
Characterization of Biomarkers of Thrombo-Inflammation in Patients with First-Diagnosed Atrial Fibrillation
 , , ,
, , ,
Abstract
1. Introduction
2. Results
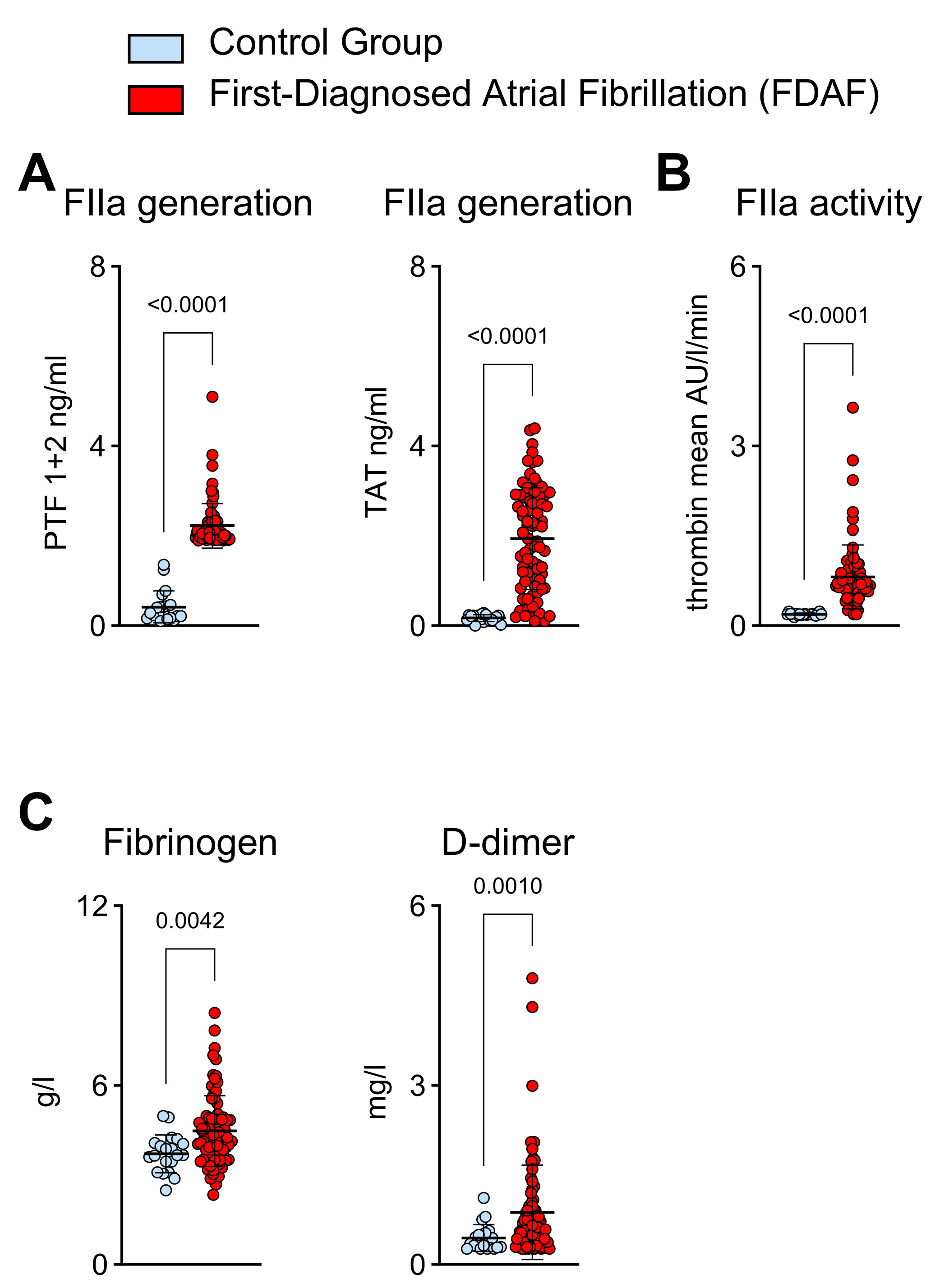
2.1. Thrombin Generation and Activity in Patients with First-Diagnosed AF
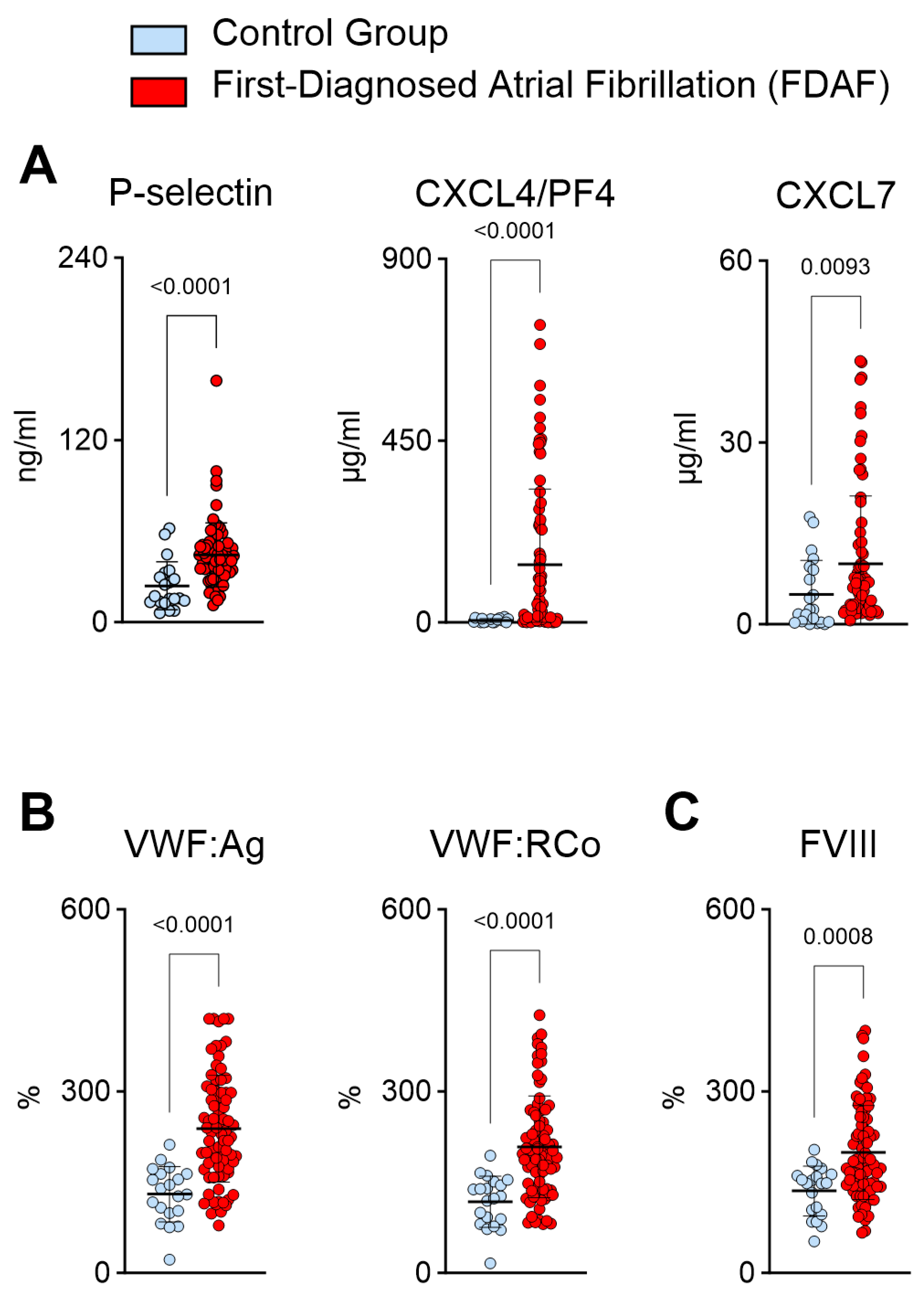
