
The Catalytic Activity of Human REV1 on Undamaged and Damaged DNA
 ,
,
Abstract
:1. Introduction
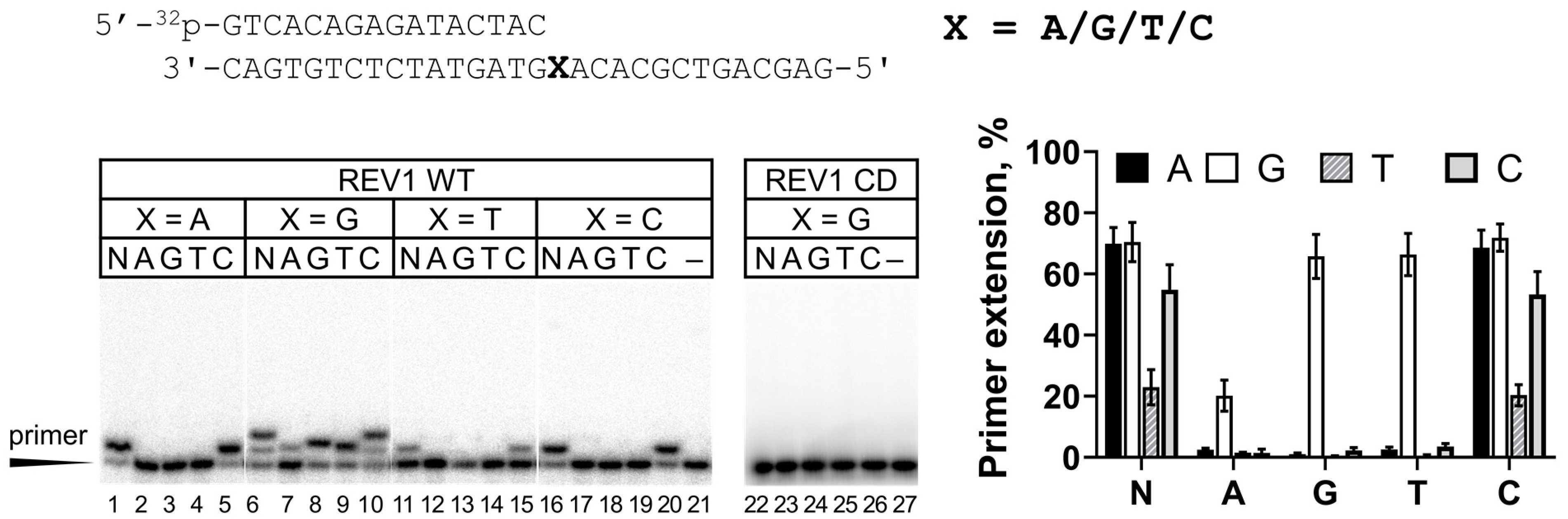
2. Results
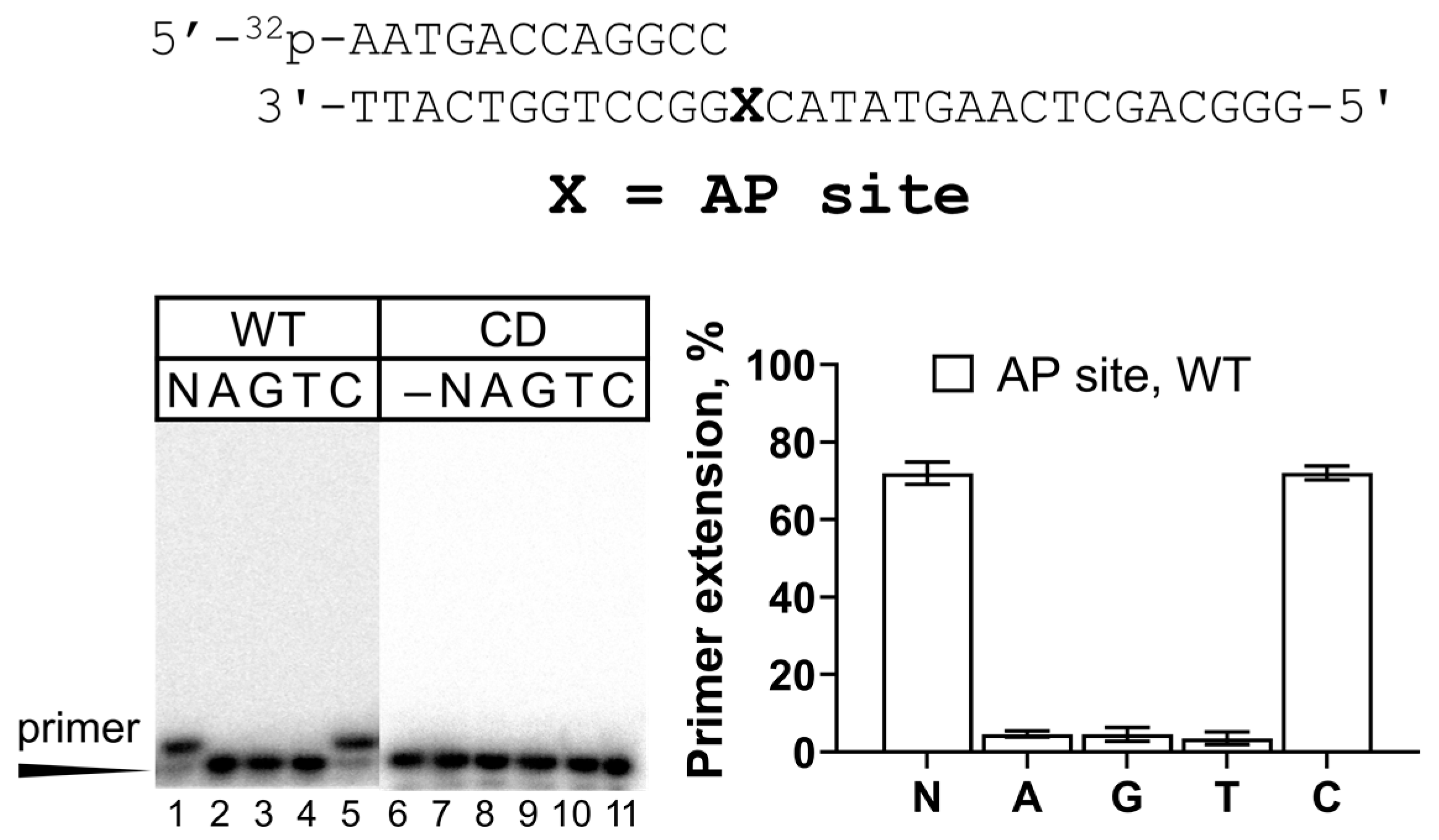
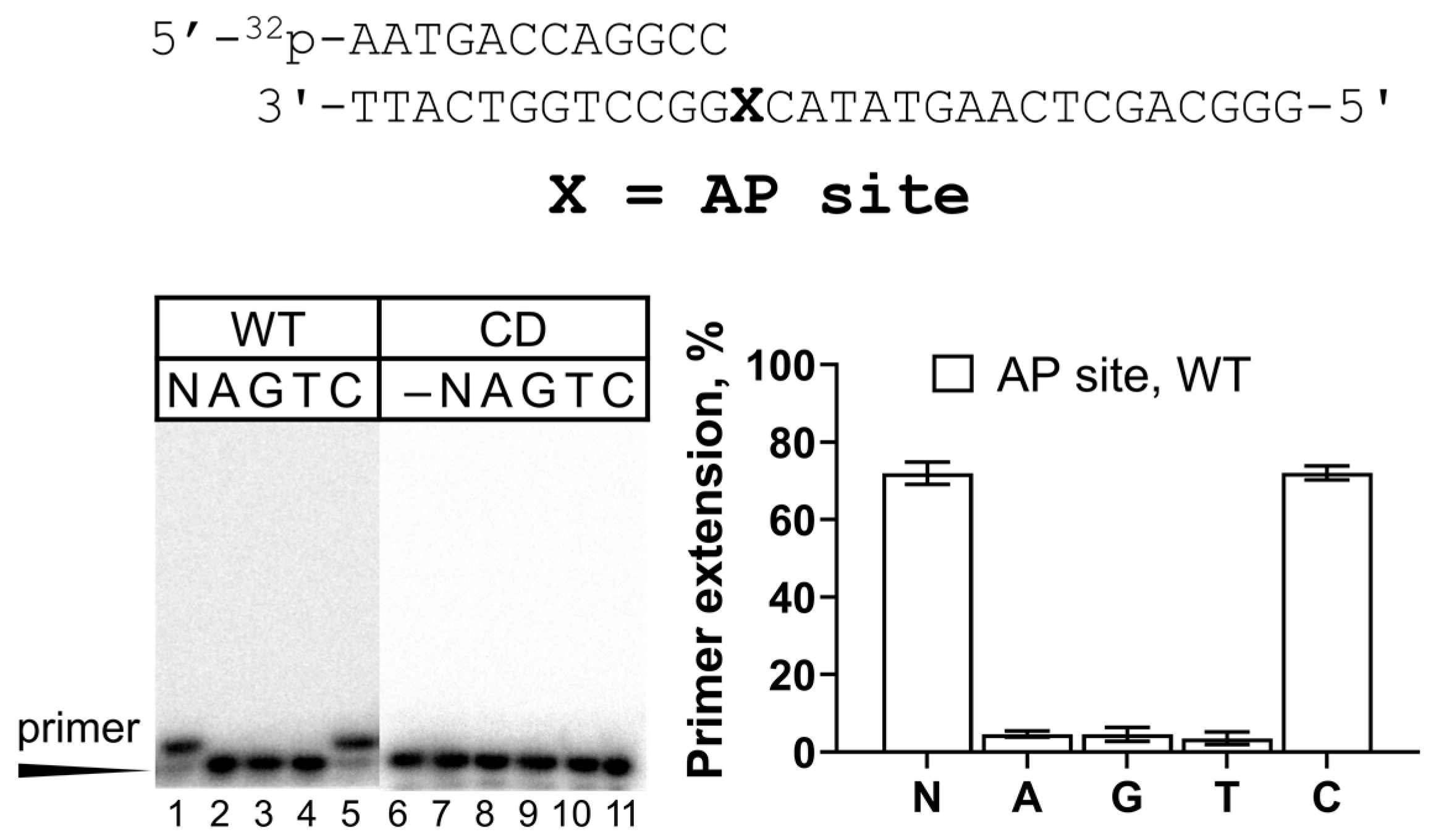
2.1. Activity of REV1 on Undamaged DNA and the AP Site
2.2. REV1 Activity on DNA with G Lesions
2.3. REV1 Activity Opposite εA and 7-DeazaA
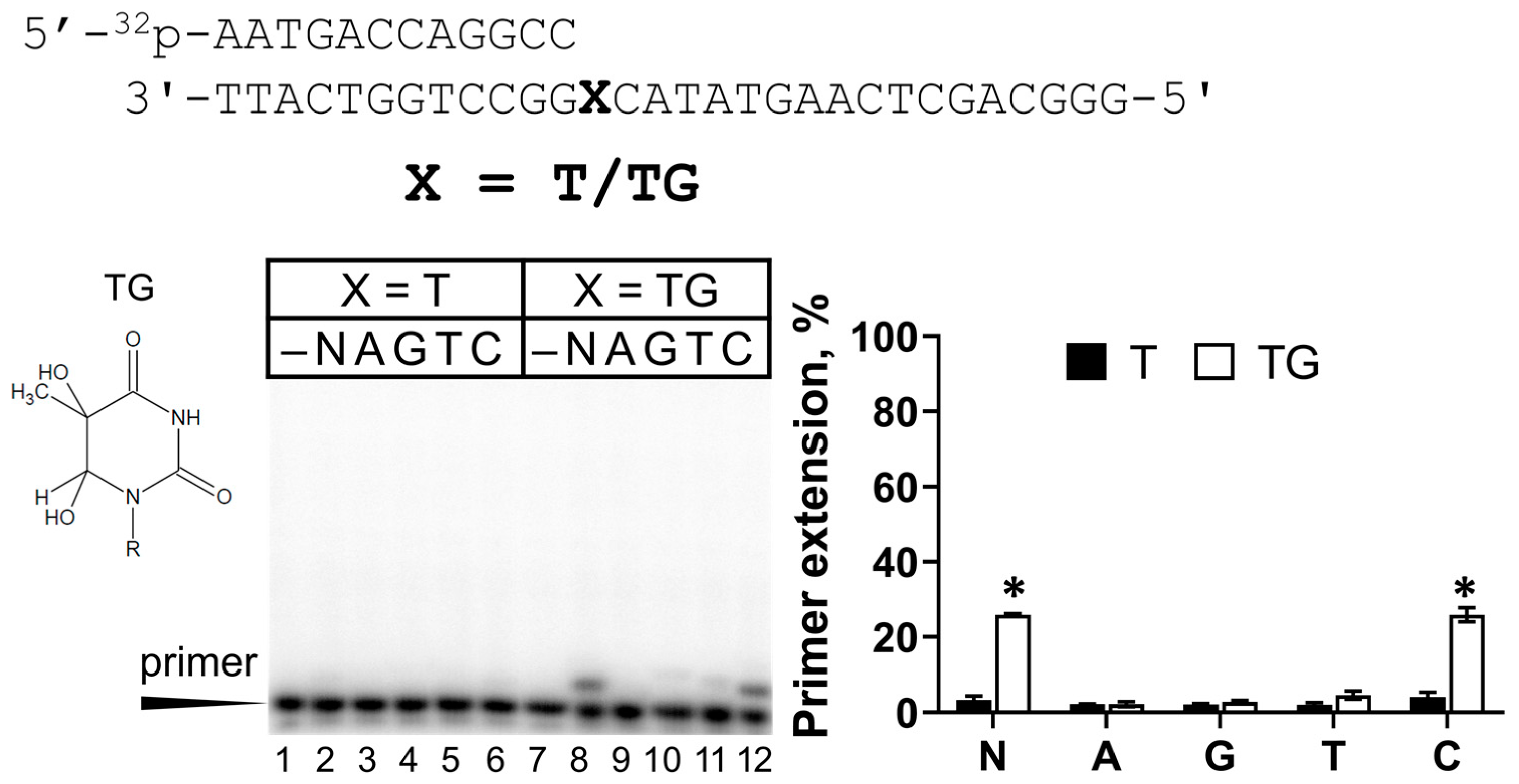
2.4. Activity of REV1 Opposite TG
3. Discussion
4. Materials and Methods
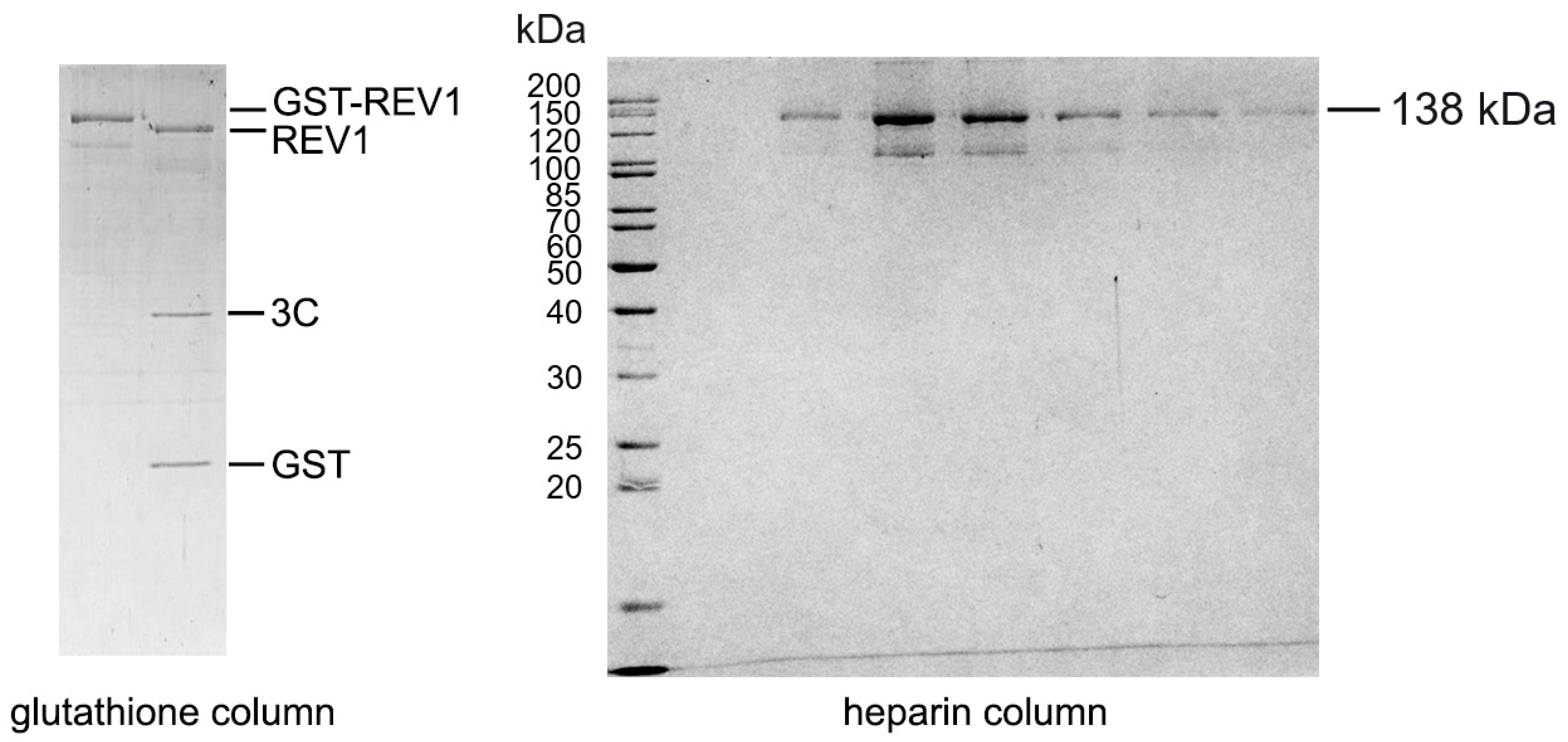
4.1. Protein Purification
4.2. DNA Substrates for the Primer Extension Assay
4.3. DNA Polymerase Reactions for the Primer Extension Assay
4.4. Steady-State Kinetics Analysis of dNMP Incorporation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
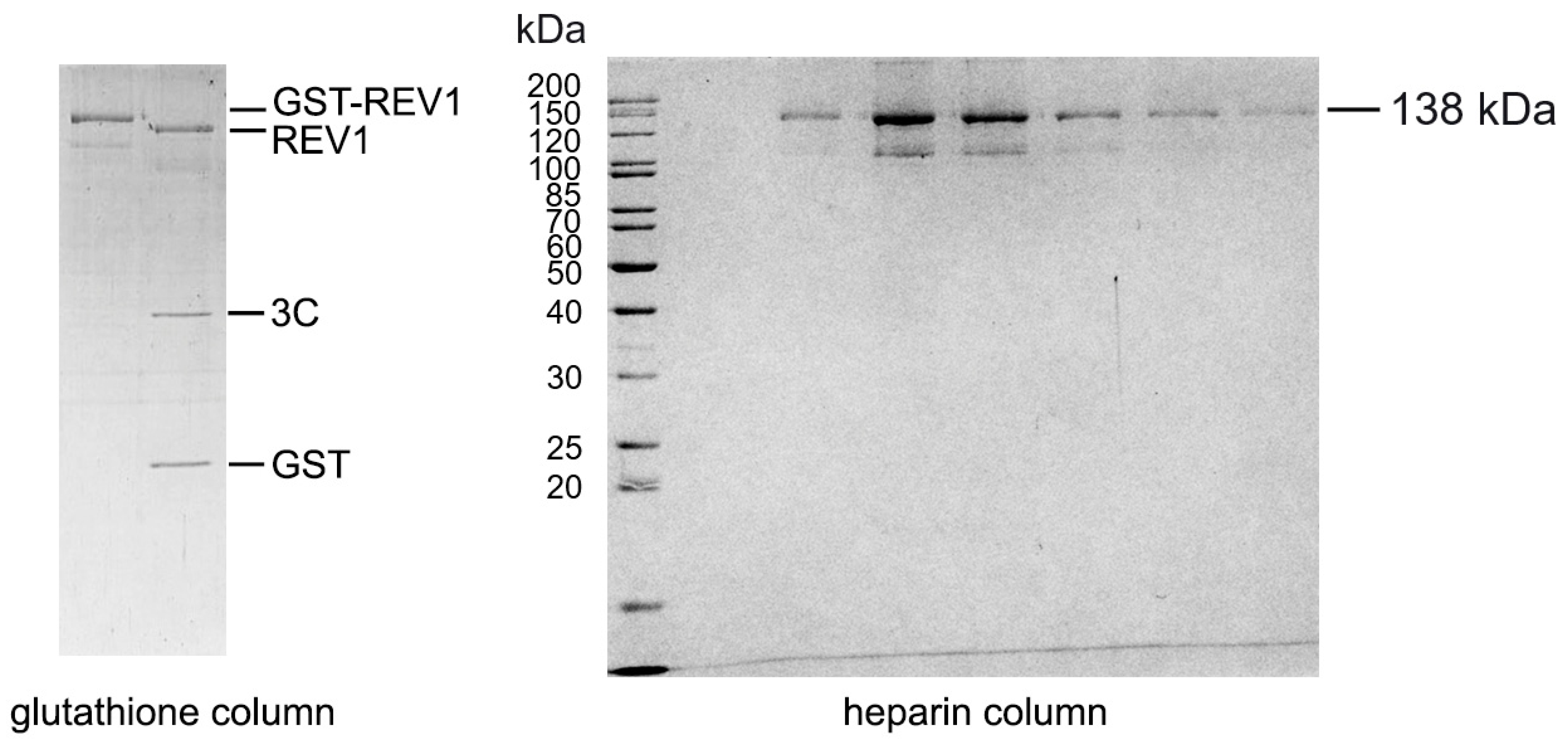
Appendix A. Cloning and Purification of Human REV1
References
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-Family of DNA Polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.A.; Korzhnev, D.M. The Rev1-Polζ translesion synthesis mutasome: Structure, interactions and inhibition. Enzymes 2019, 45, 139–181. [Google Scholar] [CrossRef] [PubMed]
- Bezalel-Buch, R.; Cheun, Y.K.; Roy, U.; Schärer, O.D.; Burgers, P.M. Bypass of DNA interstrand crosslinks by a Rev1–DNA polymerase ζ complex. Nucleic Acids Res. 2020, 48, 8461–8473. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Waters, L.S.; Walker, G.C. Novel conserved motifs in Rev1 C-terminus are required for mutagenic DNA damage tolerance. DNA Repair 2008, 7, 1455–1470. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Unk, I.; Johnson, R.E.; Johansson, E.; Burgers, P.M.; Prakash, S.; Prakash, L. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 2001, 15, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.-L.; Simpson, L.J.; Sale, J.E. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005, 33, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Prakash, S.; Prakash, L. Yeast Rev1 Protein Is a G Template-specific DNA Polymerase. J. Biol. Chem. 2002, 277, 15546–15551. [Google Scholar] [CrossRef] [PubMed]
