


Exploring the Pathophysiology of ATP-Dependent Potassium Channels in Insulin Resistance



Abstract
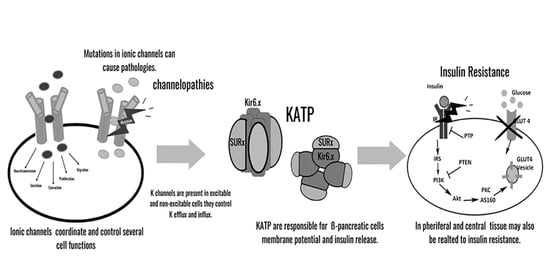
{kind=link}
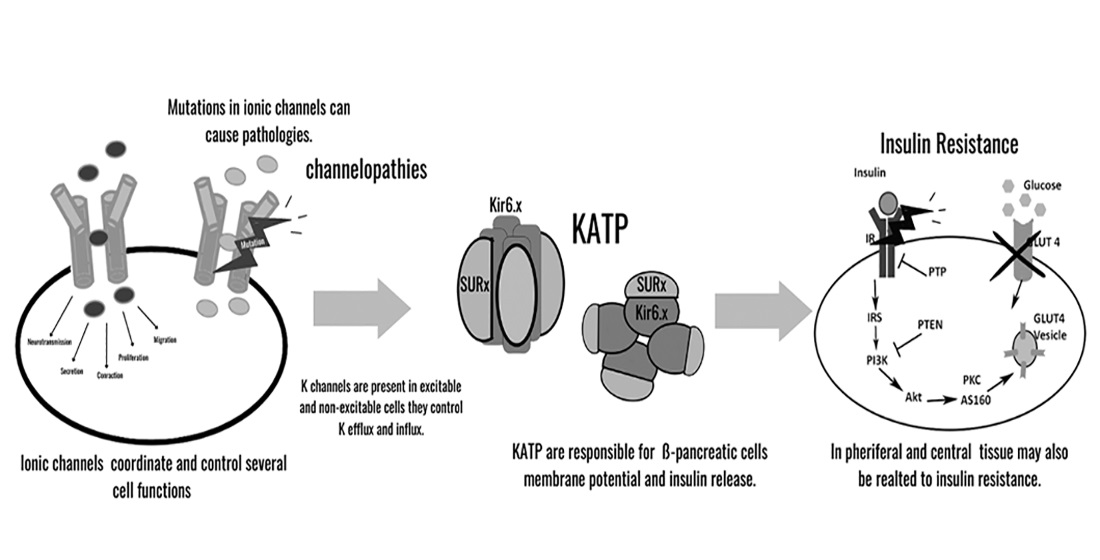
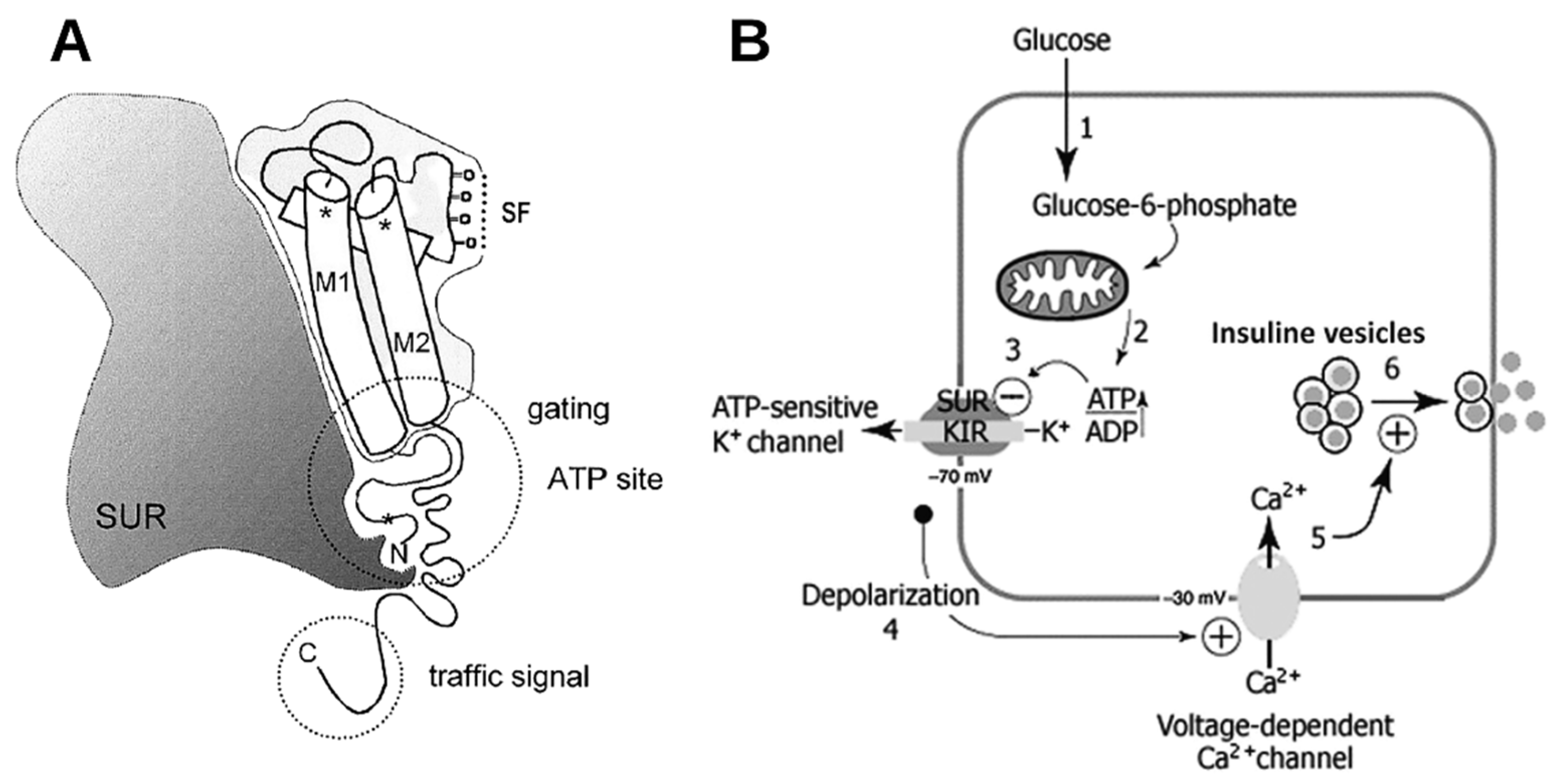{kind=link}
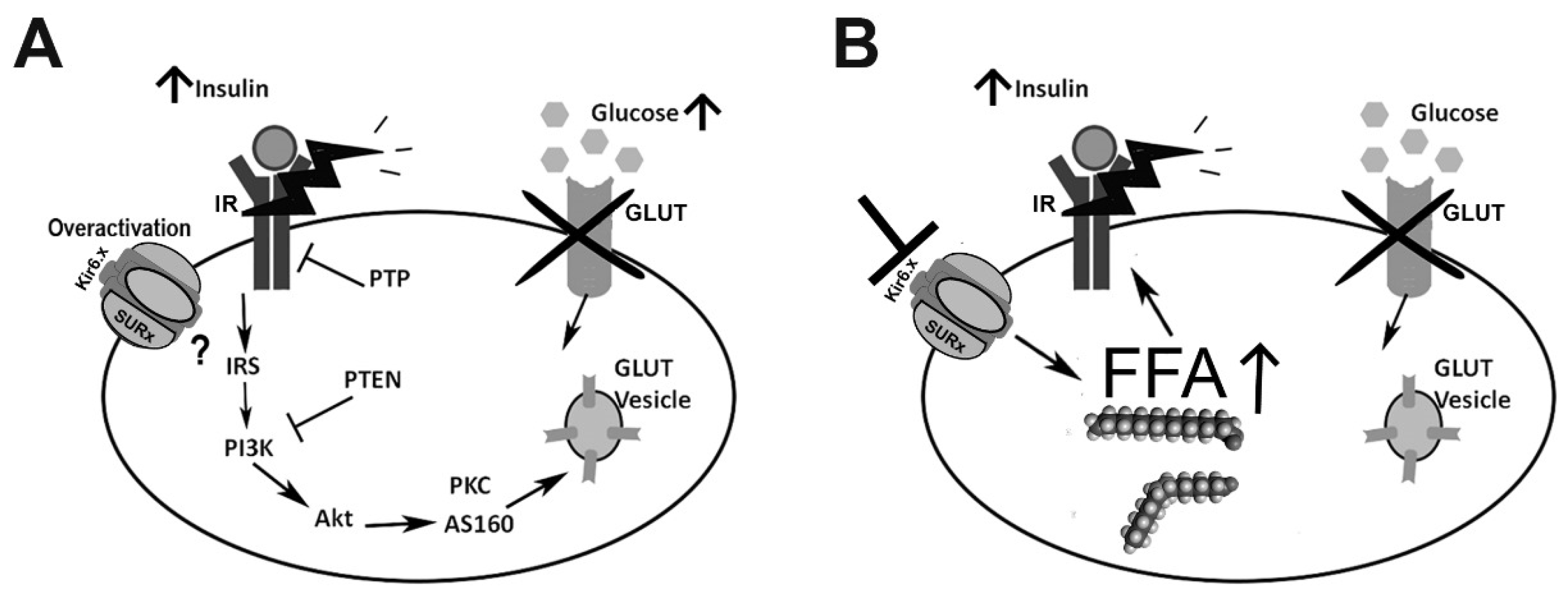{kind=link}
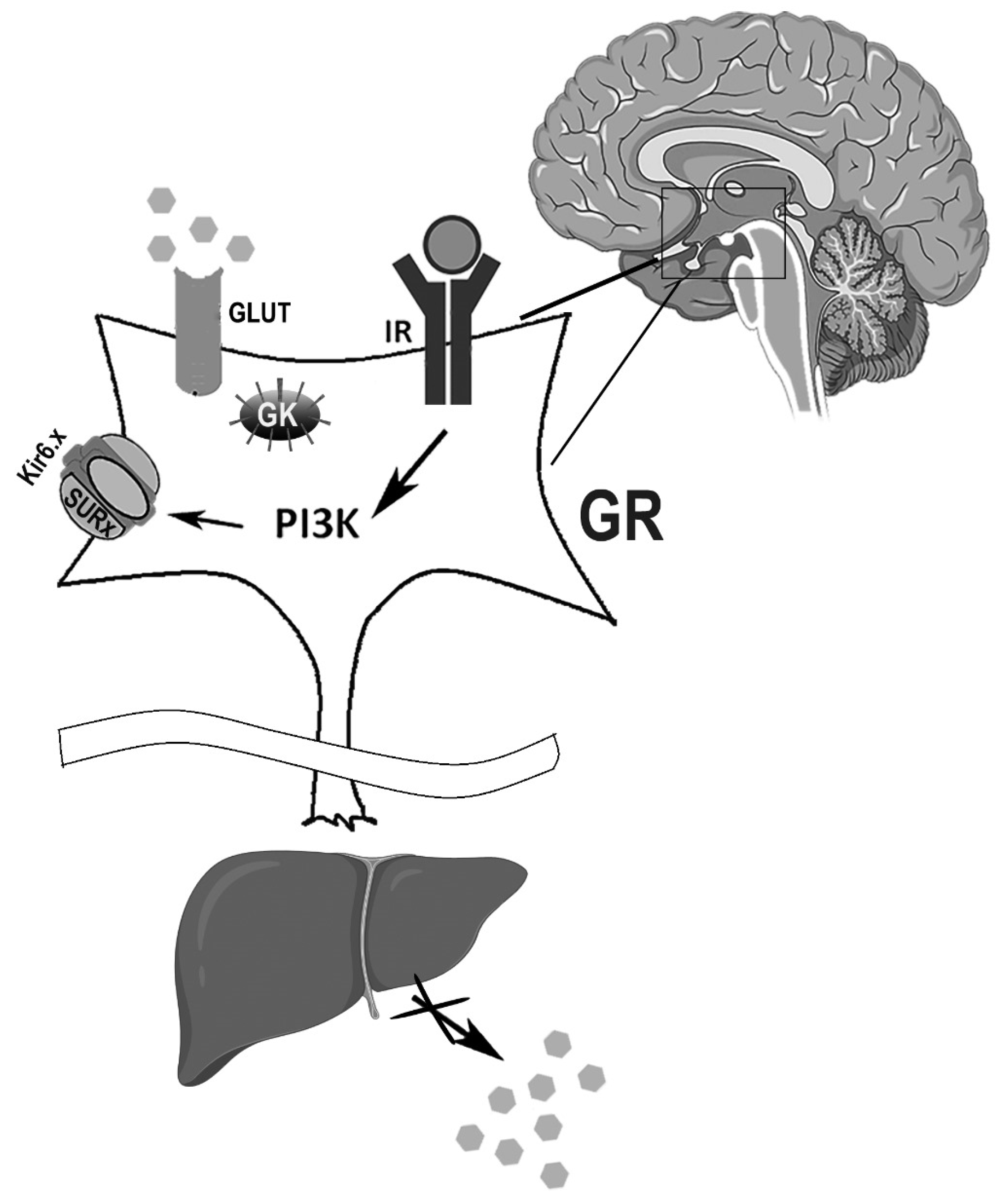{kind=link}
1. Introduction
- Voltage-gated potassium (Kv) channels: channels with six transmembrane motifs and one pore;
- Potassium inward rectifier channels (KIRs): channels with two transmembrane motifs and one pore;
- Weak potassium inward rectifier (K2P) channels: channels with four transmembrane motifs and two pores.
2. ATP-Dependent Potassium Channels (KATPs): Characteristics and Regulation
3. KATP Channel Activity and Functional Regulation
4. Genetic Variability in Human KATP Channels: Insights and Implications in Insulin Resistance
5. KATP Channels’ Role in Insulin Resistance in Peripheral Tissues
6. Beyond the Blood–Brain Barrier: Understanding the Effects of Insulin Resistance on the Brain
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kulbacka, J.; Choromanska, A.; Rossowska, J.; Wezgowiec, J.; Saczko, J.; Rols, M.P. Cell Membrane Transport Mechanisms: Ion Channels and Electrical Properties of Cell Membranes. Adv. Anat. Embryol. Cell Biol. 2017, 227, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Napolitano, C.; Priori, S.G. Genetics of Ion-Channel Disorders. Curr. Opin. Cardiol. 2012, 27, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Coates, L. Ion Permeation in Potassium Ion Channels. Acta Crystallogr. D Struct. Biol. 2020, 76, 326–331. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of Potassium Channels. Cell Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Salkoff, L.; Butler, A.; Ferreira, G.; Santi, C.; Wei, A. High-Conductance Potassium Channels of the SLO Family. Nat. Rev. Neurosci. 2006, 7, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Stocker, M. Ca2+-Activated K+ Channels: Molecular Determinants and Function of the SK Family. Nat. Rev. Neurosci. 2004, 5, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. Chem. Biol. Drug Des. 2014, 83, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kanters, J.K.; Fanoe, S.; Larsen, L.A.; Bloch Thomsen, P.E.; Toft, E.; Christiansen, M. T Wave Morphology Analysis Distinguishes between KvLQT1 and HERG Mutations in Long QT Syndrome. Heart Rhythm. 2004, 1, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Li, D.; Edwards, N.; Hynes, A.M.; Wood, K.; Al-Hamed, M.; Wroe, A.C.; Reaich, D.; Moochhala, S.H.; Welling, P.A.; et al. Identification of Compound Heterozygous KCNJ1 Mutations (Encoding ROMK) in a Kindred with Bartter’s Syndrome and a Functional Analysis of Their Pathogenicity. Physiol. Rep. 2013, 1, e00160. [Google Scholar] [CrossRef]
- Sakmann, B.; Trube, G. Conductance Properties of Single Inwardly Rectifying Potassium Channels in Ventricular Cells from Guinea-Pig Heart. J. Physiol. 1984, 347, 641–657. [Google Scholar] [CrossRef]
