Lahmani, F.; Zehnacker-Rentien, A. Effect of Substitution on the Photoinduced Intramolecular Proton Transfer in Salicylic Acid. J. Phys. Chem. A 1997, 101, 6141–6147. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the β-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Desfrançois, C.; Carles, S.; Schermann, J.P. Weakly Bound Clusters of Biological Interest. Chem. Rev. 2000, 100, 3943–3962. [Google Scholar] [CrossRef] [PubMed]
- Dopfer, O.; Fujii, M. Probing Solvation Dynamics around Aromatic and Biological Molecules at the Single-Molecular Level. Chem. Rev. 2016, 116, 5432–5463. [Google Scholar] [CrossRef] [PubMed]
- Becucci, M.; Melandri, S. High-Resolution Spectroscopic Studies of Complexes Formed by Medium-Size Organic Molecules. Chem. Rev. 2016, 116, 5014–5037. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.; Muckle, M.T.; Zaleski, D.P.; Seifert, N.A.; Temelso, B.; Shields, G.C.; Kisiel, Z.; Pate, B.H. Structures of Cage, Prism, and Book Isomers of Water Hexamer from Broadband Rotational Spectroscopy. Science 2012, 336, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.; Neill, J.L.; Muckle, M.T.; Zaleski, D.P.; Peña, I.; Lopez, J.C.; Alonso, J.L.; Pate, B.H. Water–Water and Water–Solute Interactions in Microsolvated Organic Complexes. Angew. Chem. Int. Ed. 2015, 54, 979–982. [Google Scholar] [CrossRef]
- Pérez, C.; López, J.C.; Blanco, S.; Schnell, M. Water-Induced Structural Changes in Crown Ethers from Broadband Rotational Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 4053–4058. [Google Scholar] [CrossRef]
- López, J.C.; Pérez, C.; Blanco, S.; Shubert, V.A.; Temelso, B.; Shields, G.C.; Schnell, M. Water induces the same crown shapes as Li+ or Na+ in 15-crown-5 ether: A broadband rotational study. Phys. Chem. Chem. Phys. 2019, 21, 2875–2881. [Google Scholar] [CrossRef]
- Steber, A.L.; Temelso, B.; Kisiel, Z.; Schnell, M.; Pérez, C. Rotational dive into the water clusters on a simple sugar substrate. Proc. Natl. Acad. Sci. USA 2023, 120, e2214970120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Pérez, C.; Steber, A.L.; Schnell, M.; Lv, D.; Wang, G.; Zeng, X.; Zhou, M. Evolution of Solute–Water Interactions in the Benzaldehyde-(H2O)1–6 Clusters by Rotational Spectroscopy. J. Am. Chem. Soc. 2023, 145, 4119–4128. [Google Scholar] [CrossRef] [PubMed]
- Bellissent-Funel, M.C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Pinacho, P.; López, J.C. Hydrogen-Bond Cooperativity in Formamide2–Water: A Model for Water-Mediated Interactions. Angew. Chem. 2016, 128, 9477–9481. [Google Scholar] [CrossRef]
- Blanco, S.; Pinacho, P.; López, J.C. Structure and Dynamics in Formamide–(H2O)3: A Water Pentamer Analogue. J. Phys. Chem. Lett. 2017, 8, 6060–6066. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Howard, B.J. High-Resolution Microwave Spectroscopic and ab initio Studies of Propanoic Acid and Its Hydrates. J. Phys. Chem. A 2008, 112, 8208–8214. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Howard, B.J. Hydrates of trans- and gauche-Difluoroacetic Acids: A High-Resolution Microwave Spectroscopic Study. J. Phys. Chem. A 2010, 114, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Howard, B.J. The monohydrate and dihydrate of acetic acid: A high-resolution microwave spectroscopic study. Phys. Chem. Chem. Phys. 2009, 11, 366–373. [Google Scholar] [CrossRef]
- Ouyang, B.; Starkey, T.G.; Howard, B.J. High-Resolution Microwave Studies of Ring-Structured Complexes between Trifluoroacetic Acid and Water. J. Phys. Chem. A 2007, 111, 6165–6175. [Google Scholar] [CrossRef]