- Swan, M.K.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structure of the Human Rev1–DNA–dNTP Ternary Complex. J. Mol. Biol. 2009, 390, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xin, H.; Zhang, Y.; Wu, X.; Yuan, F.; Wang, Z. The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic Acids Res. 1999, 27, 4468–4475. [Google Scholar] [CrossRef]
- Masuda, Y.; Takahashi, M.; Fukuda, S.; Sumii, M.; Kamiya, K. Mechanisms of dCMP Transferase Reactions Catalyzed by Mouse Rev1 Protein. J. Biol. Chem. 2002, 277, 3040–3046. [Google Scholar] [CrossRef]
- Chan, K.; Resnick, M.A.; Gordenin, D.A. The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair 2013, 12, 878–889. [Google Scholar] [CrossRef]
- Kim, N.; Mudrak, S.V.; Jinks-Robertson, S. The dCMP transferase activity of yeast Rev1 is biologically relevant during the bypass of endogenously generated AP sites. DNA Repair 2011, 10, 1262–1271. [Google Scholar] [CrossRef]
- Otsuka, C.; Kunitomi, N.; Iwai, S.; Loakes, D.; Negishi, K. Roles of the polymerase and BRCT domains of Rev1 protein in translesion DNA synthesis in yeast in vivo. Mutat. Res. Mol. Mech. Mutagen. 2005, 578, 79–87. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Zhang, Y.; Wang, Z. The catalytic function of the Rev1 dCMP transferase is required in a lesion-specific manner for translesion synthesis and base damage-induced mutagenesis. Nucleic Acids Res. 2010, 38, 5036–5046. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Reams, C.; Simpson, L.J.; Sale, J.E. Epigenetic Instability due to Defective Replication of Structured DNA. Mol. Cell 2010, 40, 703–713. [Google Scholar] [CrossRef]