- Inagaki, N.; Gonoi, T.; Iv, J.P.C.; Wang, C.-Z.; Aguilar-Bryan, L.; Bryan, J.; Seino, S. A Family of Sulfonylurea Receptors Determines the Pharmacological Properties of ATP-Sensitive K+ Channels. Neuron 1996, 16, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Terzic, A. Quaternary Structure of KATP Channel SUR2A Nucleotide Binding Domains Resolved by Synchrotron Radiation X-Ray Scattering. J. Struct. Biol. 2010, 169, 243–251. [Google Scholar] [CrossRef][Green Version]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P.; Boyd, A.E.; González, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; Nelson, D.A. Cloning of the Beta Cell High-Affinity Sulfonylurea Receptor: A Regulator of Insulin Secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Simon, M.C.; Le Beau, M.M.; Burant, C.F. Cloning, Tissue Expression, and Chromosomal Localization of SUR2, the Putative Drug-Binding Subunit of Cardiac, Skeletal Muscle, and Vascular KATP Channels. Diabetes 1996, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Mohler, P.J. Differential Roles for SUR Subunits in KATP Channel Membrane Targeting and Regulation. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H33–H35. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, Y.; Wang, X.; Shi, W.; Jiang, C. Single Nucleotide Polymorphisms in K(ATP) Channels: Muscular Impact on Type 2 Diabetes. Diabetes 2005, 54, 1592–1597. [Google Scholar] [CrossRef][Green Version]
- Sakura, H.; Ammala, C.; Smith, P.A.; Gribble, F.M.; Ashcroft, F.M. Cloning and Functional Expression of the CDNA Encoding a Novel ATP-Sensitive Potassium Channel Subunit Expressed in Pancreatic Beta-Cells, Brain, Heart and Skeletal Muscle. FEBS Lett. 1995, 377, 338–344. [Google Scholar] [CrossRef]
- Saito, S.; Iida, A.; Sekine, A.; Miura, Y.; Ogawa, C.; Kawauchi, S.; Higuchi, S.; Nakamura, Y. Identification of 779 Genetic Variations in Eight Genes Encoding Members of the ATP-Binding Cassette, Subfamily C (ABCC/MRP/CFTR). J. Hum. Genet. 2002, 47, 147–171. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Urciuoli, W.R.; Chatterjee, S.; Fisher, A.B.; Nehrke, K.; Brookes, P.S. KIR 6.2 Is Not the Mitochondrial KATP Channel, but Is Required for Cardioprotection by Ischemic Preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1439–H1445. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef]
- Navarro, M.; Rodriquez de Fonseca, F.; Alvarez, E.; Chowen, J.A.; Zueco, J.A.; Gomez, R.; Eng, J.; Blázquez, E. Colocalization of Glucagon-like Peptide-1 (GLP-1) Receptors, Glucose Transporter GLUT-2, and Glucokinase MRNAs in Rat Hypothalamic Cells: Evidence for a Role of GLP-1 Receptor Agonists as an Inhibitory Signal for Food and Water Intake. J. Neurochem. 1996, 67, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Siddiqui, J.; Ellard, S. Mutations in the Genes Encoding the Pancreatic Beta-Cell KATP Channel Subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) in Diabetes Mellitus and Hyperinsulinism. Hum. Mutat. 2006, 27, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A.; Bokvist, K.; Arkhammar, P.; Berggren, P.O.; Rorsman, P. Delayed Rectifying and Calcium-Activated K+ Channels and Their Significance for Action Potential Repolarization in Mouse Pancreatic Beta-Cells. J. Gen. Physiol. 1990, 95, 1041–1059. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Wheeler, M.B. Voltage-Dependent K+ Channels in Pancreatic Beta Cells: Role, Regulation and Potential as Therapeutic Targets. Diabetologia 2003, 46, 1046–1062. [Google Scholar] [CrossRef]
- Dukes, I.D.; Philipson, L.H. K+ Channels: Generating Excitement in Pancreatic Beta-Cells. Diabetes 1996, 45, 845–853. [Google Scholar] [CrossRef]
