
Integrated Untargeted Metabolome, Full-Length Sequencing and Transcriptome Analyses Reveal the Mechanism of Flavonoid Biosynthesis in Blueberry (Vaccinium spp.) Fruit

 ,
,
Abstract
1. Introduction
2. Results
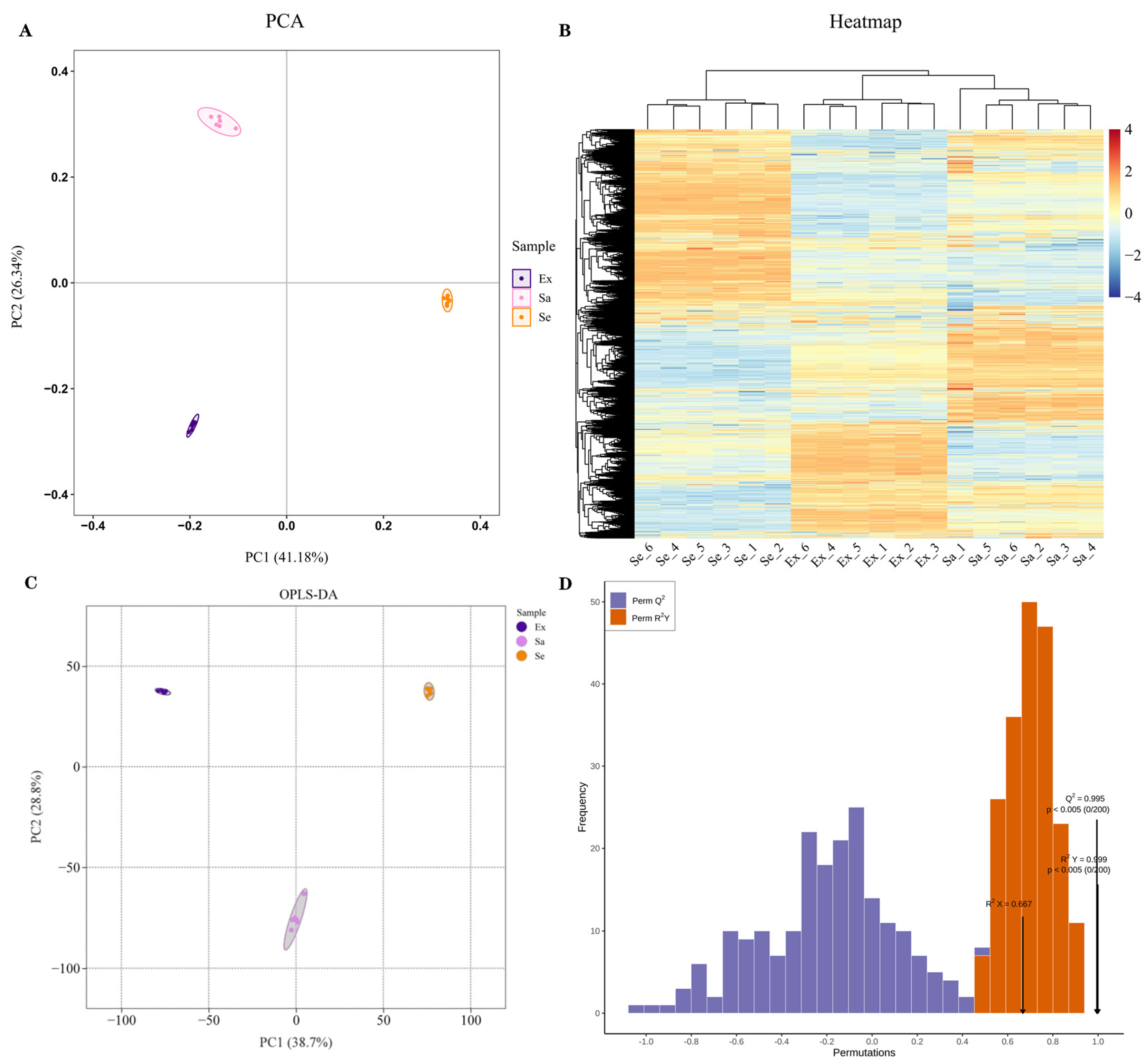
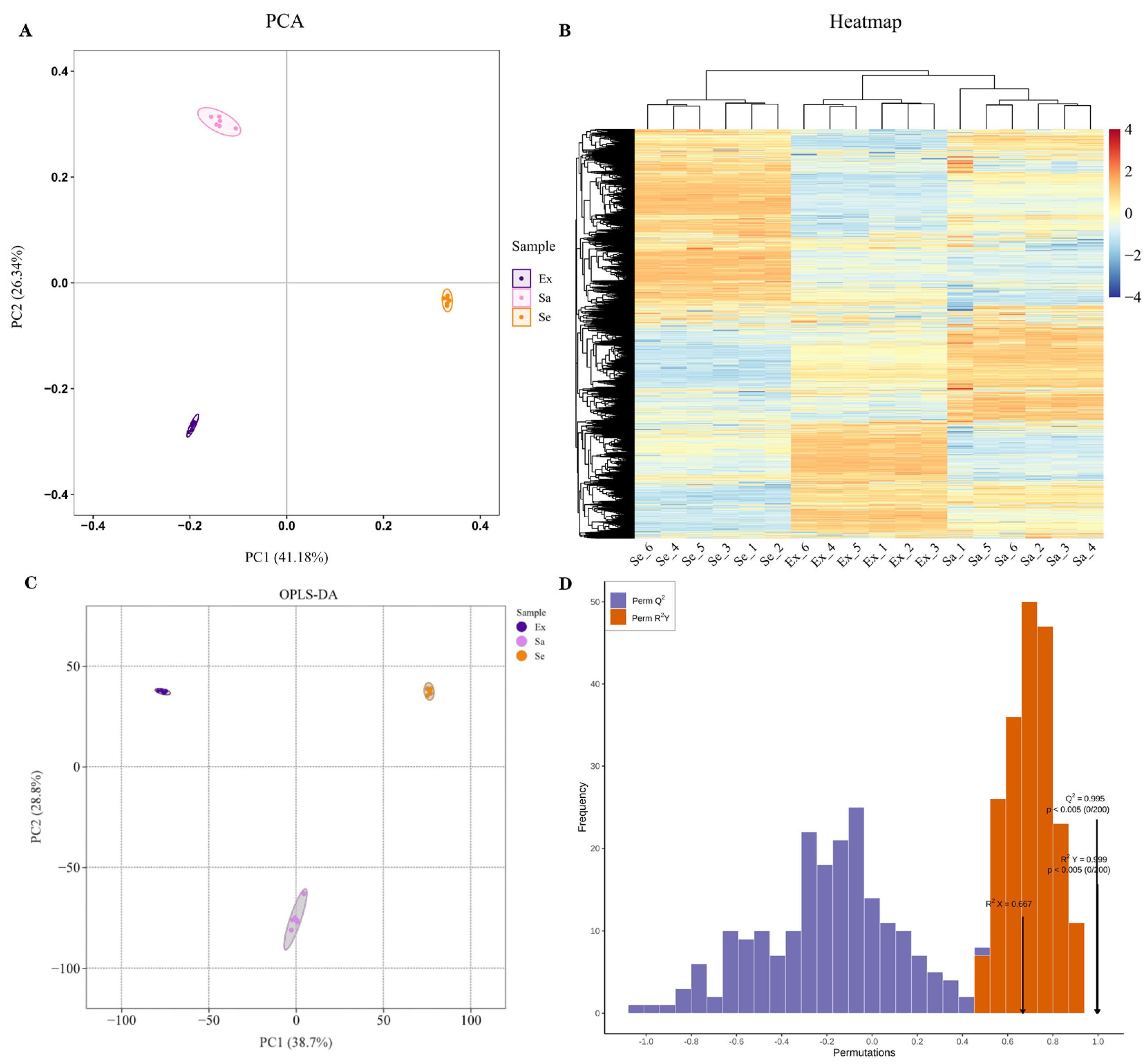
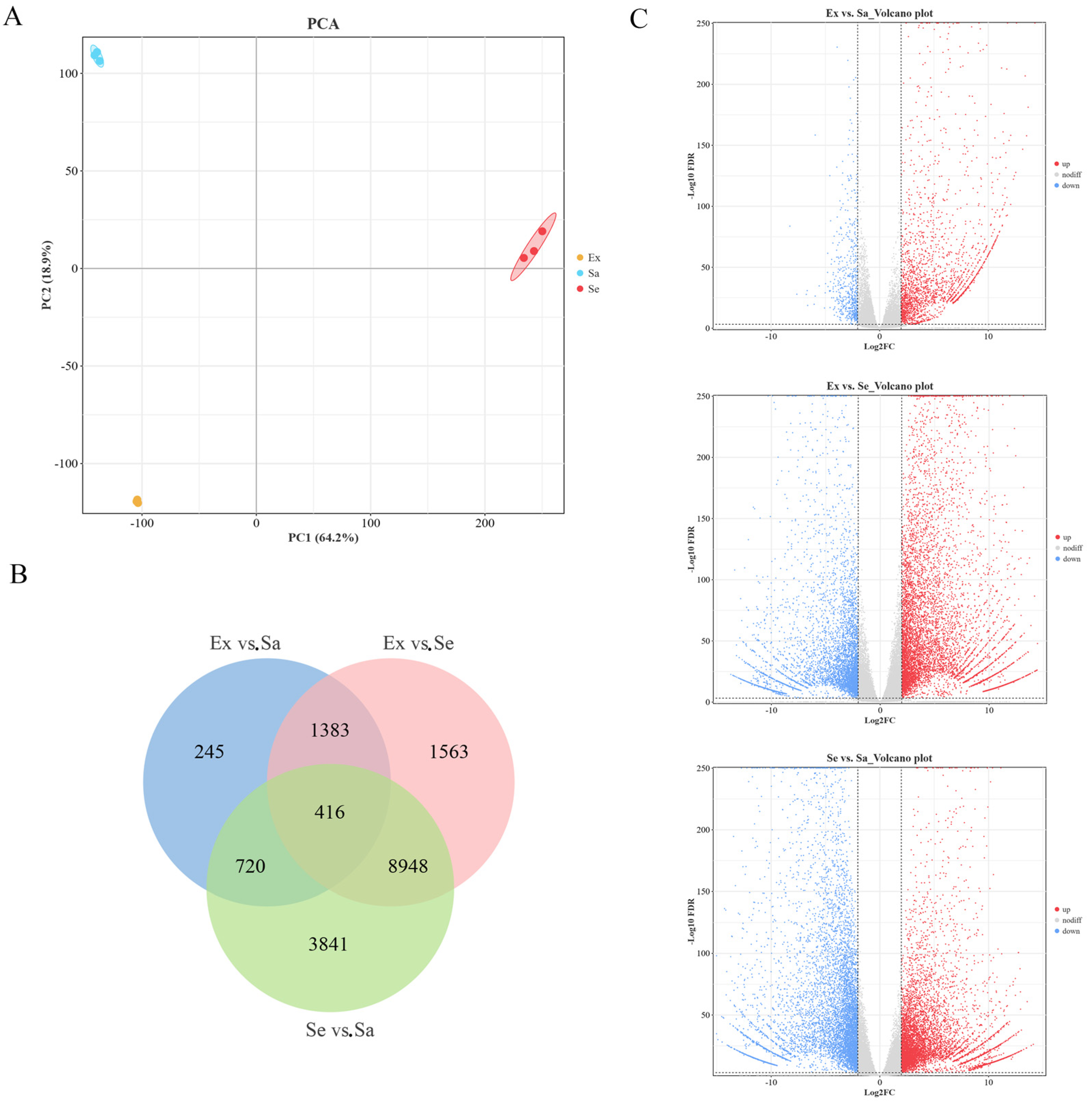