- Jansen, J.G.; Langerak, P.; Tsaalbi-Shtylik, A.; van den Berk, P.; Jacobs, H.; de Wind, N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 2006, 203, 319–323. [Google Scholar] [CrossRef]
- Prasad, R.; Poltoratsky, V.; Hou, E.W.; Wilson, S.H. Rev1 is a base excision repair enzyme with 5′-deoxyribose phosphate lyase activity. Nucleic Acids Res. 2016, 44, 10824–10833. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N. Potential Strategies to Target Protein–Protein Interactions in the DNA Damage Response and Repair Pathways. J. Med. Chem. 2017, 60, 9932–9959. [Google Scholar] [CrossRef] [PubMed]
- Sasatani, M.; Zaharieva, E.K.; Kamiya, K. The in vivo role of Rev1 in mutagenesis and carcinogenesis. Genes Environ. 2020, 42, 1–5. [Google Scholar] [CrossRef]
- Shilkin, E.S.; Boldinova, E.O.; Stolyarenko, A.D.; Goncharova, R.I.; Chuprov-Netochin, R.N.; Khairullin, R.F.; Smal, M.P.; Makarova, A.V. Translesion DNA Synthesis and Carcinogenesis. Biochemistry 2020, 85, 425–435. [Google Scholar] [CrossRef]
- Shilkin, E.S.; Boldinova, E.O.; Stolyarenko, A.D.; Goncharova, R.I.; Chuprov-Netochin, R.N.; Smal, M.P.; Makarova, A.V. Translesion DNA Synthesis and Reinitiation of DNA Synthesis in Chemotherapy Resistance. Biochemistry 2020, 85, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Haracska, L.; Prakash, S.; Prakash, L. Complex Formation of Yeast Rev1 with DNA Polymerase η. Mol. Cell Biol. 2007, 27, 8401–8408. [Google Scholar] [CrossRef]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Deoxycytidyl transferase activity of yeast REV1 protein. Nature 1996, 382, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kino, K.; Kawada, T.; Morikawa, M.; Kobayashi, T.; Miyazawa, H. Analysis of Nucleotide Insertion Opposite 2,2,4-Triamino-5(2H)-oxazolone by Eukaryotic B- and Y-Family DNA Polymerases. Chem. Res. Toxicol. 