- Rosário, L.M.; Barbosa, R.M.; Antunes, C.M.; Silva, A.M.; Abrunhosa, A.J.; Santos, R.M. Bursting Electrical Activity in Pancreatic Beta-Cells: Evidence That the Channel Underlying the Burst Is Sensitive to Ca2+ Influx through L-Type Ca2+ Channels. Pflugers Arch. 1993, 424, 439–447. [Google Scholar] [CrossRef]
- Straub, S.G.; James, R.F.; Dunne, M.J.; Sharp, G.W. Glucose Activates Both K(ATP) Channel-Dependent and K(ATP) Channel-Independent Signaling Pathways in Human Islets. Diabetes 1998, 47, 758–763. [Google Scholar] [CrossRef]
- Sato, Y.; Anello, M.; Henquin, J.C. Glucose Regulation of Insulin Secretion Independent of the Opening or Closure of Adenosine Triphosphate-Sensitive K+ Channels in Beta Cells. Endocrinology 1999, 140, 2252–2257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Straub, S.G.; Sharp, G.W. Glucose-Stimulated Signaling Pathways in Biphasic Insulin Secretion. Diabetes Metab. Res. Rev. 2002, 18, 451–463. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Bryan, J. Molecular Biology of Adenosine Triphosphate-Sensitive Potassium Channels. Endocr. Rev. 1999, 20, 101–135. [Google Scholar] [CrossRef]
- D’hahan, N.; Moreau, C.; Prost, A.L.; Jacquet, H.; Alekseev, A.E.; Terzic, A.; Vivaudou, M. Pharmacological Plasticity of Cardiac ATP-Sensitive Potassium Channels toward Diazoxide Revealed by ADP. Proc. Natl. Acad. Sci. USA 1999, 96, 12162–12167. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Tucker, S.J.; Seino, S.; Ashcroft, F.M. Tissue Specificity of Sulfonylureas: Studies on Cloned Cardiac and Beta-Cell K(ATP) Channels. Diabetes 1998, 47, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.; Aziz, Q.; Li, Y.; Specterman, M. ATP-Sensitive Potassium Channels and Their Physiological and Pathophysiological Roles. Compr. Physiol. 2018, 8, 1463–1511. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Larsen, A.P.; Torres, N.S.; Grunnet, M.; Poelzing, S. Potassium Channel Activators Differentially Modulate the Effect of Sodium Channel Blockade on Cardiac Conduction. Acta Physiol. 2013, 207, 280–289. [Google Scholar] [CrossRef]
- Nichols, C.G.; Makhina, E.N.; Pearson, W.L.; Sha, Q.; Lopatin, A.N. Inward Rectification and Implications for Cardiac Excitability. Circ. Res. 1996, 78, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ashford, M.L.; Bond, C.T.; Blair, T.A.; Adelman, J.P. Cloning and Functional Expression of a Rat Heart KATP Channel. Nature 1994, 370, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose Induces Closure of Single Potassium Channels in Isolated Rat Pancreatic Beta-Cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Proks, P.; Corkey, B.E.; Ashcroft, F.M. Mechanism of Cloned ATP-Sensitive Potassium Channel Activation by Oleoyl-CoA. J. Biol. Chem. 1998, 273, 26383–26387. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Tarasov, A.; Ashcroft, F.M. Mapping the Architecture of the ATP-Binding Site of the KATP Channel Subunit Kir6.2. J. Physiol. 2004, 557, 347–354. [Google Scholar] [CrossRef]
- Ueda, K.; Inagaki, N.; Seino, S. MgADP Antagonism to Mg2+-Independent ATP Binding of the Sulfonylurea Receptor SUR1. J. Biol. Chem. 1997, 272, 22983–22986. [Google Scholar] [CrossRef]