2.2. Plasma Biomarkers Suggest Increased Platelet Activation and Platelet–Endothelial Interactions during First-Diagnosed AF
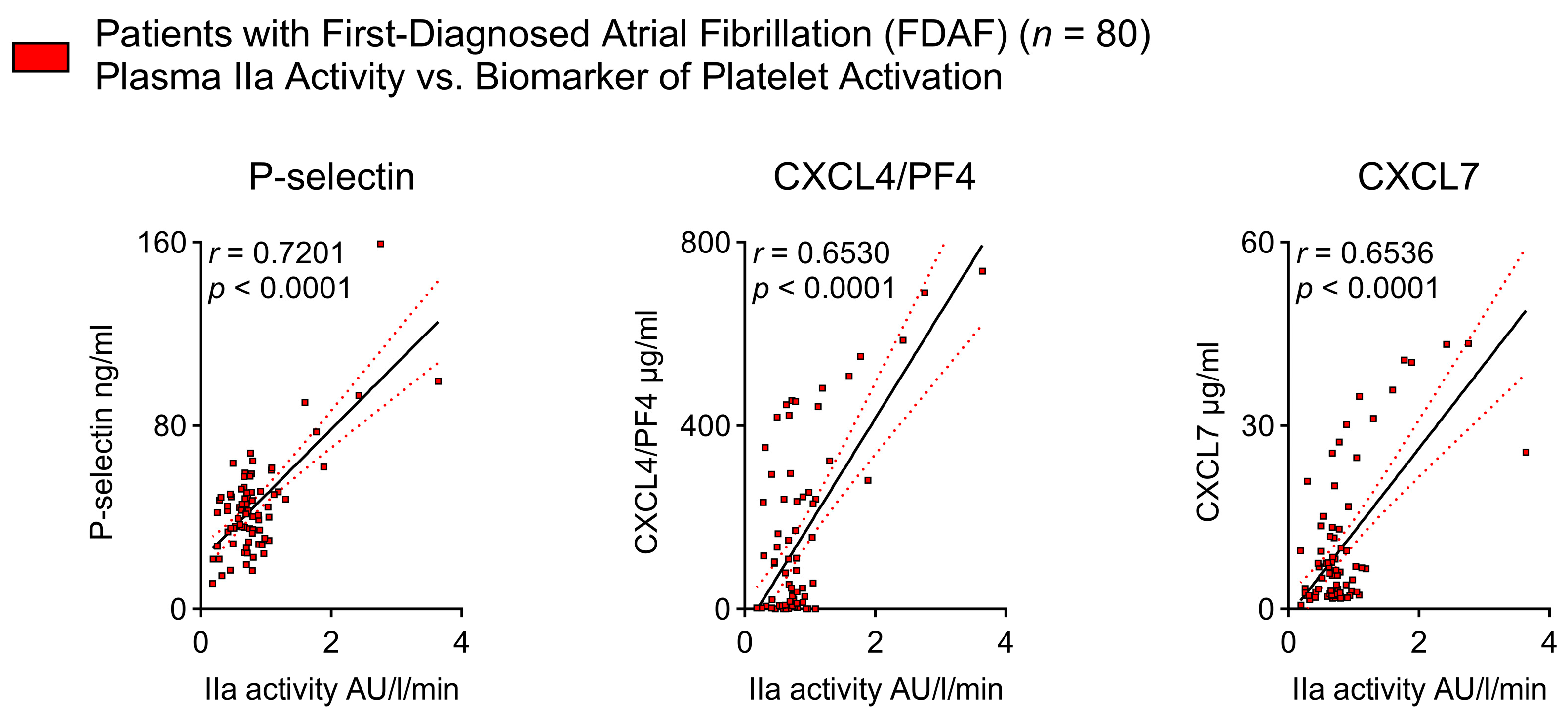
2.3. Thrombin Activity Is Associated with Platelet Activation Indicators in Patients with Early AF
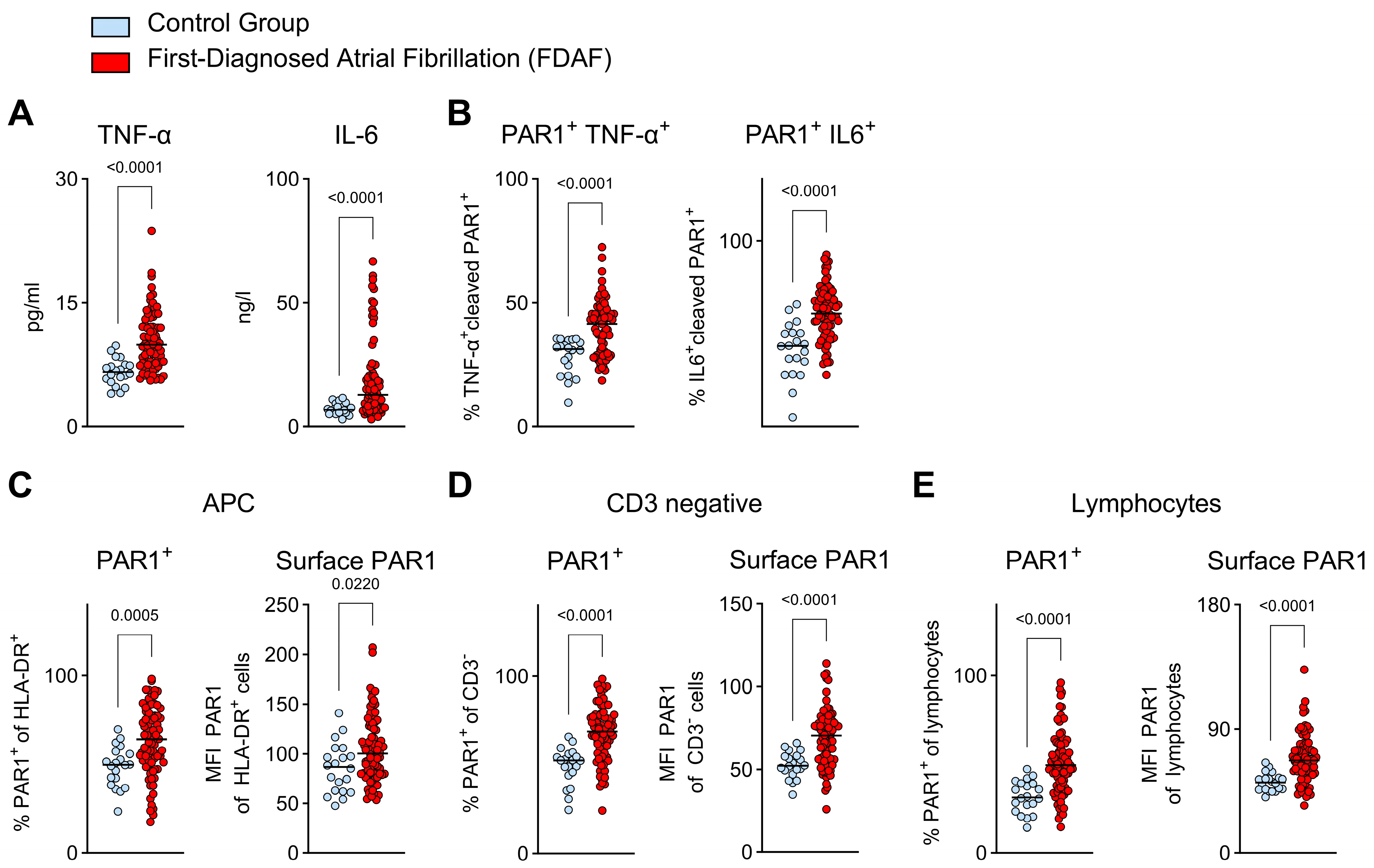
2.4. Pro-Inflammatory Immune Cell Function in Very Early AF—Indications for PAR1 Activation via Thrombin