2015, 28, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kino, K.; Kawada, T.; Oyoshi, T.; Morikawa, M.; Kobayashi, T.; Miyazawa, H. Contiguous 2,2,4-triamino-5(2H)-oxazolone obstructs DNA synthesis by DNA polymerases α, β, η, ι, κ, REV1 and Klenow Fragment exo-, but not by DNA polymerase ζ. J. Biochem. 2015, 159, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.T.; Minko, I.G.; Johnson, R.E.; Haracska, L.; Harris, T.M.; Lloyd, R.S.; Prakash, S.; Prakash, L. Efficient and Error-Free Replication past a Minor-Groove N2-Guanine Adduct by the Sequential Action of Yeast Rev1 and DNA Polymerase ζ. Mol. Cell Biol. 2004, 24, 6900–6906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.; Rechkoblit, O.; Geacintov, N.E.; Taylor, J.S.; Wang, Z. Response of human REV1 to different DNA damage: Preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002, 30, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Fowler, J.D.; Suo, Z. Kinetic Basis of Nucleotide Selection Employed by a Protein Template-Dependent DNA Polymerase. Biochemistry 2010, 49, 5504–5510. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Masuda, Y.; Kamiya, K. Specific amino acid residues are involved in substrate discrimination and template binding of human REV1 protein. Biochem. Biophys. Res. Commun. 2010, 392, 140–144. [Google Scholar] [CrossRef]
- Sherrer, S.M.; Sanman, L.E.; Xia, C.X.; Bolin, E.R.; Malik, C.K.; Efthimiopoulos, G.; Basu, A.K.; Suo, Z. Kinetic Analysis of the Bypass of a Bulky DNA Lesion Catalyzed by Human Y-Family DNA Polymerases. Chem. Res. Toxicol. 2012, 25, 730–740. [Google Scholar] [CrossRef]