- Kozak, J.A.; Logothetis, D.E. A Calcium-Dependent Chloride Current in Insulin-Secreting Beta TC-3 Cells. Pflugers Arch. 1997, 433, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, W.F.; Satin, L.S.; Cook, D.L. Inactivation Kinetics and Pharmacology Distinguish Two Calcium Currents in Mouse Pancreatic B-Cells. J. Membr. Biol. 1991, 119, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.L.; Hales, C.N. Intracellular ATP Directly Blocks K+ Channels in Pancreatic B-Cells. Nature 1984, 311, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Findlay, I. Effects of ADP upon the ATP-Sensitive K+ Channel in Rat Ventricular Myocytes. J. Membr. Biol. 1988, 101, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Yamane, T.; Terai, T.; Katayama, Y.; Hiraoka, M. Functional Linkage of the Cardiac ATP-Sensitive K+ Channel to the Actin Cytoskeleton. Pflugers Arch. 1996, 431, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Hilgemann, D.W.; Ball, R. Regulation of Cardiac Na+,Ca2+ Exchange and KATP Potassium Channels by PIP2. Science 1996, 273, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Makielski, J.C. Anionic Phospholipids Activate ATP-Sensitive Potassium Channels. J. Biol. Chem. 1997, 272, 5388–5395. [Google Scholar] [CrossRef] [PubMed]
- Ribalet, B.; John, S.A.; Weiss, J.N. Regulation of Cloned ATP-Sensitive K Channels by Phosphorylation, MgADP, and Phosphatidylinositol Bisphosphate (PIP(2)): A Study of Channel Rundown and Reactivation. J. Gen. Physiol. 2000, 116, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Gonoi, T.; Clement, J.P.; Namba, N.; Inazawa, J.; Gonzalez, G.; Aguilar-Bryan, L.; Seino, S.; Bryan, J. Reconstitution of IKATP: An Inward Rectifier Subunit plus the Sulfonylurea Receptor. Science 1995, 270, 1166–1170. [Google Scholar] [CrossRef]
- Okuyama, Y.; Yamada, M.; Kondo, C.; Satoh, E.; Isomoto, S.; Shindo, T.; Horio, Y.; Kitakaze, M.; Hori, M.; Kurachi, Y. The Effects of Nucleotides and Potassium Channel Openers on the SUR2A/Kir6.2 Complex K+ Channel Expressed in a Mammalian Cell Line, HEK293T Cells. Pflugers Arch. 1998, 435, 595–603. [Google Scholar] [CrossRef]
- Gerbitz, K.D.; Gempel, K.; Brdiczka, D. Mitochondria and Diabetes. Genetic, Biochemical, and Clinical Implications of the Cellular Energy Circuit. Diabetes 1996, 45, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Kakei, M. ATP-Sensitive K+ Channels in Rat Pancreatic Beta-Cells: Modulation by ATP and Mg2+ Ions. J. Physiol. 1989, 416, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Findlay, I. ATP4− and ATP.Mg Inhibit the ATP-Sensitive K+ Channel of Rat Ventricular Myocytes. Pflugers Arch. 1988, 412, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G.; Shyng, S.L.; Nestorowicz, A.; Glaser, B.; Clement, J.P.; Gonzalez, G.; Aguilar-Bryan, L.; Permutt, M.A.; Bryan, J. Adenosine Diphosphate as an Intracellular Regulator of Insulin Secretion. Science 1996, 272, 1785–1787. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G. KATP Channels as Molecular Sensors of Cellular Metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Hopkins, W.F.; Fatherazi, S.; Peter-Riesch, B.; Corkey, B.E.; Cook, D.L. Two Sites for Adenine-Nucleotide Regulation of ATP-Sensitive Potassium Channels in Mouse Pancreatic β-Cells and HIT Cells. J. Membr. Biol. 1992, 129, 287–295. [Google Scholar] [CrossRef]
- Tucker, S.J.; Gribble, F.M.; Zhao, C.; Trapp, S.; Ashcroft, F.M. Truncation of Kir6.2 Produces ATP-Sensitive K+ Channels in the Absence of the Sulphonylurea Receptor. Nature 1997, 387, 179–183. [Google Scholar] [CrossRef]
