


Endoplasmic Reticulum Stress in Gliomas: Exploiting a Dual-Effect Dysfunction through Chemical Pharmaceutical Compounds and Natural Derivatives for Therapeutical Uses
 , , and
, , and
Abstract
1. Introduction
2. General Characteristics and Classification of Gliomas
3. General Characteristics of the Stress Response of the ER
The Function of the ER
4. Current Perspectives for the Treatment of Glioblastoma (GB)
4.1. Tumor Markers for the Treatment of GB
4.1.1. Chemical and Pharmacological Approaches
4.1.2. Derivatives of Natural Compounds in Treating GB
4.2. Beginning of Stress in the ER: ERAD and UPR Responses
4.2.1. PERK
4.2.2. IRE-1
4.2.3. ATF6
5. Conclusions
Funding
Conflicts of Interest
References
- Laug, D.; Glasgow, S.M.; Deneen, B. A glial blueprint for gliomagenesis. Nat. Rev. Neurosci. 2018, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.R. Brain tumors in children. N. Engl. J. Med. 2022, 386, 1922–1931. [Google Scholar] [CrossRef]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro-Oncology 2018, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.-J.; Liu, Y.; Lang, F.; Yang, C. D-2-Hydroxyglutarate in Glioma Biology. Cells 2021, 10, 2345. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol.-Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Papavassiliou, A.G.; Piperi, C. Targeting of endoplasmic reticulum (ER) stress in gliomas. Pharmacol. Res. 2020, 157, 104823. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. The FAD-and O2-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, N.M.P.; Meijer, C.; Kruyt, F.A. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kyaw, Y.Y.; Cheong, J. Functional interaction of endoplasmic reticulum stress and hepatitis B virus in the pathogenesis of liver diseases. World J. Gastroenterol. 2017, 23, 7657–7665. [Google Scholar] [CrossRef]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef]
- Garrido-Armas, M.; Corona, J.C.; Escobar, M.L.; Torres, L.; Ordóñez-Romero, F.; Hernández-Hernández, A.; Arenas-Huertero, F. Paraptosis in human glioblastoma cell line induced by curcumin. Toxicol. In Vitro 2018, 51, 63–73. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Montagnani Marelli, M. Role of endoplasmic reticulum stress in the anticancer activity of natural compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef]
- Banerjee, H.N.; Hyman, G.; Evans, S.; Manglik, V.; Gwebu, E.; Banerjee, A.; Vaughan, D.; Medley, J.; Krauss, C.; Wilkins, J. Identification of the transmembrane glucose regulated protein 78 as a biomarker for the brain cancer glioblastoma multiforme by gene expression and proteomic studies. J. Membr. Sci. Technol. 2014, 4, 1000126. [Google Scholar] [PubMed]