- Sherrer, S.M.; Fiala, K.A.; Fowler, J.D.; Newmister, S.A.; Pryor, J.M.; Suo, Z. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 2010, 39, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Tokarsky, E.J.; Gadkari, V.V.; Zahurancik, W.J.; Malik, C.K.; Basu, A.K.; Suo, Z. Pre-steady-state kinetic investigation of bypass of a bulky guanine lesion by human Y-family DNA polymerases. DNA Repair 2016, 46, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Yeom, M.; Kim, I.-H.; Kim, J.-K.; Kang, K.; Eoff, R.L.; Guengerich, F.P.; Choi, J.-Y. Effects of Twelve Germline Missense Variations on DNA Lesion and G-Quadruplex Bypass Activities of Human DNA Polymerase REV1. Chem. Res. Toxicol. 2016, 29, 367–379. [Google Scholar] [CrossRef]
- Zhao, L.; Pence, M.G.; Christov, P.P.; Wawrzak, Z.; Choi, J.-Y.; Rizzo, C.J.; Egli, M.; Guengerich, F. Basis of Miscoding of the DNA Adduct N2,3-Ethenoguanine by Human Y-family DNA Polymerases. J. Biol. Chem. 2012, 287, 35516–35526. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Xie, Z.; Shen, H.; Zhao, B.; Wang, Z. Translesion synthesis of acetylaminofluorene-dG adducts by DNA polymerase is stimulated by yeast Rev1 protein. Nucleic Acids Res. 2004, 32, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Kuang, L.; Kou, H.; Xie, Z.; Zhou, Y.; Feng, X.; Wang, L.; Wang, Z. A non-catalytic function of Rev1 in translesion DNA synthesis and mutagenesis is mediated by its stable interaction with Rad5. DNA Repair 2013, 12, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Takahashi, M.; Tsunekuni, N.; Minami, T.; Sumii, M.; Miyagawa, K.; Kamiya, K. Deoxycytidyl Transferase Activity of the Human REV1 Protein Is Closely Associated with the Conserved Polymerase Domain. J. Biol. Chem. 2001, 276, 15051–15058. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Guengerich, F.P. Kinetic Analysis of Translesion Synthesis Opposite Bulky N2- and O6-Alkylguanine DNA Adducts by Human DNA Polymerase REV1. J. Biol. Chem. 2008, 283, 23645–23655. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Kamiya, K. Biochemical properties of the human REV1 protein. FEBS Lett. 2002, 520, 88–92. [Google Scholar] [CrossRef]