- Bushman, J.D.; Zhou, Q.; Shyng, S.L. A Kir6.2 Pore Mutation Causes Inactivation of ATP-Sensitive Potassium Channels by Disrupting PIP2-Dependent Gating. PLoS ONE 2013, 8, e63733. [Google Scholar] [CrossRef]
- Ribalet, B.; John, S.A.; Xie, L.H.; Weiss, J.N. ATP-Sensitive K+ Channels: Regulation of Bursting by the Sulphonylurea Receptor, PIP2 and Regions of Kir6.2. J. Physiol. 2006, 571, 303–317. [Google Scholar] [CrossRef]
- Shyng, S.; Ferrigni, T.; Nichols, C.G. Regulation of KATP Channel Activity by Diazoxide and MgADP. Distinct Functions of the Two Nucleotide Binding Folds of the Sulfonylurea Receptor. J. Gen. Physiol. 1997, 110, 643–654. [Google Scholar] [CrossRef]
- Shyng, S.L.; Cukras, C.A.; Harwood, J.; Nichols, C.G. Structural Determinants of Pip2 Regulation of Inward Rectifier KATP Channels. J. Gen. Physiol. 2000, 116, 599. [Google Scholar] [CrossRef] [PubMed]
- Reimann, F.; Tucker, S.J.; Proks, P.; Ashcroft, F.M. Involvement of the N-Terminus of Kir6.2 in Coupling to the Sulphonylurea Receptor. J. Physiol. 1999, 518 Pt 2, 325–336. [Google Scholar] [CrossRef]
- Shyng, S.L.; Nichols, C.G. Membrane Phospholipid Control of Nucleotide Sensitivity of KATP Channels. Science 1998, 282, 1138–1141. [Google Scholar] [CrossRef] [PubMed]
- Pipatpolkai, T.; Usher, S.G.; Vedovato, N.; Ashcroft, F.M.; Stansfeld, P.J. The Dynamic Interplay of PIP2 and ATP in the Regulation of the KATP Channel. J. Physiol. 2022, 600, 4503–4519. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Clauin, S.; Bellanne-Chantelot, C.; de Lonlay, P.; Harries, L.W.; Gloyn, A.L.; Ellard, S. Update of Mutations in the Genes Encoding the Pancreatic Beta-Cell K(ATP) Channel Subunits Kir6.2 (KCNJ11) and Sulfonylurea Receptor 1 (ABCC8) in Diabetes Mellitus and Hyperinsulinism. Hum. Mutat. 2009, 30, 170–180. [Google Scholar] [CrossRef]
- Nichols, C.G.; York, N.W.; Remedi, M.S. ATP-Sensitive Potassium Channels in Hyperinsulinism and Type 2 Diabetes: Inconvenient Paradox or New Paradigm? Diabetes 2022, 71, 367–375. [Google Scholar] [CrossRef]
- Nestorowicz, A.; Wilson, B.A.; Schoor, K.P.; Inoue, H.; Glaser, B.; Landau, H.; Stanley, C.A.; Thornton, P.S.; Clement, J.P.; Bryan, J.; et al. Mutations in the Sulonylurea Receptor Gene Are Associated with Familial Hyperinsulinism in Ashkenazi Jews. Hum. Mol. Genet. 1996, 5, 1813–1822. [Google Scholar] [CrossRef]
- Glamoclija, U.; Jevric-Causevic, A. Genetic Polymorphisms in Diabetes: Influence on Therapy with Oral Antidiabetics. Acta Pharm. 2010, 60, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Nicolson, T.J.; Riveline, J.P.; Taneja, T.K.; Baldwin, S.A.; Baldwin, J.M.; Charpentier, G.; Gautier, J.F.; Froguel, P.; Vaxillaire, M.; et al. A Rare Mutation in ABCC8/SUR1 Leading to Altered ATP-Sensitive K+ Channel Activity and Beta-Cell Glucose Sensing Is Associated with Type 2 Diabetes in Adults. Diabetes 2008, 57, 1595–1604. [Google Scholar] [CrossRef]
- Bonfanti, D.H.; Alcazar, L.P.; Arakaki, P.A.; Martins, L.T.; Agustini, B.C.; de Moraes Rego, F.G.; Frigeri, H.R. ATP-Dependent Potassium Channels and Type 2 Diabetes Mellitus. Clin. Biochem. 2015, 48, 476–482. [Google Scholar] [CrossRef]