2.1. Untargeted Metabolomic Analysis: Differentially Expressed Metabolites in Blueberry Fruit Tissues
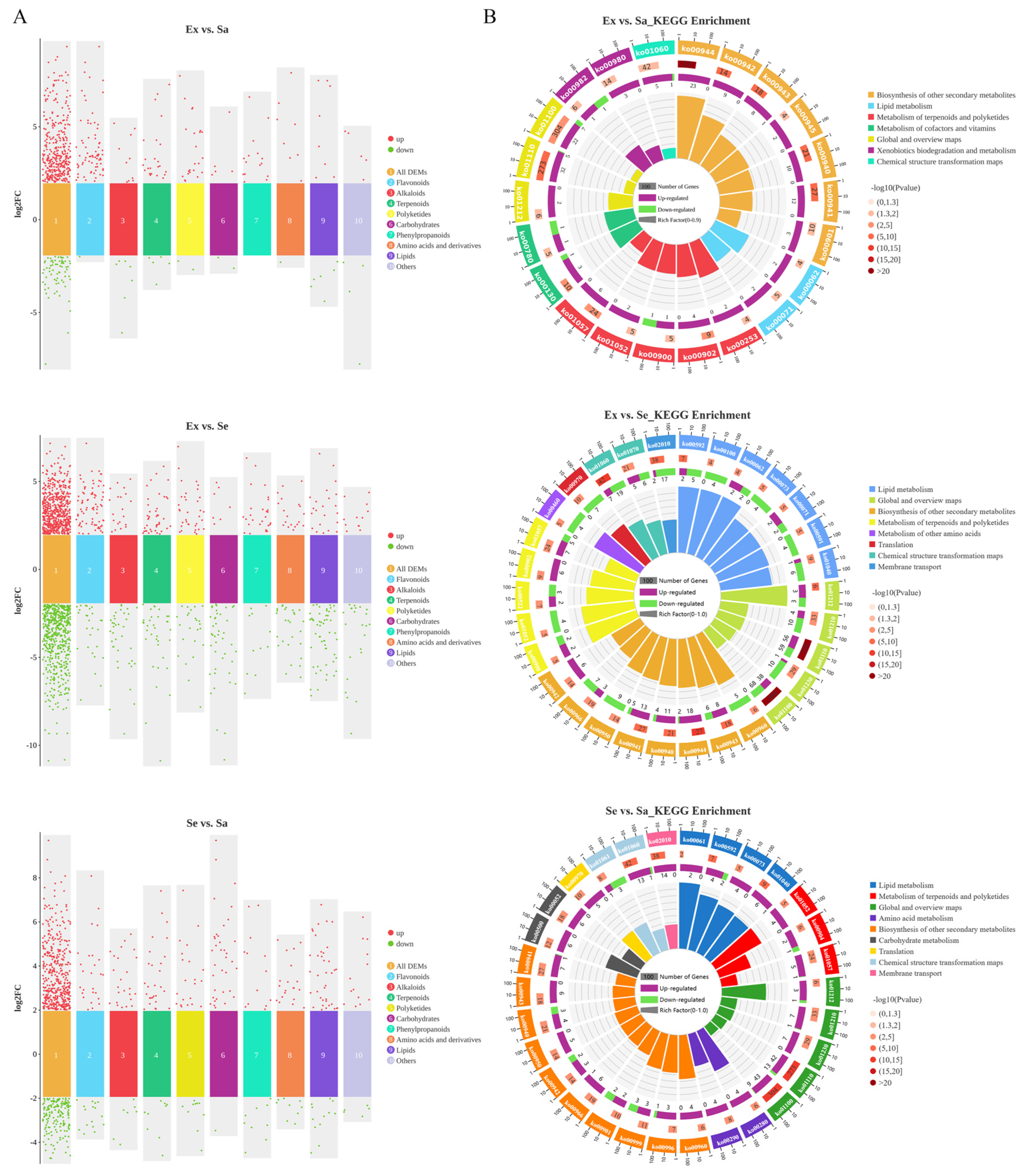
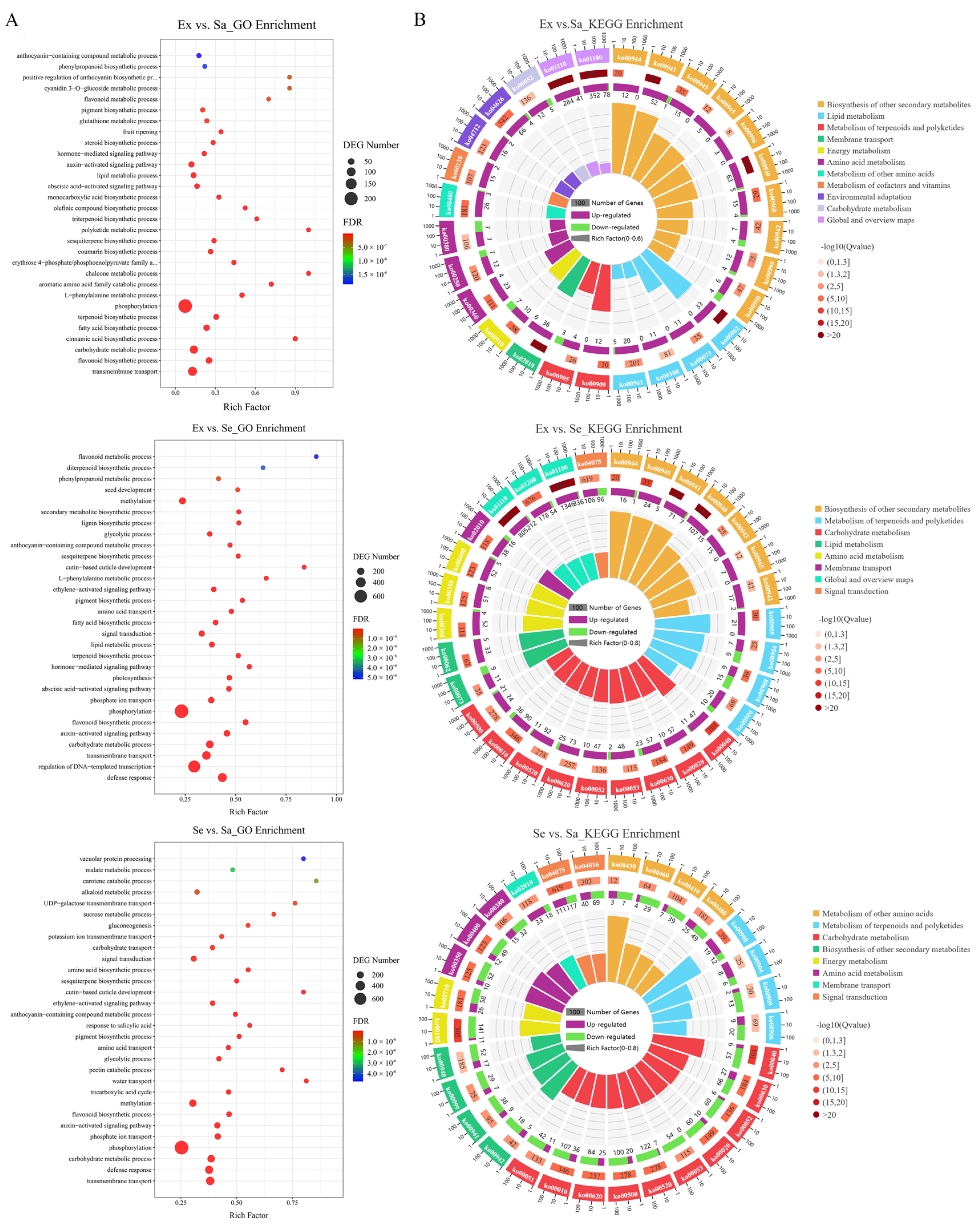