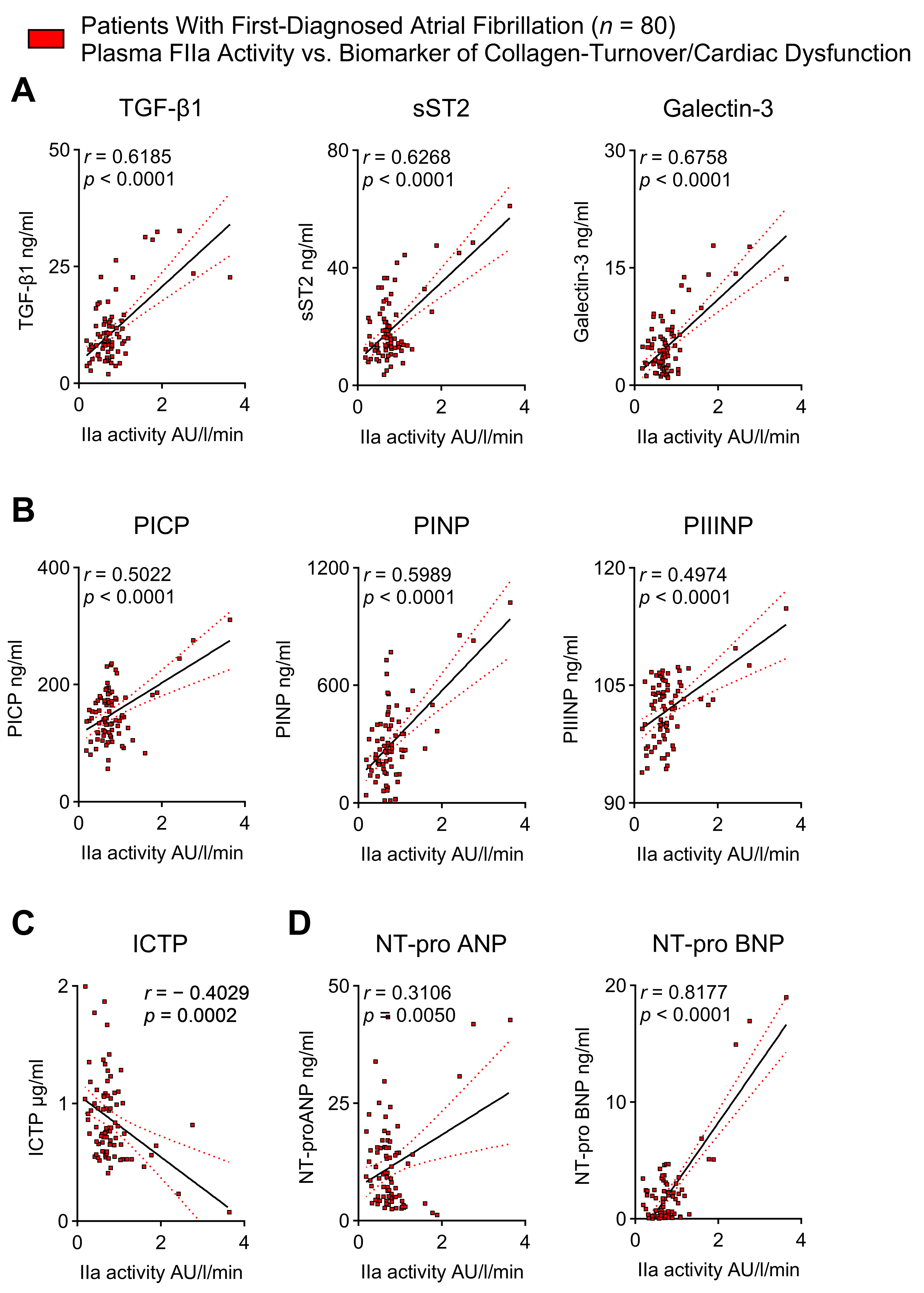
2.5. Thrombin Activity Correlates with Biomarkers of Cardiac Fibrosis and Cardiac Dysfunction in Patients with First-Diagnosed AF
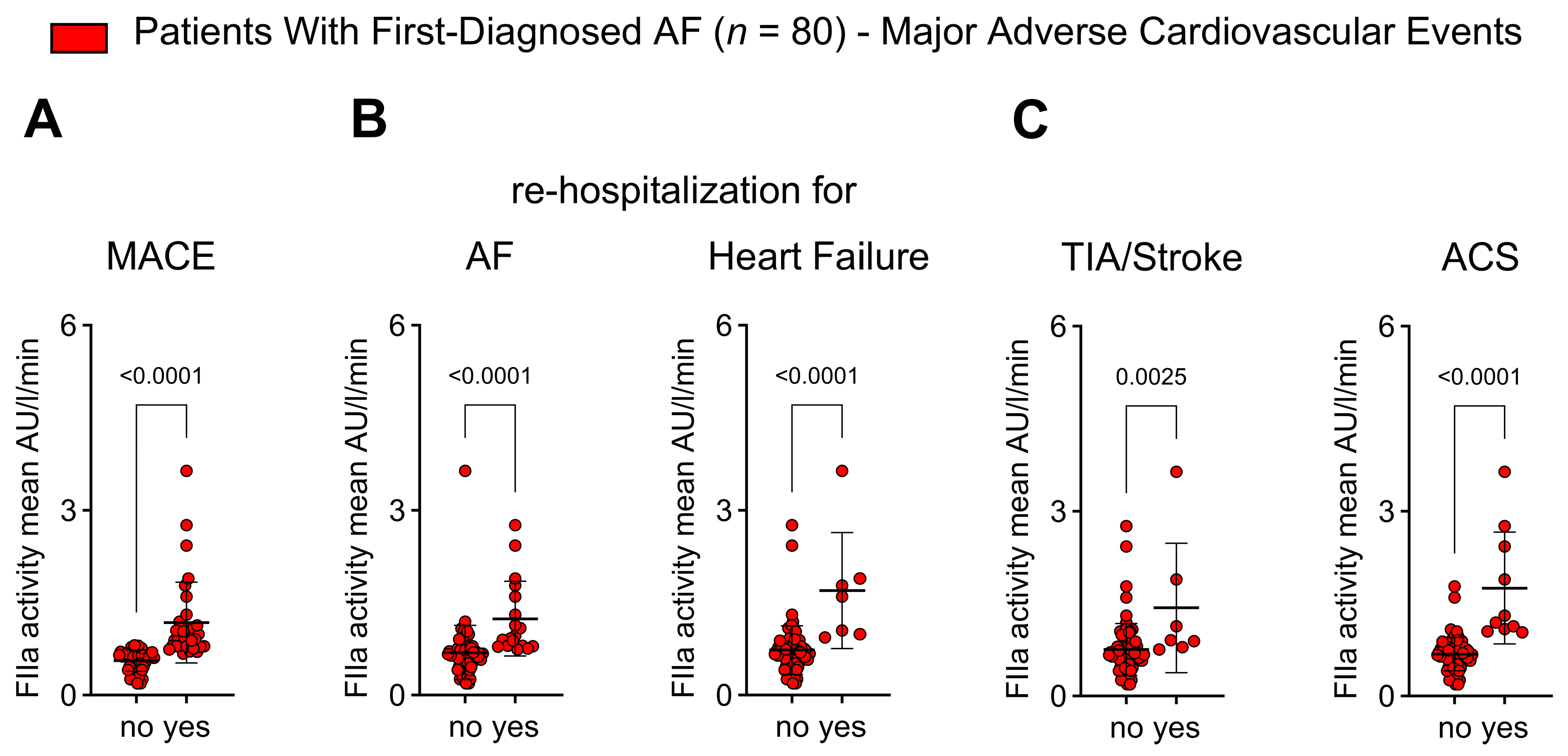
2.6. Thrombin Activity Is Associated with Adverse Outcomes in Patients with First Diagnosis of AF
3. Discussion
- Increased FIIa generation and FIIa activity in patients with FDAF.
- Platelet activation biomarkers correlate with FIIa activity.
- Pro-inflammatory immune cell function in very early AF is linked to PAR1 activation via FIIa.
- Plasma indicators of cardiac fibrosis and cardiac dysfunction are related to FIIa activity.
- FIIa activity is higher in patients with FDAF who experience adverse outcomes.