- Nair, D.T.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Protein-Template-Directed Synthesis across an Acrolein-Derived DNA Adduct by Yeast Rev1 DNA Polymerase. Structure 2008, 16, 239–245. [Google Scholar] [CrossRef]
- Taggart, D.J.; Fredrickson, S.W.; Gadkari, V.V.; Suo, Z. Mutagenic Potential of 8-Oxo-7,8-dihydro-2′-deoxyguanosine Bypass Catalyzed by Human Y-Family DNA Polymerases. Chem. Res. Toxicol. 2014, 27, 931–940. [Google Scholar] [CrossRef]
- Howell, C.A.; Prakash, S.; Washington, M.T. Pre-Steady-State Kinetic Studies of Protein-Template-Directed Nucleotide Incorporation by the Yeast Rev1 Protein. Biochemistry 2007, 46, 13451–13459. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Johnson, R.E.; Prakash, L.; Prakash, S. DNA polymerase θ accomplishes translesion synthesis opposite 1,N6-ethenodeoxyadenosine with a remarkably high fidelity in human cells. Minerva Anestesiol. 2019, 33, 282–287. [Google Scholar] [CrossRef]
- Guo, C.; Fischhaber, P.L.; Luk-Paszyc, M.J.; Masuda, Y.; Zhou, J.; Kamiya, K.; Kisker, C.; Friedberg, E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003, 22, 6621–6630. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Lim, S.; Kim, E.-J.; Jo, A.; Guengerich, F.P. Translesion Synthesis across Abasic Lesions by Human B-Family and Y-Family DNA Polymerases α, δ, η, ι, κ, and REV1. J. Mol. Biol. 2010, 404, 34–44. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Hodge, R.P.; Hackfeld, L.C.; Park, J.; Choudhury, J.R.; Prakash, S.; Prakash, L. Genetic control of predominantly error-free replication through an acrolein-derived minor-groove DNA adduct. J. Biol. Chem. 2018, 293, 2949–2958. [Google Scholar] [CrossRef]