- Martin, G.M.; Sung, M.W.; Shyng, S.L. Pharmacological Chaperones of ATP-Sensitive Potassium Channels: Mechanistic Insight from CryoEM Structures. Mol. Cell Endocrinol. 2020, 502, 110667. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. ATP-Sensitive Potassium Channelopathies: Focus on Insulin Secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ozawa, T.; Yoshida, T.; Umezawa, Y. A Fluorescent Indicator for Tyrosine Phosphorylation-Based Insulin Signaling Pathways. Anal. Chem. 1999, 71, 3948–3954. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Pearson, E.R.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.C.L.; Molnes, J.; et al. Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes. N. Engl. J. Med. 2004, 350, 1838–1849. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Vila, M.C.; Watson, J.E.; Nair, G.; Pollet, R.J.; Standaert, M.; Farese, R. V Sulfonylurea-Stimulated Glucose Transport Association with Diacylglycerollike Activation of Protein Kinase C in BC3H1 Myocytes. Diabetes 1990, 39, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.J.; Standaert, M.L.; Pollet, R.J. Direct Effects of Sulfonylurea Agents on Glucose Transport in the BC3H-1 Myocyte. Diabetes 1987, 36, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Maloff, B.L.; Lockwood, D.H. In Vitro Effects of a Sulfonylurea on Insulin Action in Adipocytes. Potentiation of Insulin-Stimulated Hexose Transport. J. Clin. Investig. 1981, 68, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Moller, D.; Flier, J.S.; Nayak, R.C.; Smith, R.J. Coordinate Regulation of Glucose Transporter Function, Number, and Gene Expression by Insulin and Sulfonylureas in L6 Rat Skeletal Muscle Cells. J. Clin. Investig. 1989, 84, 62–67. [Google Scholar] [CrossRef]
- Barrett-Jolley, R.; Davies, N.W. Kinetic Analysis of the Inhibitory Effect of Glibenclamide on KATP Channels of Mammalian Skeletal Muscle. J. Membr. Biol. 1997, 155, 257–262. [Google Scholar] [CrossRef]
- Kramer, J.H.; Lampson, W.G.; Schaffer, S.W. Effect of Tolbutamide on Myocardial Energy Metabolism. Am. J. Physiol. 1983, 245, H313–H319. [Google Scholar] [CrossRef]
- Daniels, E.L.; Lewis, S.B. Acute Tolbutamide Administration Alone or Combined with Insulin Enhances Glucose Uptake in the Perfused Rat Hindlimb. Endocrinology 1982, 110, 1840–1842. [Google Scholar] [CrossRef] [PubMed]
- Tsiani, E.; Ramlal, T.; Leiter, L.A.; Klip, A.; Fantus, I.G. Stimulation of Glucose Uptake and Increased Plasma Membrane Content of Glucose Transporters in L6 Skeletal Muscle Cells by the Sulfonylureas Gliclazide and Glyburide. Endocrinology 1995, 136, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Pulido, N.; Casla, A.; Suárez, A.; Casanova, B.; Arrieta, F.J.; Rovira, A. Sulphonylurea Stimulates Glucose Uptake in Rats through an ATP-Sensitive K+ Channel Dependent Mechanism. Diabetologia 1996, 39, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Nagashima, K.; Tashiro, F.; Kotake, K.; Yoshitomi, H.; Tamamoto, A.; Gonoi, T.; Iwanaga, T.; Miyazaki, J.; Seino, S. Defective Insulin Secretion and Enhanced Insulin Action in KATP Channel-Deficient Mice. Proc. Natl. Acad. Sci. USA 1998, 95, 10402–10406. [Google Scholar] [CrossRef] [PubMed]