- Thomas, J.; Sukhorukov, O.; Jäger, W.; Xu, Y. Direct Spectroscopic Detection of the Orientation of Free OH Groups in Methyl Lactate–(Water)1,2 Clusters: Hydration of a Chiral Hydroxy Ester. Angew. Chem. Int. Ed. 2014, 53, 1156–1159. [Google Scholar] [CrossRef]
- Brutschy, B. The Structure of Microsolvated Benzene Derivatives and the Role of Aromatic Substituents. Chem. Rev. 2000, 100, 3891–3920. [Google Scholar] [CrossRef]
- Salmon, R.A.; Schiller, C.L.; Harris, G.W. Evaluation of the Salicylic Acid—Liquid Phase Scrubbing Technique to Monitor Atmospheric Hydroxyl Radicals. J. Atmospheric Chem. 2004, 48, 81–104. [Google Scholar] [CrossRef]
- López, J.C.; Macario, A.; Blanco, S. Conformational equilibria in o -anisic acid and its monohydrated complex: The prevalence of the trans-COOH form. Phys. Chem. Chem. Phys. 2019, 21, 6844–6850. [Google Scholar] [CrossRef] [PubMed]
- Melandri, S.; Giuliano, B.M.; Maris, A.; Favero, L.B.; Ottaviani, P.; Velino, B.; Caminati, W. Methylsalicylate: A Rotational Spectroscopy Study. J. Phys. Chem. A 2007, 111, 9076–9079. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.; López, J.C.; Blanco, S. How Solvents Change the Conformational Landscape in Molecules with Weak Intramolecular Interactions: Methyl 2-Methoxybenzoate. In Proceedings of the 76th International Symposium on Molecular Spectroscopy, Champaign and Urbana, IL, USA, 21–25 June 2021. [Google Scholar] [CrossRef]
- Ghosh, S.; Thomas, J.; Huang, W.; Xu, Y.; Jäger, W. Rotational Spectra of Two Hydrogen-Bonded Methyl Salicylate Monohydrates: Relative Stability and Tunneling Motions. J. Phys. Chem. Lett. 2015, 6, 3126–3131. [Google Scholar] [CrossRef]
- Schnitzler, E.G.; Jäger, W. The benzoic acid–water complex: A potential atmospheric nucleation precursor studied using microwave spectroscopy and ab initio calculations. Phys. Chem. Chem. Phys. 2014, 16, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Priem, D.; Ha, T.-K.; Bauder, A. Rotational spectra and structures of three hydrogen-bonded complexes between formic acid and water. J. Chem. Phys. 2000, 113, 169–175. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding, 1st ed.; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Watson, J.K.G. Vibrational Spectra and Structure, During, J.R., Ed.; Elsevier: Amsterdam, The Netherlands, 1977; Volume 6.
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Pinacho, P.; López, J.C.; Kisiel, Z.; Blanco, S. Microsolvation of ethyl carbamate conformers: Effect of carrier gas on the formation of complexes. Phys. Chem. Chem. Phys. 2020, 22, 18351–18360. [Google Scholar] [CrossRef] [PubMed]
- Pinacho, P.; López, J.C.; Kisiel, Z.; Blanco, S. The effect of microsolvation on the structure, nuclear quadrupole coupling and internal rotation: The methyl carbamate⋯(H2O)1-3 complexes. J. Chem. Phys. 2024, accepted. [Google Scholar]
- Törring, T.; Papoušek, D.; Aliev, M.R. Molecular Vibrational and Rotational Spectra. In Studies in Physical and Theoretical Chemistry; Elsevier: Amsterdam, The Netherlands; Oxford, UK; New York, NY, USA, 1982; Volume 17. [Google Scholar]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra. In Techniques of Chemistry, 3rd ed.; Wiley: New York, NY, USA, 1984. [Google Scholar]
- Onda, M.; Asai, M.; Takise, K.; Kuwae, K.; Hayami, K.; Kuroe, A.; Mori, M.; Miyazaki, H.; Suzuki, N.; Yamaguchi, I. Microwave spectrum of benzoic acid. J. Mol. Struct. 1999, 482–483, 301–303. [Google Scholar] [CrossRef]
- Larsen, N.W. Microwave spectra of the six mono-13C-substituted phenols and of some monodeuterated species of phenol. Complete substitution structure and absolute dipole moment. J. Mol. Struct. 1979, 51, 175–190. [Google Scholar] [CrossRef]
- Kraitchman, J. Determination of Molecular Structure from Microwave Spectroscopic Data. Am. J. Phys. 1953, 21, 17–24. [Google Scholar] [CrossRef]