3.1. Plasma Biomarkers Indicate a Prothrombotic and Hypercoagulable State in Patients with First-Diagnosed AF
3.2. Biomarkers of Inflammation and Cardiac Fibrosis in Early AF Are Linked to Thrombin
3.3. Clinical Implications
3.4. Limitations
4. Materials and Methods
4.1. Patient Studies
4.2. Laboratory Assays, Incl’ ELISA
4.3. Flow Cytometry
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2020, 42, 373–498. [Google Scholar] [CrossRef]
- Goette, A.; Borof, K.; Breithardt, G.; Camm, A.J.; Crijns, H.; Kuck, K.H.; Wegscheider, K.; Kirchhof, P. Presenting Pattern of Atrial Fibrillation and Outcomes of Early Rhythm Control Therapy. J. Am. Coll. Cardiol. 2022, 80, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.F.; Chan, Y.H.; Chiang, C.E.; Tuan, T.C.; Liao, J.N.; Chen, T.J.; Lip, G.Y.H.; Chen, S.A. Early Rhythm Control and the Risks of Ischemic Stroke, Heart Failure, Mortality, and Adverse Events When Performed Early (<3 Months): A Nationwide Cohort Study of Newly Diagnosed Patients with Atrial Fibrillation. Thromb. Haemost. 2022, 122, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Breitenstein, A.; Glanzmann, M.; Falk, V.; Maisano, F.; Stämpfli, S.F.; Holy, E.W.; Finlay, M.; Ling, L.H.; Schilling, R.J.; Lüscher, T.F.; et al. Increased prothrombotic profile in the left atrial appendage of atrial fibrillation patients. Int. J. Cardiol. 2015, 185, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Motoki, H.; Tomita, T.; Aizawa, K.; Kasai, H.; Izawa, A.; Kumazaki, S.; Tsutsui, H.; Koyama, J.; Ikeda, U. Coagulation activity is increased in the left atria of patients with paroxysmal atrial fibrillation during the non-paroxysmal period. Comparison with chronic atrial fibrillation. Circ. J. 2009, 73, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Y.; Gupta, D.; Lip, G.Y.H. Atrial fibrillation and the prothrombotic state: Revisiting Virchow’s triad in 2020. Heart 2020, 106, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Tilly, M.J.; Geurts, S.; Pezzullo, A.M.; Bramer, W.M.; de Groot, N.M.S.; Kavousi, M.; de Maat, M.P.M. The association of coagulation and atrial fibrillation: A systematic review and meta-analysis. EP Eur. 2022, 25, 28–39. [Google Scholar] [CrossRef]
- Bukowska, A.; Hammwöhner, M.; Corradi, D.; Mahardhika, W.; Goette, A. Atrial thrombogenesis in atrial fibrillation. Herzschrittmachertherapie + Elektrophysiologie 2018, 29, 76–83. [Google Scholar] [CrossRef] [PubMed]
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; Ten Cate-Hoek, A.J.; Ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef]
- Friebel, J.; Witkowski, M.; Wegner, M.; Blöbaum, L.; Lammel, S.; Schencke, P.A.; Jakobs, K.; Puccini, M.; Reißner, D.; Steffens, D.; et al. Cytotoxic CD8+ T Cells Are Involved in the Thrombo-Inflammatory Response during First-Diagnosed Atrial Fibrillation. Cells 2022, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Zacharias, I.; Weinert, S.; Skopp, K.; Hartmann, C.; Huth, C.; Goette, A. Coagulation factor Xa induces an inflammatory signalling by activation of protease-activated receptors in human atrial tissue. Eur. J. Pharmacol. 2013, 718, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, T.; Grune, J.; Landmesser, U.; Attanasio, P. Detection and modification of biomarkers of inflammation determining successful rhythm control in patients with atrial fibrillation. Biomarkers 2023, 28, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Posma, J.J.; Posthuma, J.J.; Spronk, H.M. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. 2016, 14, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rivera, I.; López, E.; López-Colomé, A.M. Diversification of PAR signaling through receptor crosstalk. Cell. Mol. Biol. Lett. 2022, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Weithauser, A.; Rauch, U. Role of protease-activated receptors for the innate immune response of the heart. Trends Cardiovasc. Med. 2014, 24, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Date, T.; Ikegami, M.; Hongo, K.; Fujisaki, M.; Katoh, D.; Yoshino, T.; Anzawa, R.; Nagoshi, T.; Yamashita, S.; et al. An immunohistochemical analysis of tissue thrombin expression in the human atria. PLoS ONE 2013, 8, e65817. [Google Scholar] [CrossRef] [PubMed]
- Friebel, J.; Moritz, E.; Witkowski, M.; Jakobs, K.; Strässler, E.; Dörner, A.; Steffens, D.; Puccini, M.; Lammel, S.; Glauben, R.; et al. Pleiotropic Effects of the Protease-Activated Receptor 1 (PAR1) Inhibitor, Vorapaxar, on Atherosclerosis and Vascular Inflammation. Cells 2021, 10, 3517. [Google Scholar] [CrossRef] [PubMed]
- Friebel, J.; Weithauser, A.; Witkowski, M.; Rauch, B.H.; Savvatis, K.; Dörner, A.; Tabaraie, T.; Kasner, M.; Moos, V.; Bösel, D.; et al. Protease-activated receptor 2 deficiency mediates cardiac fibrosis and diastolic dysfunction. Eur. Heart J. 2019, 40, 3318–3332. [Google Scholar] [CrossRef] [PubMed]
- Friebel, J.; Witkowski, M.; Rauch, U. Treating the unstable atherosclerotic plaque by targeting activated factor X—Anticoagulation and beyond. Circ. J. 2015, 79, 2329–2331. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, E.; Posma, J.J.N.; Spronk, H.M.H.; Ten Cate, H. Tissue factor (:Factor VIIa) in the heart and vasculature: More than an envelope. Thromb. Res. 2018, 168, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Kremers, B.M.M.; Ten Cate, H.; Spronk, H.M.H. Pleiotropic effects of the hemostatic system. J. Thromb. Haemost. 2018, 16, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Ten Cate, H.; Guzik, T.J.; Eikelboom, J.; Spronk, H.M.H. Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: Mechanistic insights and implications for anti-thrombotic treatment. Cardiovasc. Res. 2021, 117, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, T.; Seppelt, C.; Abdelwahed, Y.S.; Meteva, D.; Wolfram, C.; Stapmanns, P.; Erbay, A.; Zanders, L.; Nelles, G.; Musfeld, J.; et al. Culprit plaque morphology determines inflammatory risk and clinical outcomes in acute coronary syndrome. Eur. Heart J. 2023, 44, 3911–3925. [Google Scholar] [CrossRef] [PubMed]