- Wasada, T. Adenosine Triphosphate-Sensitive Potassium (K(ATP)) Channel Activity Is Coupled with Insulin Resistance in Obesity and Type 2 Diabetes Mellitus. Intern. Med. 2002, 41, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Echwald, S.M.; Hansen, T.; Urhammer, S.A.; Clausen, J.O.; Pedersen, O. Amino Acid Polymorphisms in the ATP-Regulatable Inward Rectifier Kir6.2 and Their Relationships to Glucose- and Tolbutamide-Induced Insulin Secretion, the Insulin Sensitivity Index, and NIDDM. Diabetes 1997, 46, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Wasada, T.; Watanabe, C.; Nakagami, T.; Iwamoto, Y. Adenosine Triphosphate-Sensitive Potassium Channels Are Involved in Insulin-Mediated Glucose Transport in Humans. Metabolism 1999, 48, 432–436. [Google Scholar] [CrossRef]
- Wasada, T.; Yano, T.; Ohta, M.; Yui, N.; Iwamoto, Y. ATP-Sensitive Potassium Channels Modulate Glucose Transport in Cultured Human Skeletal Muscle Cells. Endocr. J. 2001, 48, 369–375. [Google Scholar] [CrossRef]
- Linde, C.; Löffler, C.; Quast, U. Inhibition by Protein Kinase C of the 86Rb+ Efflux and Vasorelaxation Induced by P1075, a K(ATP) Channel Opener, in Rat Isolated Aorta. Naunyn Schmiedebergs Arch. Pharmacol. 1997, 356, 425–432. [Google Scholar] [CrossRef]
- Standridge, M.; Alemzadeh, R.; Zemel, M.; Koontz, J.; Moustaid-Moussa, N. Diazoxide Down-Regulates Leptin and Lipid Metabolizing Enzymes in Adipose Tissue of Zucker Rats. FASEB J. 2000, 14, 455–460. [Google Scholar] [CrossRef]
- Shi, H.; Moustaid-Moussa, N.; Wilkison, W.O.; Zemel, M.B. Role of the Sulfonylurea Receptor in Regulating Human Adipocyte Metabolism. FASEB J. 1999, 13, 1833–1838. [Google Scholar] [CrossRef]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered Fat Storage and Mobilization in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Chen, G.; Lee, Y.; Unger, R.H. Tissue Triglycerides, Insulin Resistance, and Insulin Production: Implications for Hyperinsulinemia of Obesity. Am. J. Physiol. 1997, 273, E708–E713. [Google Scholar] [CrossRef]
- Kelley, D.E.; Mandarino, L.J. Fuel Selection in Human Skeletal Muscle in Insulin Resistance: A Reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Ordway, R.W.; Walsh, J.V.; Singer, J.J. Arachidonic Acid and Other Fatty Acids Directly Activate Potassium Channels in Smooth Muscle Cells. Science 1989, 244, 1176–1179. [Google Scholar] [CrossRef]
- Larsson, O.; Deeney, J.T.; Bränström, R.; Berggren, P.O.; Corkey, B.E. Activation of the ATP-Sensitive K+ Channel by Long Chain Acyl-CoA. A Role in Modulation of Pancreatic Beta-Cell Glucose Sensitivity. J. Biol. Chem. 1996, 271, 10623–10626. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E.; Dunn-Meynell, A.A.; Routh, V.H. Brain Glucose Sensing and Body Energy Homeostasis: Role in Obesity and Diabetes. Am. J. Physiol. 1999, 276, R1223–R1231. [Google Scholar] [CrossRef]
- Kow, L.M.; Pfaff, D.W. Actions of Feeding-Relevant Agents on Hypothalamic Glucose-Responsive Neurons in Vitro. Brain Res. Bull. 1985, 15, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Oomura, Y. Glucose Responding Neurons in the Nucleus Tractus Solitarius of the Rat: In Vitro Study. Brain Res. 1984, 307, 109–116. [Google Scholar] [CrossRef]
- Oomura, Y.; Ooyama, H.; Sugimori, M.; Nakamura, T.; Yamada, Y. Glucose Inhibition of the Glucose-Sensitive Neurone in the Rat Lateral Hypothalamus. Nature 1974, 247, 284–286. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Rawson, N.E.; Levin, B.E. Distribution and Phenotype of Neurons Containing the ATP-Sensitive K+ Channel in Rat Brain. Brain Res. 1998, 814, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ashford, M.L.; Boden, P.R.; Treherne, J.M. Tolbutamide Excites Rat Glucoreceptive Ventromedial Hypothalamic Neurones by Indirect Inhibition of ATP-K+ Channels. Br. J. Pharmacol. 1990, 101, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Jetton, T.L.; Liang, Y.; Pettepher, C.C.; Zimmerman, E.C.; Cox, F.G.; Horvath, K.; Matschinsky, F.M.; Magnuson, M.A. Analysis of Upstream Glucokinase Promoter Activity in Transgenic Mice and Identification of Glucokinase in Rare Neuroendocrine Cells in the Brain and Gut. J. Biol. Chem. 1994, 269, 3641–3654. [Google Scholar] [CrossRef] [PubMed]