- Roberts, N.B.; Alqazzaz, A.; Hwang, J.R.; Qi, X.; Keegan, A.D.; Kim, A.J.; Winkles, J.A.; Woodworth, G.F. Oxaliplatin disrupts pathological features of glioma cells and associated macrophages independent of apoptosis induction. J. Neurooncol. 2018, 140, 497–507. [Google Scholar] [CrossRef] [PubMed]
- WANG, S.; Wei, W.; Yuan, Y.; Sun, B.; Yang, D.; Liu, N.; Zhao, X. Chimeric antigen receptor T cells targeting cell surface GRP78 efficiently kill glioblastoma and cancer stem cells. J. Transl. Med. 2023, 21, 493. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, Z.; Zheng, Y.; Chen, Z.; Yue, X.; Bian, E.; Zhao, B. Identification of an endoplasmic reticulum stress-related signature associated with clinical prognosis and immune therapy in glioma. BMC Neurol. 2022, 22, 192. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ba, Y.; Li, C.; Xu, G. Inactivation of CACNA1H induces cell apoptosis by initiating endoplasmic reticulum stress in glioma. Transl. Neurosci. 2023, 14, 20220285. [Google Scholar] [CrossRef] [PubMed]
- Jian, S.; Chen, L.; Minxue, L.; Hongmin, C.; Ronghua, T.; Xiaoxuan, F.; Binbin, Z.; Shiwen, G. Tanshinone I induces apoptosis and protective autophagy in human glioblastoma cells via a reactive oxygen species-dependent pathway. Int. J. Mol. Med. 2020, 45, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, W.; Shergalis, A.; Xu, J.; Delaney, A.M.; Calcaterra, A.; Pal, A.; Ljungman, M.; Neamati, N.; Rehemtulla, A. Activation of the unfolded protein response via inhibition of protein disulfide isomerase decreases the capacity for DNA repair to sensitize glioblastoma to radiotherapy. Cancer Res. 2019, 79, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Wang, W.; Golden, E.B.; Thomas, S.; Sivakumar, W.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H. Green tea epigallocatechin gallate enhances therapeutic efficacy of temozolomide in orthotopic mouse glioblastoma models. Cancer Lett. 2011, 302, 100–108. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef]
- Noack, J.; Choi, J.; Richter, K.; Kopp-Schneider, A.; Régnier-Vigouroux, A. A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis. 2014, 5, e1425. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Zhang, C.; Chen, L.; Chen, B.-D.; Li, Q.-Z.; Zhang, X.-J.; Li, W.-P. Radicol, a Novel Trinorguaiane-Type Sesquiterpene, Induces Temozolomide-Resistant Glioma Cell Apoptosis via ER Stress and Akt/mTOR Pathway Blockade. Phytother. Res. 2017, 31, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, Y.-R.; Chen, W.; Chen, M.-H.; Wang, H.; Wang, X.-D.; Sun, L.-L.; Wang, F.-Z.; Wang, D.