- Leistner, D.M.; Kränkel, N.; Meteva, D.; Abdelwahed, Y.S.; Seppelt, C.; Stähli, B.E.; Rai, H.; Skurk, C.; Lauten, A.; Mochmann, H.C.; et al. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: Results from the prospective translational OPTICO-ACS study. Eur. Heart J. 2020, 41, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Meteva, D.; Vinci, R.; Seppelt, C.; Abdelwahed, Y.S.; Pedicino, D.; Nelles, G.; Skurk, C.; Haghikia, A.; Rauch-Kröhnert, U.; Gerhardt, T.; et al. Toll-like receptor 2, hyaluronan, and neutrophils play a key role in plaque erosion: The OPTICO-ACS study. Eur. Heart J. 2023, 44, 3892–3907. [Google Scholar] [CrossRef] [PubMed]
- Baaten, C.C.F.M.J.; Nagy, M.; Bergmeier, W.; Spronk, H.M.H.; van der Meijden, P.E.J. Platelet biology and function: Plaque erosion vs. rupture. Eur. Heart J. 2023, 45, 18–31. [Google Scholar] [CrossRef]
- Blöbaum, L.; Witkowski, M.; Wegner, M.; Lammel, S.; Schencke, P.A.; Jakobs, K.; Puccini, M.; Reißner, D.; Steffens, D.; Landmesser, U.; et al. Intestinal Barrier Dysfunction and Microbial Translocation in Patients with First-Diagnosed Atrial Fibrillation. Biomedicines 2023, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.B.; Marinelli, E.A.; Arbelo, E.; Boriani, G.; Boveda, S.; Buckley, C.M.; Camm, A.J.; Casadei, B.; Chua, W.; Dagres, N.; et al. Early diagnosis and better rhythm management to improve outcomes in patients with atrial fibrillation: The 8th AFNET/EHRA consensus conference. EP Eur. 2022, 25, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.A.; Belgore, F.; Li-Saw-Hee, F.L.; Conway, D.S.; Blann, A.D.; Lip, G.Y. Is the hypercoagulable state in atrial fibrillation mediated by vascular endothelial growth factor? Stroke 2002, 33, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Kusak, P.; Czarnecka, D.; Gissel, M.; Plens, K.; Butenas, S.; Undas, A. Activated factor IX, factor XI and tissue factor identify patients with permanent atrial fibrillation treated with warfarin who are at risk of ischemic stroke. Arch. Med. Sci. 2016, 12, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Willoughby, S.R.; Schultz, C.; Alasady, M.; Rangnekar, G.; Dang, J.; Gan, C.; Lau, D.H.; Roberts-Thomson, K.C.; Young, G.D.; et al. Thrombogenic Risk in Patients With Atrial Fibrillation: Importance of Comorbid Conditions and Intracardiac Changes. JACC Clin. Electrophysiol. 2015, 1, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; D’Agostino, R.B.; Silbershatz, H.; Lipinska, I.; Massaro, J.; Levy, D.; Benjamin, E.J.; Wolf, P.A.; Tofler, G.H. Hemostatic state and atrial fibrillation (the Framingham Offspring Study). Am. J. Cardiol. 2001, 87, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Ohara, K.; Inoue, H.; Nozawa, T.; Hirai, T.; Iwasa, A.; Okumura, K.; Lee, J.D.; Shimizu, A.; Hayano, M.; Yano, K. Accumulation of risk factors enhances the prothrombotic state in atrial fibrillation. Int. J. Cardiol. 2008, 126, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Roldán, V.; Marín, F.; García, A.; Tello-Montoliu, A.; Lip, G.Y. Is an advanced age an additive risk factor to the prothrombotic or hypercoagulable state in atrial fibrillation? Int. J. Cardiol. 2006, 110, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Friebel, J.; Tabaraie, T.; Grabitz, S.; Dörner, A.; Taghipour, L.; Jakobs, K.; Stratmann, B.; Tschoepe, D.; Landmesser, U.; et al. Metformin Is Associated with Reduced Tissue Factor Procoagulant Activity in Patients with Poorly Controlled Diabetes. Cardiovasc. Drugs Ther. 2021, 35, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303. [Google Scholar] [CrossRef]
- Witkowski, M.; Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; et al. Vascular endothelial tissue factor contributes to trimethylamine N-oxide-enhanced arterial thrombosis. Cardiovasc. Res. 2022, 118, 2367–2384. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Witkowski, M.; Saffarzadeh, M.; Friebel, J.; Tabaraie, T.; Ta Bao, L.; Chakraborty, A.; Dörner, A.; Stratmann, B.; Tschoepe, D.; et al. Vascular miR-181b controls tissue factor-dependent thrombogenicity and inflammation in type 2 diabetes. Cardiovasc. Diabetol. 2020, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Ünlü, B.; Bogdanov, V.Y.; Versteeg, H.H. Interplay between alternatively spliced Tissue Factor and full length Tissue Factor in modulating coagulant activity of endothelial cells. Thromb. Res. 2017, 156, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Østerud, B.; Unruh, D.; Olsen, J.O.; Kirchhofer, D.; Owens, A.P., 3rd; Bogdanov, V.Y. Procoagulant and proinflammatory effects of red blood cells on lipopolysaccharide-stimulated monocytes. J. Thromb. Haemost. 2015, 13, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, V.Y.; Versteeg, H.H. “Soluble Tissue Factor” in the 21st Century: Definitions, Biochemistry, and Pathophysiological Role in Thrombus Formation. Semin. Thromb. Hemost. 2015, 41, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nakamura, K.; Fukushima-Kusano, K.; Ohta, K.; Matsubara, H.; Hamuro, T.; Yutani, C.; Ohe, T. Tissue factor expression in atrial endothelia associated with nonvalvular atrial fibrillation: Possible involvement in intracardiac thrombogenesis. Thromb. Res. 2003, 111, 137–142. [Google Scholar] [CrossRef]
- Elias, A.; Khoury, Y.; Shehadeh, F.; Ron, G.; Boulos, M.; Nashashibi, J.; Zukermann, R.; Elias, M.; Gepstein, L.; Suleiman, M. Elevated thrombin generation levels in the left atrial appendage in patients with atrial fibrillation. Res. Pract. Thromb. Haemost. 2023, 7, 100127. [Google Scholar] [CrossRef] [PubMed]