- Costain, C.C. Further comments on the accuracy of rs substitution structures. Trans. Am. Crystallogr. Assoc. 1966, 2, 157–164. [Google Scholar]
- Rudolph, H.D. Contribution to the systematics of r0-derived molecular structure determinations from rotational parameters. Struct. Chem. 1991, 2, 581–588. [Google Scholar] [CrossRef]
- Desyatnyk, O.; Pszczółkowski, L.; Thorwirth, S.; Krygowski, T.M.; Kisiel, Z. The rotational spectra, electric dipole moments and molecular structures of anisole and benzaldehyde. Phys. Chem. Chem. Phys. 2005, 7, 1708–1715. [Google Scholar] [CrossRef]
- Watson, J.K.G.; Roytburg, A.; Ulrich, W. Least-Squares Mass-Dependence Molecular Structures. J. Mol. Spectrosc. 1999, 196, 102–119. [Google Scholar] [CrossRef] [PubMed]
- Kisiel, Z. Least-squares mass-dependence molecular structures for selected weakly bound intermolecular clusters. J. Mol. Spectrosc. 2003, 218, 58–67. [Google Scholar] [CrossRef]
- Portalone, G.; Schultz, G.; Domenicano, A.; Hargittai, I. Molecular structure and ring distortion of phenol. An electron diffraction study. Chem. Phys. Lett. 1992, 197, 482–488. [Google Scholar] [CrossRef]
- Schnitzler, E.G.; Seifert, N.A.; Kusuma, I.; Jäger, W. Rotational Spectroscopy of p-Toluic Acid and Its 1:1 Complex with Water. J. Phys. Chem. A 2017, 121, 8625–8631. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Dyke, T.R.; Mack, K.M.; Muenter, J.S. The structure of water dimer from molecular beam electric resonance spectroscopy. J. Chem. Phys. 1977, 66, 498–510. [Google Scholar] [CrossRef]
- Cruzan, J.D.; Braly, L.B.; Liu, K.; Brown, M.G.; Loeser, J.G.; Saykally, R.J. Quantifying Hydrogen Bond Cooperativity in Water: VRT Spectroscopy of the Water Tetramer. Science 1996, 271, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Keutsch, F.N.; Cruzan, J.D.; Saykally, R.J. The Water Trimer. Chem. Rev. 2003, 103, 2533–2578. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S. Formamide, Water, and their Complexes: A Microwave Spectroscopy Study. In Proceedings of the 73rd International Symposium on Molecular Spectroscopy, Champaign and Urbana, IL, USA, 18–22 June 2018. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Pinacho, P.; Blanco, S.; López, J.C. The complete conformational panorama of formanilide–water complexes: The role of water as a conformational switch. Phys. Chem. Chem. Phys. 2019, 21, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Plusquellic, D.F. User Guide to JB95.exe Spectral Fitting Program v1.02.4, 1/30/01; NIST: Gaithersburg, MD, USA, 2008. Available online: https://www.nist.gov/publications/user-guide-jb95exe-spectral-fitting-program-v1024-13001 (accessed on 8 February 2024).
- Kisiel, Z.; Pszczółkowski, L.; Medvedev, I.R.; Winnewisser, M.; De Lucia, F.C.; Herbst, E. Rotational spectrum of trans–trans diethyl ether in the ground and three excited vibrational states. J. Mol. Spectrosc. 2005, 233, 231–243. [Google Scholar] [CrossRef]
- “PROSPE-Programs for ROtational SPEctroscopy”. [Online]. Available online: http://info.ifpan.edu.pl/~kisiel/prospe.htm (accessed on 6 March 2024).
- Pickett, H.M. The fitting and prediction of vibration-rotation spectra with spin interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.; Weinhold, F. NBO 5.9: Natural Atomic Orbital and Natural Bond Orbital Analysis; Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, USA. 2009. Available online: https://www2.chem.wisc.edu/~nbo7/ (accessed on 1 April 2024).
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. “Gaussian 16 Rev. C.01.” Wallingford, CT, USA. 2016. Available online: https://gaussian.com/relnotes/ (accessed on 1 April 2024).