-C. Fluoxetine synergizes with temozolomide to induce the CHOP-dependent endoplasmic reticulum stress-related apoptosis pathway in glioma cells. Oncol. Rep. 2016, 36, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Kang, Y.J.; Yoon, M.J.; Kim, E.H.; Kim, S.U.; Kwon, T.K.; Kim, I.A.; Choi, K.S. Amiodarone sensitizes human glioma cells but not astrocytes to TRAIL-induced apoptosis via CHOP-mediated DR5 upregulation. Neuro-Oncology 2011, 13, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Qiu, Y.; Yang, L.; Peng, L.; Xia, Z.; Hou, L.-N.; Fang, C.; Qi, H.; Chen, H.-Z. Desipramine induces apoptosis in rat glioma cells via endoplasmic reticulum stress-dependent CHOP pathway. J. Neurooncol. 2011, 101, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-N.; Kim, S.-H.; Kim, K.-Y.; Ji, J.-H.; Seo, Y.-K.; Yu, H.S.; Ahn, S.-C. Salinomycin induces endoplasmic reticulum stress-mediated autophagy and apoptosis through generation of reactive oxygen species in human glioma U87MG cells. Oncol. Rep. 2017, 37, 3321–3328. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Wang, H.; Jia, Y.; Pan, H.; Ding, H. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother. Pharmacol. 2017, 79, 1031–1041. [Google Scholar] [CrossRef]
- Qu, C.; Ma, J.; Liu, X.; Xue, Y.; Zheng, J.; Liu, L.; Liu, J.; Li, Z.; Zhang, L.; Liu, Y. Dihydroartemisinin exerts anti-tumor activity by inducing mitochondrion and endoplasmic reticulum apoptosis and autophagic cell death in human glioblastoma cells. Front. Cell. Neurosci. 2017, 11, 310. [Google Scholar] [CrossRef]
- White, M.C.; Johnson, G.G.; Zhang, W.; Hobrath, J.V.; Piazza, G.A.; Grimaldi, M. Sulindac sulfide inhibits sarcoendoplasmic reticulum Ca2+ ATPase, induces endoplasmic reticulum stress response, and exerts toxicity in glioma cells: Relevant similarities to and important differences from celecoxib. J. Neurosci. Res. 2013, 91, 393–406. [Google Scholar] [CrossRef]
- Jang, E.; Kim, I.Y.; Kim, H.; Lee, D.M.; Seo, D.Y.; Lee, J.A.; Choi, K.S.; Kim, E. Quercetin and chloroquine synergistically kill glioma cells by inducing organelle stress and disrupting Ca2+ homeostasis. Biochem. Pharmacol. 2020, 178, 114098. [Google Scholar] [CrossRef]
- Chou, Y.-C.; Chang, M.-Y.; Wang, M.-J.; Harnod, T.; Hung, C.-H.; Lee, H.-T.; Shen, C.-C.; Chung, J.-G. PEITC induces apoptosis of Human Brain Glioblastoma GBM8401 Cells through the extrinsic-and intrinsic-signaling pathways. Neurochem. Int. 2015, 81, 32–40. [Google Scholar] [CrossRef]
- Virrey, J.J.; Liu, Z.; Cho, H.-Y.; Kardosh, A.; Golden, E.B.; Louie, S.G.; Gaffney, K.J.; Petasis, N.A.; Schönthal, A.H.; Chen, T.C. Antiangiogenic activities of 2, 5-dimethyl-celecoxib on the tumor vasculature. Mol. Cancer Ther. 2010, 9, 631–641. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro-Oncology 2013, 15, 1186–1199. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiao, Y.; Liu, Y.; Zhang, M.; Wang, Z.; Li, Y.; Li, T.; Zhao, X.; Wang, D. Sinomenine hydrochloride inhibits the metastasis of human glioblastoma cells by suppressing the expression of matrix metalloproteinase-2/-9 and reversing the endogenous and exogenous epithelial-mesenchymal transition. Int. J. Mol. Sci. 2018, 19, 844. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gundelach, J.H.; Bram, R.J. Cycloheximide promotes paraptosis induced by inhibition of cyclophilins in glioblastoma multiforme. Cell Death Dis. 2017, 8, e2807. [Google Scholar] [CrossRef]
- Romero-Hernández, M.A.; Eguía-Aguilar, P.; Perézpeña-DiazConti, M.; Rodríguez-Leviz, A.; Sadowinski-Pine, S.; Velasco-Rodríguez, L.A.; Cáceres-Cortés, J.R.; Arenas-Huertero, F. Toxic effects induced by curcumin in human astrocytoma cell lines. Toxicol. Mech. Methods 2013, 23, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.-J.; Wang, S.-H.; Chen, K.-C.; Kuei, H.-P.; Shih, Y.-L.; Hou, S.-Y.; Chiu, W.-T.; Hsiao, S.-H.; Shih, C.-M. Evodiamine, a plant alkaloid, induces calcium/JNK-mediated autophagy and calcium/mitochondria-mediated apoptosis in human glioblastoma cells. Chem. Biol. Interact. 2013, 205, 20–28. [Google Scholar] [CrossRef]
- Kavitha, C.V.; Jain, A.K.; Agarwal, C.; Pierce, A.; Keating, A.; Huber, K.M.; Serkova, N.J.; Wempe, M.F.; Agarwal, R.; Deep, G. Asiatic acid induces endoplasmic reticulum stress and apoptotic death in glioblastoma multiforme cells both in vitro and in vivo. Mol. Carcinog. 2015, 54, 1417–1429. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.; Xue, Y.; Li, Z.; Qu, C.; Liu, Y. Enhanced antitumor effect of shikonin by inhibiting Endoplasmic Reticulum Stress via JNK/c-Jun pathway in human glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2015, 466, 103–110. [Google Scholar] [CrossRef]
- Tsai, C.-F.; Yeh, W.-L.; Huang, S.M.; Tan, T.-W.; Lu, D.-Y. Wogonin induces reactive oxygen species production and cell apoptosis in human glioma cancer cells. Int. J. Mol. Sci. 2012, 13, 9877–9892. [Google Scholar] [CrossRef]
- Tsai, S.-F.; Tao, M.; Ho, L.-I.; Chiou, T.-W.; Lin, S.-Z.; Su, H.-L.; Harn, H.-J. Isochaihulactone-induced DDIT3 causes ER stress-PERK independent apoptosis in glioblastoma multiforme cells. Oncotarget 2017, 8, 4051–4061. [Google Scholar] [CrossRef]
- Ma, X.; Yu, M.; Hao, C.; Yang, W. Shikonin induces tumor apoptosis in glioma cells via endoplasmic reticulum stress, and Bax/Bak mediated mitochondrial outer membrane permeability. J. Ethnopharmacol. 2020, 263, 113059. [Google Scholar] [CrossRef]