- Seaquist, E.R.; Damberg, G.S.; Tkac, I.; Gruetter, R. The Effect of Insulin on in Vivo Cerebral Glucose Concentrations and Rates of Glucose Transport/Metabolism in Humans. Diabetes 2001, 50, 2203–2209. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Figlewicz, D.P.; Baskin, D.G.; Woods, S.C.; Porte, D. Insulin in the Brain: A Hormonal Regulator of Energy Balance. Endocr. Rev. 1992, 13, 387–414. [Google Scholar] [CrossRef]
- Hörsch, D.; Kahn, C.R. Region-Specific MRNA Expression of Phosphatidylinositol 3-Kinase Regulatory Isoforms in the Central Nervous System of C57BL/6J Mice. J. Comp. Neurol. 1999, 415, 105–120. [Google Scholar] [CrossRef]
- Choudhury, A.I.; Heffron, H.; Smith, M.A.; Al-Qassab, H.; Xu, A.W.; Selman, C.; Simmgen, M.; Clements, M.; Claret, M.; Maccoll, G.; et al. The Role of Insulin Receptor Substrate 2 in Hypothalamic and Beta Cell Function. J. Clin. Investig. 2005, 115, 940–950. [Google Scholar] [CrossRef]
- Pagotto, U. Where Does Insulin Resistance Start? The Brain. Diabetes Care 2009, 32 (Suppl. 2), S174–S177. [Google Scholar] [CrossRef]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing Hypothalamic Insulin Receptors Causes Hyperphagia and Insulin Resistance in Rats. Nat. Neurosci. 2002, 5, 566–572. [Google Scholar] [CrossRef]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic Insulin Signaling Is Required for Inhibition of Glucose Production. Nat. Med. 2002, 8, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) Channels Control Hepatic Glucose Production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Tschritter, O.; Preissl, H.; Hennige, A.M.; Stumvoll, M.; Porubska, K.; Frost, R.; Marx, H.; Klosel, B.; Lutzenberger, W.; Birbaumer, N.; et al. The Cerebrocortical Response to Hyperinsulinemia Is Reduced in Overweight Humans: A Magnetoencephalographic Study. Proc. Natl. Acad. Sci. USA 2006, 103, 12103–12108. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Rivera, N.S.; Barrera-Oviedo, D. Exploring the Pathophysiology of ATP-Dependent Potassium Channels in Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 4079. https://doi.org/10.3390/ijms25074079
Rodríguez-Rivera NS, Barrera-Oviedo D. Exploring the Pathophysiology of ATP-Dependent Potassium Channels in Insulin Resistance. International Journal of Molecular Sciences. 2024; 25(7):4079. https://doi.org/10.3390/ijms25074079
Chicago/Turabian StyleRodríguez-Rivera, Nidia Samara, and Diana Barrera-Oviedo. 2024. "Exploring the Pathophysiology of ATP-Dependent Potassium Channels in Insulin Resistance" International Journal of Molecular Sciences 25, no. 7: 4079. https://doi.org/10.3390/ijms25074079
APA StyleRodríguez-Rivera, N. S., & Barrera-Oviedo, D. (2024). Exploring the Pathophysiology of ATP-Dependent Potassium Channels in Insulin Resistance. International Journal of Molecular Sciences, 25(7), 4079. https://doi.org/10.3390/ijms25074079