- Gerhards, M.; Schmitt, M.; Kleinermanns, K.; Stahl, S. The structure of phenol(H2O) obtained by microwave spectroscopy. J. Chem. Phys. 1996, 104, 967–971. [Google Scholar] [CrossRef]
- Vallejo, J.L.G.; Heredia, J.D.; Gordon, M.S. Bonding analysis of water clusters using quasi-atomic orbitals. Phys. Chem. Chem. Phys. 2021, 23, 18734–18743. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fitted Param. 1 | (MW) | (MW + MMW) 2 | (MMW, ref. [11]) 3 | SA-I—Theor. |
|---|---|---|---|---|
| A/MHz | 2340.25075(42) 4 | 2340.25105(17) | 2340.248(2) | 2336.30 |
| B/MHz | 1223.76931(31) | 1223.76941(13) | 1223.79(2) | 1221.78 |
| C/MHz | 803.89784(31) | 803.89788(18) | 803.90(1) | 802.24 |
| Pcc/uÅ2 | 0.12962(19) | 0.12960(10) | 0.127 | 0.00 |
| ΔJ/kHz | 0.1091(90) | 0.11722(67) | 0.049(6) | 0.1897 |
| ΔJK/kHz | −0.230(28) | −0.2536(26) | 0.130(9) | −0.3939 |
| ΔK/kHz | 0.134(20) | 0.1505(44) | - | 0.2128 |
| δJ/kHz | −0.0303(44) | −0.03392(37) | - | 0.0843 |
| δK/kHz | 0.069(14) | 0.0812(18) | - | −0.1244 |
| N | 64 | 114 | 50 | - |
| σ/kHz | 6.8 | 11.1 | 23.0 | - |
| Fitted Param. 1 | SA-w | I-w-a | II-w-a | SA-w2 | I-w2-a | II-w2-a |
|---|---|---|---|---|---|---|
| A/MHz | 2315.34456(45) 2 | 2313.15 | 2311.93 | 1769.91699(48) | 1780.40 | 1759.99 |
| B/MHz | 645.08107(14) | 648.76 | 647.51 | 451.04489(18) | 453.67 | 450.32 |
| C/MHz | 505.03872(14) | 507.09 | 506.29 | 360.15478(16) | 362.04 | 359.17 |
| Pcc/uÅ 2 | 0.51743(24) | 0.42 | 0.44 | 1.38688(57) | 0.96 | 1.17 |
| ΔJ/kHz | 0.0845(37) | 0.017 | 0.016 | 0.0763(27) | 0.008 | 0.007 |
| ΔJK/kHz | −0.0757(32) | 0.106 | 0.104 | −0.0699(26) | 0.064 | 0.092 |
| ΔK/kHz | - | 0.066 | 0.056 | - | 0.072 | 0.072 |
| δJ/kHz | −0.0274(15) | 0.004 | 0.004 | −0.0333(12) | 0.001 | 0.001 |
| δK/kHz | - | 0.068 | 0.066 | - | 0.049 | 0.049 |
| µa/µb/µc/D | +/+/- | 0.6/1.0/1.2 | 0.7/0.6/1.2 | +/+/- | 0.5/0.9/0.0 | 0.8/0.8/0.0 |
| ΔEDFT/cm−1 | - | 0.0 3 | 890.6 | - | 0.0 4 | 639.9 |
| N | 84 | - | - | 126 | - | - |
| σ/kHz | 5.4 | - | - | 7.7 | - | - |
| Fitted Param. 1 | SA-w3 (ν = 0) | SA-w3 (ν = 1) | I-w3-a | I-w3-b | I-w3-c |
|---|---|---|---|---|---|
| A/MHz | 1412.46036(98) 2 | 1412.42970(98) | 1412.10 | 1436.05 | 1421.44 |
| B/MHz | 315.84360(19) | 315.84610(22) | 319.03 | 317.16 | 327.39 |
| C/MHz | 260.77801(14) | 260.78823(15) | 263.04 | 262.98 | 284.18 |
| Pcc/uÅ2 | 9.9634(11) | 9.9989(12) | 10.33 | 11.82 | 60.41 |
| ΔJ/kHz | 0.01785(63) | 0.018 | 0.016 | 0.029 | |
| ΔJK/kHz | −0.1706(86) | −0.091 | −0.077 | −0.163 | |
| ΔK/kHz | 1.431(79) | 0.846 | 0.812 | 1.225 | |
| d1/kHz | −0.00297(44) | −0.001 | −0.002 | −0.001 | |
| d2/kHz | 0.00197(39) | −0.001 | −0.001 | 0.001 | |