- Zhang, L.; Tong, X.; Zhang, J.; Huang, J.; Wang, J. DAW22, a natural sesquiterpene coumarin isolated from Ferula ferulaeoides (Steud.) Korov. that induces C6 glioma cell apoptosis and endoplasmic reticulum (ER) stress. Fitoterapia 2015, 103, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; López-Alonso, H.; Martínez, P.; Millán, A.; Cagide, E.; Vieytes, M.R.; Vega, F.V.; Botana, L.M. Yessotoxin induces ER-stress followed by autophagic cell death in glioma cells mediated by mTOR and BNIP3. Cell. Signal. 2014, 26, 419–432. [Google Scholar] [CrossRef]
- Kim, I.Y.; Kwon, M.; Choi, M.-K.; Lee, D.; Lee, D.M.; Seo, M.J.; Choi, K.S. Ophiobolin A kills human glioblastoma cells by inducing endoplasmic reticulum stress via disruption of thiol proteostasis. Oncotarget 2017, 8, 106740. [Google Scholar] [CrossRef]
- Kim, T.H.; Song, J.; Kim, S.-H.; Parikh, A.K.; Mo, X.; Palanichamy, K.; Kaur, B.; Yu, J.; Yoon, S.O.; Nakano, I. Piperlongumine treatment inactivates peroxiredoxin 4, exacerbates endoplasmic reticulum stress, and preferentially kills high-grade glioma cells. Neuro-Oncology 2014, 16, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, X. RETRACTED: Bufothionine Promotes Apoptosis via Triggering ER Stress and Synergizes with Temozolomide in Glioblastoma Multiforme Cells. Anat. Rec. 2019, 302, 1950–1957. [Google Scholar] [CrossRef]
- Kumar, V.; Radin, D.; Leonardi, D. Probing the oncolytic and chemosensitizing effects of dihydrotanshinone in an in vitro glioblastoma model. Anticancer Res. 2017, 37, 6025–6030. [Google Scholar] [PubMed]
- Hebert, D.N.; Bernasconi, R.; Molinari, M. ERAD substrates: Which way out? Semin. Cell Dev. Biol. 2010, 21, 526–532. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tanaka, K.; Inoue, K.; Kakizuka, A. Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J. Biol. Chem. 2002, 277, 47358–47365. [Google Scholar] [CrossRef]
- Kamhi-Nesher, S.; Shenkman, M.; Tolchinsky, S.; Fromm, S.V.; Ehrlich, R.; Lederkremer, G.Z. A novel quality control compartment derived from the endoplasmic reticulum. Mol. Biol. Cell 2001, 12, 1711–1723. [Google Scholar] [CrossRef]
- Obacz, J.; Avril, T.; Le Reste, P.-J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma—From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10, eaal2323. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, P.; Luo, S.; Skarnes, W.C.; Sui, G.; Seto, E.; Shi, Y.; Lee, A.S. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: Activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 2005, 25, 4529–4540. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Conti, S.; Facchiano, A.; Filippini, A.; Ziparo, E. Cancer microenvironment and endoplasmic reticulum stress response. Mediators Inflamm. 2015, 2015, 417281. [Google Scholar] [CrossRef]