- Matsue, Y.; Suzuki, M.; Abe, M.; Ono, M.; Seya, M.; Nakamura, T.; Iwatsuka, R.; Mizukami, A.; Toyama, K.; Kumasaka, L.; et al. Endothelial dysfunction in paroxysmal atrial fibrillation as a prothrombotic state. Comparison with permanent/persistent atrial fibrillation. J. Atheroscler. Thromb. 2011, 18, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Soto, A.G.; Coronel, L.J.; Goss, A.; van Ryn, J.; Trejo, J. Characterization of thrombin-bound dabigatran effects on protease-activated receptor-1 expression and signaling in vitro. Mol. Pharmacol. 2015, 88, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Negreva, M.; Zarkova, A.; Prodanova, K.; Petrov, P. Paroxysmal Atrial Fibrillation: Insight Into the Intimate Mechanisms of Coagulation. Cardiol. Res. 2020, 11, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Takeshita, K.; Inden, Y.; Ishii, H.; Cheng, X.W.; Yamamoto, K.; Murohara, T. Platelet activation and induction of tissue factor in acute and chronic atrial fibrillation: Involvement of mononuclear cell-platelet interaction. Thromb. Res. 2011, 128, e113–e118. [Google Scholar] [CrossRef]
- Akar, J.G.; Jeske, W.; Wilber, D.J. Acute onset human atrial fibrillation is associated with local cardiac platelet activation and endothelial dysfunction. J. Am. Coll. Cardiol. 2008, 51, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Pourtau, L.; Sellal, J.M.; Lacroix, R.; Poncelet, P.; Bernus, O.; Clofent-Sanchez, G.; Hocini, M.; Haïssaguerre, M.; Dignat-George, F.; Sacher, F.; et al. Platelet function and microparticle levels in atrial fibrillation: Changes during the acute episode. Int. J. Cardiol. 2017, 243, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Willoughby, S.R.; Schultz, C.; Gan, C.; Alasady, M.; Lau, D.H.; Leong, D.P.; Brooks, A.G.; Young, G.D.; Kistler, P.M.; et al. Effect of atrial fibrillation on atrial thrombogenesis in humans: Impact of rate and rhythm. J. Am. Coll. Cardiol. 2013, 61, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Sekiguchi, A.; Iwasaki, Y.K.; Sagara, K.; Hatano, S.; Iinuma, H.; Aizawa, T.; Fu, L.T. Thrombomodulin and tissue factor pathway inhibitor in endocardium of rapidly paced rat atria. Circulation 2003, 108, 2450–2452. [Google Scholar] [CrossRef] [PubMed]
- Sohara, H.; Amitani, S.; Kurose, M.; Miyahara, K. Atrial fibrillation activates platelets and coagulation in a time-dependent manner: A study in patients with paroxysmal atrial fibrillation. J. Am. Coll. Cardiol. 1997, 29, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Li-Saw-Hee, F.L.; Blann, A.D.; Gurney, D.; Lip, G.Y. Plasma von Willebrand factor, fibrinogen and soluble P-selectin levels in paroxysmal, persistent and permanent atrial fibrillation. Effects of cardioversion and return of left atrial function. Eur. Heart J. 2001, 22, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.; Chin, B.S.; Blann, A.D.; Lip, G.Y. A study of platelet activation in paroxysmal, persistent and permanent atrial fibrillation. Blood Coagul. Fibrinolysis 2002, 13, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Jabati, S.; Fareed, J.; Liles, J.; Otto, A.; Hoppensteadt, D.; Bontekoe, J.; Phan, T.; Walborn, A.; Syed, M. Biomarkers of Inflammation, Thrombogenesis, and Collagen Turnover in Patients with Atrial Fibrillation. Clin. Appl. Thromb. Hemost. 2018, 24, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Watson, T.; Shantsila, E.; Lip, G.Y. Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet 2009, 373, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Luo, S.Q.; Liu, S.T.; Gong, L.; Zhong, P.; Wang, Z.Z.; Lip, G.Y.; Zou, Y.X.; Guo, W.H. Clinical Features Associated with ‘Normal Range’ Fibrin D-Dimer Levels in Atrial Fibrillation Patients with Left Atrial Thrombus. Clin. Appl. Thromb. Hemost. 2022, 28, 10760296221133380. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; de Jong, A.M.; Crijns, H.J.; Schotten, U.; Van Gelder, I.C.; Ten Cate, H. Pleiotropic effects of factor Xa and thrombin: What to expect from novel anticoagulants. Cardiovasc. Res. 2014, 101, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Jumeau, C.; Rupin, A.; Chieng-Yane, P.; Mougenot, N.; Zahr, N.; David-Dufilho, M.; Hatem, S.N. Direct Thrombin Inhibitors Prevent Left Atrial Remodeling Associated with Heart Failure in Rats. JACC Basic Transl. Sci. 2016, 1, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Cubedo, J. Hypercoagulability and atrial fibrillation: A two-way road? Eur. Heart J. 2017, 38, 51–52. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Spronk, H.M.; De Jong, A.M.; Verheule, S.; De Boer, H.C.; Maass, A.H.; Lau, D.H.; Rienstra, M.; van Hunnik, A.; Kuiper, M.; Lumeij, S.; et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur. Heart J. 2017, 38, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Inoue, H.; Hirai, T.; Iwasa, A.; Okumura, K.; Lee, J.D.; Shimizu, A.; Hayano, M.; Yano, K. D-dimer level influences thromboembolic events in patients with atrial fibrillation. Int. J. Cardiol. 2006, 109, 59–65. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Wang, W.; Qu, J.; Croft, L.; Degen, J.L.; Coller, B.S.; Ahamed, J. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012, 119, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFβ. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef] [PubMed]
- Rui, S.; Yuan, Y.; Du, C.; Song, P.; Chen, Y.; Wang, H.; Fan, Y.; Armstrong, D.G.; Deng, W.; Li, L. Comparison and Investigation of Exosomes Derived from Platelet-Rich Plasma Activated by Different Agonists. Cell. Transplant. 2021, 30, 9636897211017833. [Google Scholar] [CrossRef] [PubMed]
- Koretsune, Y.; Yamashita, T.; Akao, M.; Atarashi, H.; Ikeda, T.; Okumura, K.; Shimizu, W.; Suzuki, S.; Tsutsui, H.; Toyoda, K.; et al. Coagulation Biomarkers and Clinical Outcomes in Elderly Patients with Nonvalvular Atrial Fibrillation: ANAFIE Subcohort Study. JACC Asia 2023, 3, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Li-Saw-Hee, F.L.; Blann, A.D.; Lip, G.Y. Effects of fixed low-dose warfarin, aspirin-warfarin combination therapy, and dose-adjusted warfarin on thrombogenesis in chronic atrial fibrillation. Stroke 2000, 31, 828–833. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conway, D.S.; Pearce, L.A.; Chin, B.S.; Hart, R.G.; Lip, G.Y. Plasma von Willebrand factor and soluble p-selectin as indices of endothelial damage and platelet activation in 1321 patients with nonvalvular atrial fibrillation: Relationship to stroke risk factors. Circulation 2002, 106, 1962–1967. [Google Scholar] [CrossRef] [PubMed]