- Du, H.; Wang, P.; Li, L.; Wang, Y. Repair and translesion synthesis of O6-alkylguanine DNA lesions in human cells. J. Biol. Chem. 2019, 294, 11144–11153. [Google Scholar] [CrossRef]
- Nair, D.T.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Rev1 Employs a Novel Mechanism of DNA Synthesis Using a Protein Template. Science 2005, 309, 2219–2222. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; McElhinny, S.A.N.; Watts, B.E.; Kunkel, T.A.; Burgers, P.M. Ribonucleotide incorporation by yeast DNA polymerase ζ. DNA Repair 2014, 18, 63–67. [Google Scholar] [CrossRef]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef]
- Boldinova, E.O.; Ignatov, A.; Kulbachinskiy, A.; Makarova, A.V. The active site residues Gln55 and Arg73 play a key role in DNA damage bypass by S. cerevisiae Pol η. Sci. Rep. 2018, 8, 10314. [Google Scholar] [CrossRef] [PubMed]
- Kazachenko, K.Y.; Miropolskaya, N.A.; Gening, L.V.; Tarantul, V.Z.; Makarova, A.V. Alternative splicing at exon 2 results in the loss of the catalytic activity of mouse DNA polymerase iota in vitro. DNA Repair 2017, 50, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Boldinova, E.O.; Belousova, E.A.; Lavrik, O.I. In vitro lesion bypass by human PrimPol. DNA Repair 2018, 70, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Boldinova, E.O.; Yudkina, A.V.; Shilkin, E.S.; Gagarinskaya, D.I.; Baranovskiy, A.G.; Tahirov, T.H.; Zharkov, D.O.; Makarova, A.V. Translesion activity of PrimPol on DNA with cisplatin and DNA–protein cross-links. Sci. Rep. 2021, 11, 17588. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template Nucleotide | kcat, min−1 | KM, µM | kcat/KM | Finc |
|---|---|---|---|---|
| G | 4.86 ± 0.40 | 0.04 ± 0.01 | 133.29 ± 32.53 | 1 |
| 8-oxoG | 3.20 ± 0.57 | 10.13 ± 3.02 | 0.36 ± 0.08 | 2.7∙10−3 (↓366 fold) |
| G | 3.97 ± 0.38 | 0.06 ± 0.01 | 68.66 ± 0.61 | 1 |
| O6-meG | 7.22 ± 1.74 | 2.92 ± 1.21 | 2.87 ± 0.65 | 4.2∙10−2 (↓24 fold) |