- Lee, H.K.; Xiang, C.; Cazacu, S.; Finniss, S.; Kazimirsky, G.; Lemke, N.; Lehman, N.L.; Rempel, S.A.; Mikkelsen, T.; Brodie, C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro-Oncology 2008, 10, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Suyama, K.; Watanabe, M.; Sakabe, K.; Okada, Y.; Matsuyama, D.; Kuroiwa, M.; Mochida, J. Overexpression of GRP78 protects glial cells from endoplasmic reticulum stress. Neurosci. Lett. 2011, 504, 271–276. [Google Scholar] [CrossRef]
- Bao, L.; Luo, Q.; Zhang, J.; Lao, Z. GRP78 overexpression as an unfavorable outcome in glioma patients. Int. J. Clin. Exp. Pathol. 2018, 11, 420–426. [Google Scholar]
- Mitra, S.; Ryoo, H.D. The unfolded protein response in metazoan development. J. Cell Sci. 2019, 132, jcs217216. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.A.; Osborne, J.C., Jr.; Safer, B.; Powell, G.M.; Merrick, W.C. Characteristics of eukaryotic initiation factor 2 and its subunits. J. Biol. Chem. 1980, 255, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Popko, B. Animal models in the study of the unfolded protein response. Methods Enzymol. 2011, 491, 91–109. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Zheng, W.; Hu, Q.; Cheng, Z.; Guo, F. Silencing of both ATF4 and PERK inhibits cell cycle progression and promotes the apoptosis of differentiating chondrocytes. Int. J. Mol. Med. 2017, 40, 101–111. [Google Scholar] [CrossRef]
- Prischi, F.; Nowak, P.R.; Carrara, M.; Ali, M.M. Phosphoregulation of Ire1 RNase splicing activity. Nat. Commun. 2014, 5, 3554. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, S.; Sha, H.; Liu, Z.; Yang, L.; Xue, Z.; Chen, H.; Qi, L. Emerging roles for XBP1, a sUPeR transcription factor. Gene Expr. J. Liver Res. 2010, 15, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Tirasophon, W.; Lee, K.; Callaghan, B.; Welihinda, A.; Kaufman, R.J. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000, 14, 2725–2736. [Google Scholar] [CrossRef]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Ernst, H.; Duncan, R.F.; Hershey, J.W. Cloning and sequencing of complementary DNAs encoding the alpha-subunit of translational initiation factor eIF-2. Characterization of the protein and its messenger RNA. J. Biol. Chem. 1987, 262, 1206–1212. [Google Scholar] [CrossRef]
- Zhu, C.; Johansen, F.-E.; Prywes, R. Interaction of ATF6 and serum response factor. Mol. Cell. Biol. 1997, 17, 4957–4966. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.-Y.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. FBiochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Tan, B.C.-M.; Cheng, Y.-Y.; Chen, J.-S.; Lee, S.-C. Differential regulation of CHOP translation by phosphorylated eIF4E under stress conditions. Nucleic Acids Res. 2010, 38, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 Facilitates Ribosomal Bypass of an Inhibitory Upstream ORF to Enhance CHOP Translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, M.; Ron, D.; Miller, C.P.; Habener, J.F. C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc. Natl. Acad. Sci. USA 1993, 90, 4679–4683. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)–ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]