- Shea, B.S.; Probst, C.K.; Brazee, P.L.; Rotile, N.J.; Blasi, F.; Weinreb, P.H.; Black, K.E.; Sosnovik, D.E.; Van Cott, E.M.; Violette, S.M.; et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2017, 2, e86608. [Google Scholar] [CrossRef] [PubMed]
- Petzold, T.; Thienel, M.; Dannenberg, L.; Mourikis, P.; Helten, C.; Ayhan, A.; M’Pembele, R.; Achilles, A.; Trojovky, K.; Konsek, D.; et al. Rivaroxaban Reduces Arterial Thrombosis by Inhibition of FXa-Driven Platelet Activation via Protease Activated Receptor-1. Circ. Res. 2020, 126, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D.; Ageno, W.; Castellucci, L.A.; Chiasakul, T.; Eslick, R.; Ferreiro, J.L.; Gailani, D.; Gorog, D.A.; Lip, G.Y.H.; Raffini, L.; et al. Recommendation on the nomenclature for anticoagulants: Updated communication from the International Society on Thrombosis and Haemostasis Scientific and Standardization Commitee on the Control of Anticoagulation. J. Thromb. Haemost. 2023, 21, 1381–1384. [Google Scholar] [CrossRef] [PubMed]
- Koulas, I.; Spyropoulos, A.C. A Review of FXIa Inhibition as a Novel Target for Anticoagulation. Hamostaseologie 2023, 43, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Ninni, S.; Nattel, S. Factor XIa inhibition in atrial fibrillation: Insights and knowledge gaps emerging from the PACIFIC-AF trial. Cardiovasc. Res. 2023, 119, e111–e114. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Toennis, T.; Goette, A.; Camm, A.J.; Diener, H.C.; Becher, N.; Bertaglia, E.; Blomstrom Lundqvist, C.; Borlich, M.; Brandes, A.; et al. Anticoagulation with Edoxaban in Patients with Atrial High-Rate Episodes. N. Engl. J. Med. 2023, 389, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, J.H.; Diederichsen, S.Z.; Højberg, S.; Krieger, D.W.; Graff, C.; Kronborg, C.; Olesen, M.S.; Nielsen, J.B.; Holst, A.G.; Brandes, A.; et al. Implantable loop recorder detection of atrial fibrillation to prevent stroke (The LOOP Study): A randomised controlled trial. Lancet 2021, 398, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Hatem, S.N.; Cohen, A. Atrial fibrillation and stroke: Are we looking in the right direction? Cardiovasc. Res. 2022, 118, e4–e5. [Google Scholar] [CrossRef] [PubMed]
- Freestone, B.; Chong, A.Y.; Blann, A.D.; Lip, G.Y. The effects of direct current cardioversion for persistent atrial fibrillation on indices of endothelial damage/dysfunction. Thromb. Res. 2006, 118, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Willoughby, S.R.; Schultz, C.; Chakrabarty, A.; Alasady, M.; Lau, D.H.; Roberts-Thomson, K.C.; Worthley, M.I.; Young, G.D.; Sanders, P. Successful catheter ablation decreases platelet activation and improves endothelial function in patients with atrial fibrillation. Heart Rhythm 2014, 11, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Makowski, M.; Smorag, I.; Makowska, J.; Bissinger, A.; Grycewicz, T.; Paśnik, J.; Kidawa, M.; Lubiński, A.; Zielińska, M.; Baj, Z. Platelet reactivity and mean platelet volume as risk markers of thrombogenesis in atrial fibrillation. Int. J. Cardiol. 2017, 235, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, D.; Uysal, B.A.; Aksoy, F.; Kaya, S.; Icli, A.; Ceyhan, B.M.; Ozaydin, M. Strict heart rate control attenuates prothrombotic state and platelet activity in patients with non-valvular permanent atrial fibrillation. Clin. Hemorheol. Microcirc. 2014, 56, 219–229. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group | Patients with FDAF | ||
|---|---|---|---|
| (n = 20) | (n = 80) | p-Value | |
| Male/Female | 50%/50% | 62.5%/37.5% | n.s. |
| CHA2DS2-VASc | 3.45 | 3.98 | n.s. |
| History of Heart Failure | 20% | 26% | n.s. |
| Hypertension | 85% | 87.5% | n.s. |
| Age (years) | |||
| <65 | 45% | 27.5% | n.s. |
| 65–75 | 30% | 32.5% | n.s. |
| >75 | 25% | 40% | n.s. |
| Diabetes | 25% | 30% | n.s. |
| History of TIA/Stroke | 5% | 10% | n.s. |
| Body Weight (kg) | 82.95 | 85.93 | n.s. |
| BMI kg/m2 | 27.97 | 27.55 | n.s. |
| Previous or Ongoing Anticoagulation | 0% | 0% | n.s. |
| ASA | 10% | 10% | n.s. |
| FDAF | |||
| Hospital discharge in SR | - | 100% | |
| Follow-up | |||
| Years | 1 | 2.99 | |
| MACE | 0% | 41.25% | |
| AF | 0% | 22.5% | |
| HF | 0% | 8.75% | |
| TIA/Stroke | 0% | 8.75% | |
| ACS | 0% | 12.5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friebel, J.; Wegner, M.; Blöbaum, L.; Schencke, P.-A.; Jakobs, K.; Puccini, M.; Ghanbari, E.; Lammel, S.; Thevathasan, T.; Moos, V.; et al. Characterization of Biomarkers of Thrombo-Inflammation in Patients with First-Diagnosed Atrial Fibrillation. Int. J. Mol. Sci. 2024, 25, 4109. https://doi.org/10.3390/ijms25074109
Friebel J, Wegner M, Blöbaum L, Schencke P-A, Jakobs K, Puccini M, Ghanbari E, Lammel S, Thevathasan T, Moos V, et al. Characterization of Biomarkers of Thrombo-Inflammation in Patients with First-Diagnosed Atrial Fibrillation. International Journal of Molecular Sciences. 2024; 25(7):4109. https://doi.org/10.3390/ijms25074109
Chicago/Turabian StyleFriebel, Julian, Max Wegner, Leon Blöbaum, Philipp-Alexander Schencke, Kai Jakobs, Marianna Puccini, Emily Ghanbari, Stella Lammel, Tharusan Thevathasan, Verena Moos, and et al. 2024. "Characterization of Biomarkers of Thrombo-Inflammation in Patients with First-Diagnosed Atrial Fibrillation" International Journal of Molecular Sciences 25, no. 7: 4109. https://doi.org/10.3390/ijms25074109
APA StyleFriebel, J., Wegner, M., Blöbaum, L., Schencke, P.-A., Jakobs, K., Puccini, M., Ghanbari, E., Lammel, S., Thevathasan, T., Moos, V., Witkowski, M., Landmesser, U., & Rauch-Kröhnert, U. (2024). Characterization of Biomarkers of Thrombo-Inflammation in Patients with First-Diagnosed Atrial Fibrillation. International Journal of Molecular Sciences, 25(7), 4109. https://doi.org/10.3390/ijms25074109