| Fbc/MHz | 0.1304(52) | - | - | - | |
| ΔE01/MHz | 717.0(5.2) | - | - | - | |
| µa/µb/µc/D | +/+/− | 0.6/0.6/0.6 | 0.1/0.0/0.3 | 1.5/1.3/0.1 | |
| ΔEDFT/cm−1 | - | 0.0 3 | 130.4 | 130.4 | |
| N | 160 | - | - | - | |
| σ/kHz | 7.0 | - | - | - | |
| Fitted Param. 1 | SA-w4 | I-w4-a | I-w4-b | I-w4-c | I-w4-d |
|---|---|---|---|---|---|
| A/MHz | 1055.8085(24) 2 | 1073.10 | 1066.29 | 1059.87 | 1093.11 |
| B/MHz | 248.19262(15) | 250.60 | 251.56 | 251.90 | 343.76 |
| C/MHz | 220.88132(19) | 222.99 | 224..02 | 224.40 | 217.21 |
| Pcc/uÅ 2 | 113.4455(21) | 110.68 | 113.23 | 115.48 | 104.46 |
| ΔJ/kHz | 0.191(46) | 0.035 | 0.035 | 0.040 | 0.036 |
| ΔJK/kHz | 0.094(10) | −0.212 | −0.150 | −0.175 | −0.186 |
| ΔK/kHz | −0.239(36) | 1.297 | 1.508 | 1.332 | 1.985 |
| d1/kHz | 0.208(44) | 0.001 | 0.001 | 0.001 | 0.001 |
| d2/kHz | −0.135(21) | 0.001 | 0.001 | 0.001 | 0.001 |
| HJ/Hz | 0.0106(20) | - | - | - | - |
| µa/µb/µc/D | +/+/- | 0.8/1.4/0.2 | 0.7/1.6/0.4 | 0.8/1.3/1.6 | 0.6/3.1/0.5 |
| ΔEDFT/cm−1 | - | 0.0 3 | 165.7 | 241.2 | 267.8 |
| N | 169 | - | - | - | - |
| σ/kHz | 6.4 | - | - | - | - |
| O⋯O/Å | SA | I-w-a | I-w2-a | I-w3-a | I-w4-a |
|---|---|---|---|---|---|
| O12⋯O14 | 2.635 | 2.620 | 2.617 | 2.617 | 2.612 |
| O13⋯Ow1 | - | 2.710 | 2.648 | 2.616 | 2.576 |
| O14⋯Ow2 | - | 2.779 | 2.766 | 2.764 | 2.694 |
| Ow1⋯Ow2 | - | - | 2.682 | - | 2.835 |
| Ow1⋯Ow3 | - | - | - | 2.691 | 2.740 |
| Ow2⋯Ow3 | - | - | - | 2.698 | - |
| Ow3⋯Ow4 | - | - | - | - | 2.745 |
| Ow2⋯Ow4 | - | - | - | - | 2.781 |
| O⋯O w-dimer 1 | 2.976 | ||||
| O⋯O w-trimer 2 | 2.85 | ||||
| O⋯O w-tetramer 3 | 2.78 |
| I-w-a | I-w2-a | I-w3-a | I-w4-a | |
|---|---|---|---|---|
| De/kJ mol−1 | 41.34 | 88.70 | 123.34 | 168.87 |
| De/HB/kJ mol−1 | 20.67 | 29.57 | 30.84 | 28.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macario, A.; López, J.C.; Blanco, S. Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study. Int. J. Mol. Sci. 2024, 25, 4074. https://doi.org/10.3390/ijms25074074
Macario A, López JC, Blanco S. Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study. International Journal of Molecular Sciences. 2024; 25(7):4074. https://doi.org/10.3390/ijms25074074
Chicago/Turabian StyleMacario, Alberto, Juan Carlos López, and Susana Blanco. 2024. "Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study" International Journal of Molecular Sciences 25, no. 7: 4074. https://doi.org/10.3390/ijms25074074
APA StyleMacario, A., López, J. C., & Blanco, S. (2024). Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study. International Journal of Molecular Sciences, 25(7), 4074. https://doi.org/10.3390/ijms25074074