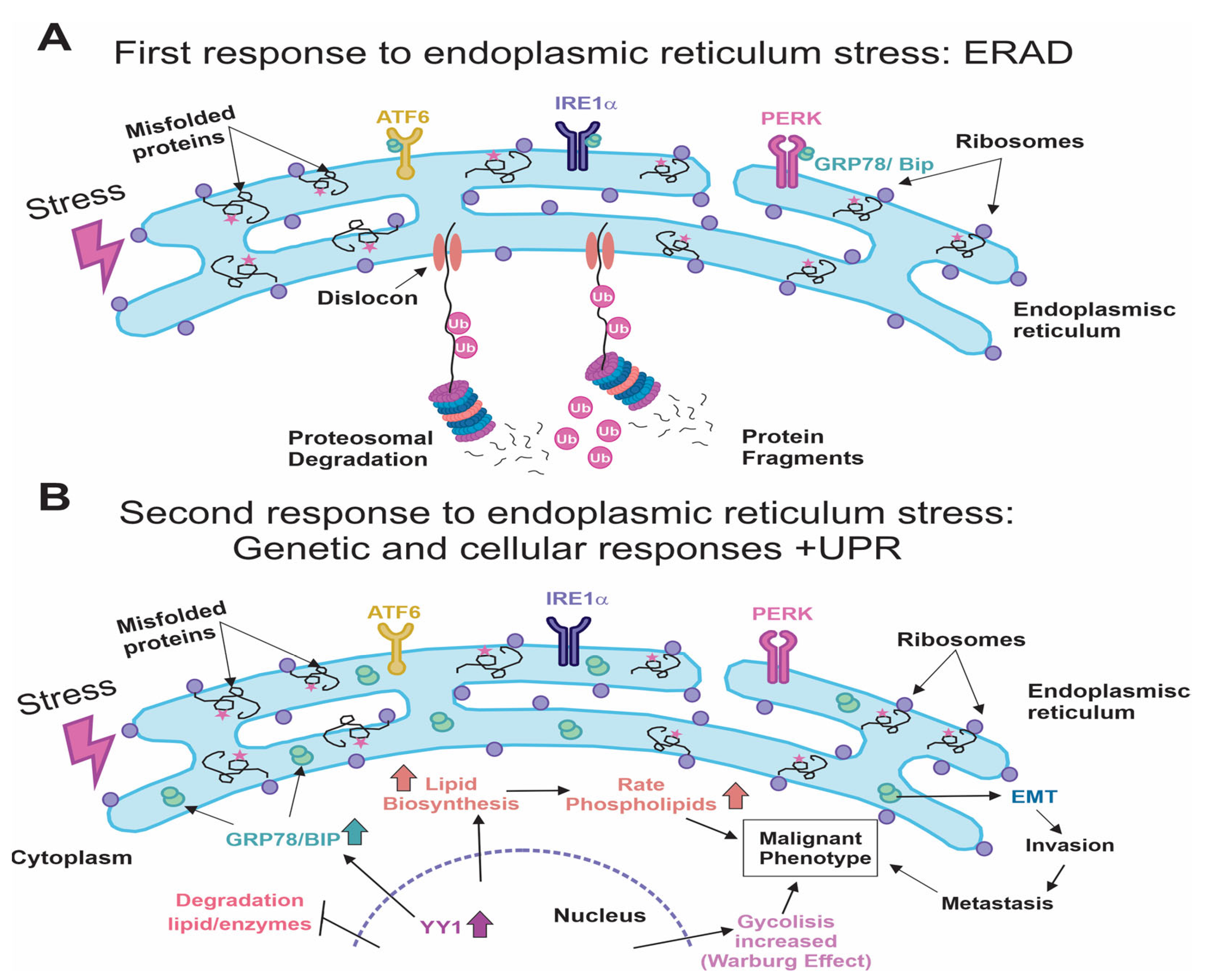{kind=link}
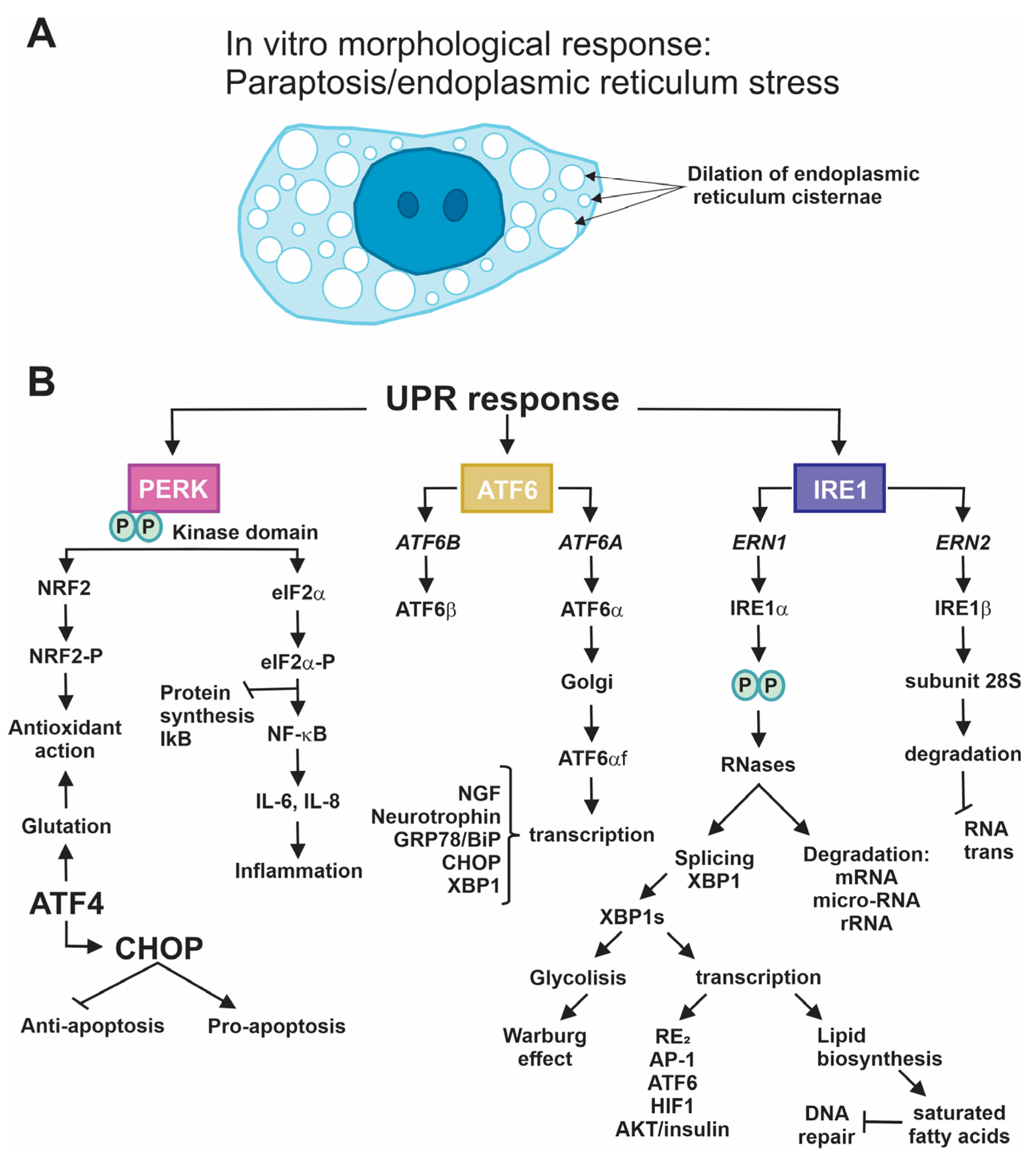{kind=link}
| Compound | Effects | Ref |
|---|---|---|
| Temozolomide (TMZ) | Citotoxic, DNA-alkylating. Proliferation inhibitor. | [28] |
| Chloroquine | Increases the cytotoxicity of TMZ. Ubiquitination of GRP78, CHOP, PARP. Inhibition of apoptosis regulated by GRP78. Promotes ERS. | [29] |
| Sphingosine kinase inhibitors (SKIs) | Accumulation of dhSph and dhCer, ROS. ERS induction. Cell death regulated by caspase 3. | [30] |
| Radicol | Apoptosis induction via ERS. Akt/mTOR blockade in TMZ-resistant tumor cells. | [31] |
| Fluoxetine Desipramine Amiodarone | Synergy with TMZ. Induction of apoptosis by CHOP. | [32,33] |
| Salinomycin Tanshinone I | Generation of ROS. Apoptosis and autophagy of tumor cells. | [35,43] |
| Luteolin | Mitochondrial dysfunction triggered by apoptosis. | [36] |
| Dihydroartemisinin | Induction of apoptosis and autophagy by ER and mitochondria. | [37] |
| Sulindac-Sulfide | Mitochondrial release of Ca2+ ions. ERSR induction. Cellular toxicity. | [37] |
| Quercetin and Chloroquine | Disruption of Ca2+ homeostasis. Induction of caspase-dependent cell death. Increase in autolysosomes and lysosomes. Cell death due to mitochondrial stress and ER. | [39] |
| Phenethyl isothiocyanate (PEITC) | Induction of apoptosis by release of ROS and Ca2+. Inhibits tumor cell growth. | [40] |
| Celecoxib (2,5-Dimetil Celecoxib) | Antiangiogenic effect. Reduces tumor size and microvessel density. Suppresses proliferation of tumor endothelial cells. Increases sensitivity of radiotherapy. | [41] |
| Sinomenine hydrochloride (SH) | Suppression of MMP-2/-9 expression. Reduces metastasis. Stops cell cycle. Inhibits expression of NFkB-p65. Promotes ERSR and autophagy. EMT inhibition. | [43] |
| Luteolin | Cell death due to paraptosis and vacuolization. Induction of NIM811. Inhibits cyclophilin anchoring. Early activation of ERSR. Autophagy. mTOR signaling. | [44] |
| Curcumin | 100 µM: death from methuosis. 50 µM: death by paraptosis. Induces ERSR, resulting in microRNA degradation. Activation of the p53-Bcl-2 and insulin-AKT pathways. AKT gene expression decreases. | [45] |
| Deri. Nat. Compounds | Effects | Ref |
| Evodiamine | Derived from the dried fruit of Evodia rutaecarpa. Ca2+-regulated autophagy and mitochondrial apoptosis. | [46] |
| Asiatic acid (AsA) | Reduced viability of GB cells. Expression of Bcl2 without cytotoxic effects for nontumor cells. | [47] |