| A | 3.49 ± 0.11 | 10.60 ± 1.92 | 0.35 ± 0.07 | 1 |
| 7-deazaA | 0.51 ± 0.05 | 56.81 ± 5.01 | 0.009 ± 0.001 | 2.6∙10−2 (↓39 fold) |
| A | 1.87 ± 0.25 | 72.02 ± 11.69 | 0.03 ± 0.01 | 1 |
| εA | 3.19 ± 0.20 | 2.18 ± 0.27 | 1.53 ± 0.28 | 54 (↑54 fold) |
| T | 0.09 ± 0.01 | 301.73 ± 27.45 | (2.88 ± 0.07) × 10−4 | 1 |
| TG | 0.53 ± 0.06 | 1.08 ± 0.06 | 0.50 ± 0.06 | 1719 (↑1719 fold) |
| 1 | Primer 16: 5′-32P-GTCACAGAGATACTAC-3′ Template: 3′-CAGTGTCTCTATGATGXACACGCTGACGAG-5′ where X = A, G, T, C, 8-oxoG, 7-deazaG and 7-deazaA |
| 2 | Primer 12 (GCC): 5′-32P-AATGACCAGGCC-3′ Template: 3′-TTACTGGTCCGGXCTTATGAACTCGACGGG-5′ where X = A and εA |
| 3 | Primer 12 (GCC): 5′-32P-AATGACCAGGCC-3′ Template: 3′-TTACTGGTCCGGXCATATGAACTCGACGGG-5′ where X = T, TG, and AP site (a tetrahydrofuran analog) |
| 4 | Primer 12 (GCA): 5′-32P-AATGACCAGGCA-3′ Template: 3′-TTACTGGTCCGTXCATATGAACTCGACGGG-5′ where X = G and O6-meG |
| 5 | Primer 15: 5′-32P-AGAAGGGATAGATGA-3′ Template: 3′-TCTTCCCTATCTACTXXTTCCTCTTCCTCC-5′ where XX = GG, and 1,2-GG CisPt CL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolyarenko, A.D.; Novikova, A.A.; Shilkin, E.S.; Poltorachenko, V.A.; Makarova, A.V. The Catalytic Activity of Human REV1 on Undamaged and Damaged DNA. Int. J. Mol. Sci. 2024, 25, 4107. https://doi.org/10.3390/ijms25074107
Stolyarenko AD, Novikova AA, Shilkin ES, Poltorachenko VA, Makarova AV. The Catalytic Activity of Human REV1 on Undamaged and Damaged DNA. International Journal of Molecular Sciences. 2024; 25(7):4107. https://doi.org/10.3390/ijms25074107
Chicago/Turabian StyleStolyarenko, Anastasia D., Anna A. Novikova, Evgeniy S. Shilkin, Valentin A. Poltorachenko, and Alena V. Makarova. 2024. "The Catalytic Activity of Human REV1 on Undamaged and Damaged DNA" International Journal of Molecular Sciences 25, no. 7: 4107. https://doi.org/10.3390/ijms25074107