| Wogonin | Derived from Scutellaria baicalensis Georgi. ERSR induction; tumor cell apoptosis. ROS elevation. | [49] |
| Shikonin | Derived from the roots of Lithospermum erythrorhizon. Cytotoxic effect; reduced cellular sensitivity. Caspase-regulated apoptosis. | [50] |
| Isochaihulactone (K8) | Extracted from Bupleurum scorzonerifolium. Increases ERSR; DDIT3 expression. NAG-1-mediated inflammatory response. Activation of caspases 3 and 9; apoptosis. | [51] |
| DAW22 | Coumarin sesquiterpene derived from Ferula ferulaeoides (Steud) Korov. Activation of caspases 3, 8, 9 and 12; mitochondria-mediated apoptosis and ERSR. Expression of XBP-1, elevation of GRP78 and CHOP. PERK and eIF2α phosphorylation, IRE1α increase. | [52] |
| Yessotoxin | Polycyclic ether extracted from dinoflagellates. Cell death due to paraptosis. Altered lipid metabolism. UPR induction; PERK and EIF2α phosphorylation. mTOR inhibition. | [53] |
| Ophiobolin A | Disrupts sulfhydryl homeostasis. ERSR activation; CHOP induction. Apoptotic vacuoles in ER. | [54] |
| Piperlongumine | Repression of PRDX4 expression. Increased ROS. ERSR activation. | [55] |
| Bufothionine Dihydrotanshinone Epigallocatechin 3-gallate | Use in synergy with TMZ. Increased chemosensitivity to TMZ. GRP78 inhibition. Survival in murine models. | [28,56,57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-López, D.; Zaragoza-Ojeda, M.; Eguía-Aguilar, P.; Arenas-Huertero, F. Endoplasmic Reticulum Stress in Gliomas: Exploiting a Dual-Effect Dysfunction through Chemical Pharmaceutical Compounds and Natural Derivatives for Therapeutical Uses. Int. J. Mol. Sci. 2024, 25, 4078. https://doi.org/10.3390/ijms25074078
García-López D, Zaragoza-Ojeda M, Eguía-Aguilar P, Arenas-Huertero F. Endoplasmic Reticulum Stress in Gliomas: Exploiting a Dual-Effect Dysfunction through Chemical Pharmaceutical Compounds and Natural Derivatives for Therapeutical Uses. International Journal of Molecular Sciences. 2024; 25(7):4078. https://doi.org/10.3390/ijms25074078
Chicago/Turabian StyleGarcía-López, Daniel, Montserrat Zaragoza-Ojeda, Pilar Eguía-Aguilar, and Francisco Arenas-Huertero. 2024. "Endoplasmic Reticulum Stress in Gliomas: Exploiting a Dual-Effect Dysfunction through Chemical Pharmaceutical Compounds and Natural Derivatives for Therapeutical Uses" International Journal of Molecular Sciences 25, no. 7: 4078. https://doi.org/10.3390/ijms25074078
APA StyleGarcía-López, D., Zaragoza-Ojeda, M., Eguía-Aguilar, P., & Arenas-Huertero, F. (2024). Endoplasmic Reticulum Stress in Gliomas: Exploiting a Dual-Effect Dysfunction through Chemical Pharmaceutical Compounds and Natural Derivatives for Therapeutical Uses. International Journal of Molecular Sciences, 25(7), 4078. https://doi.org/10.3390/ijms25074078