2.2. Classification Statistics and KEGG Enrichment Analysis of DEMs
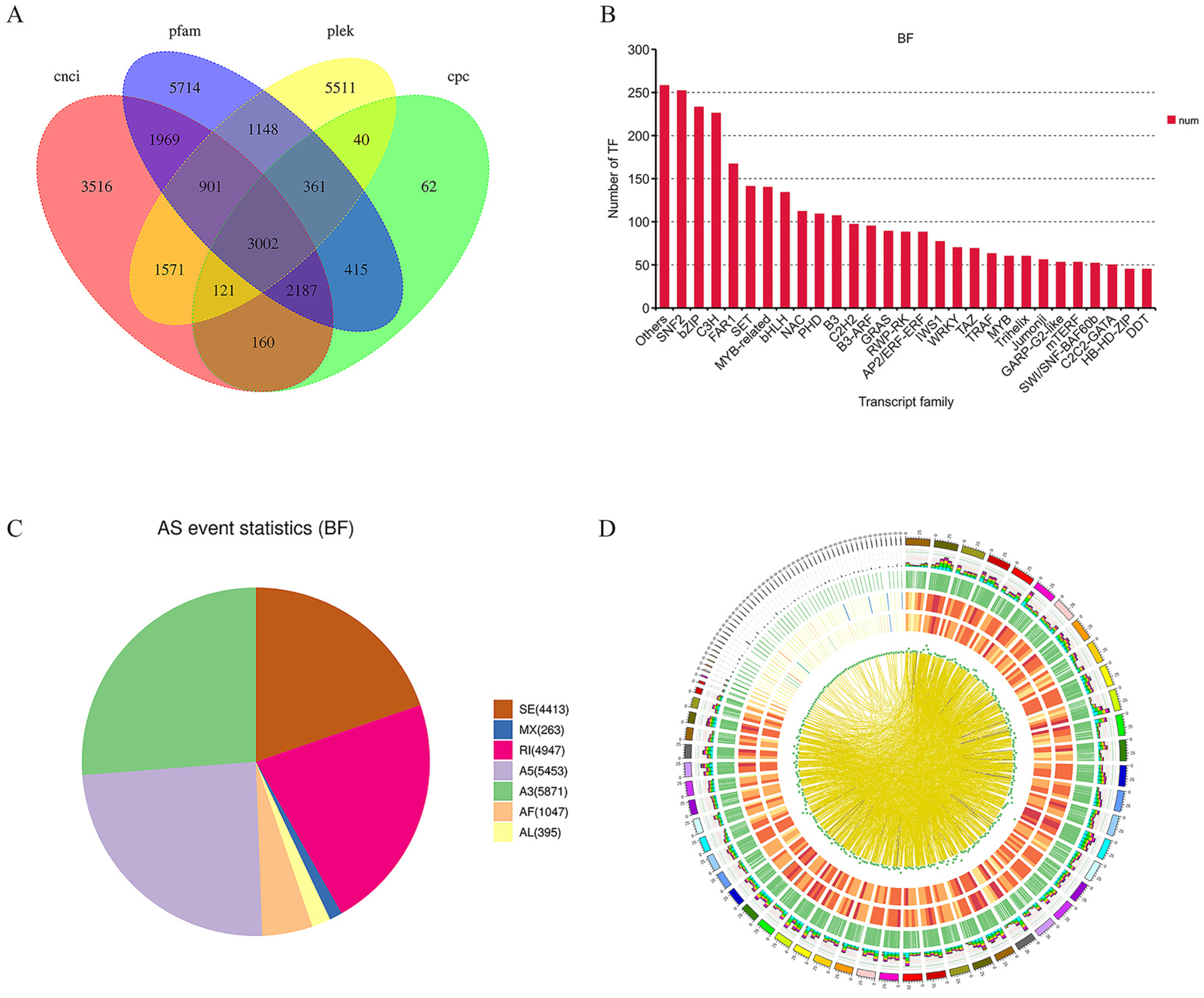
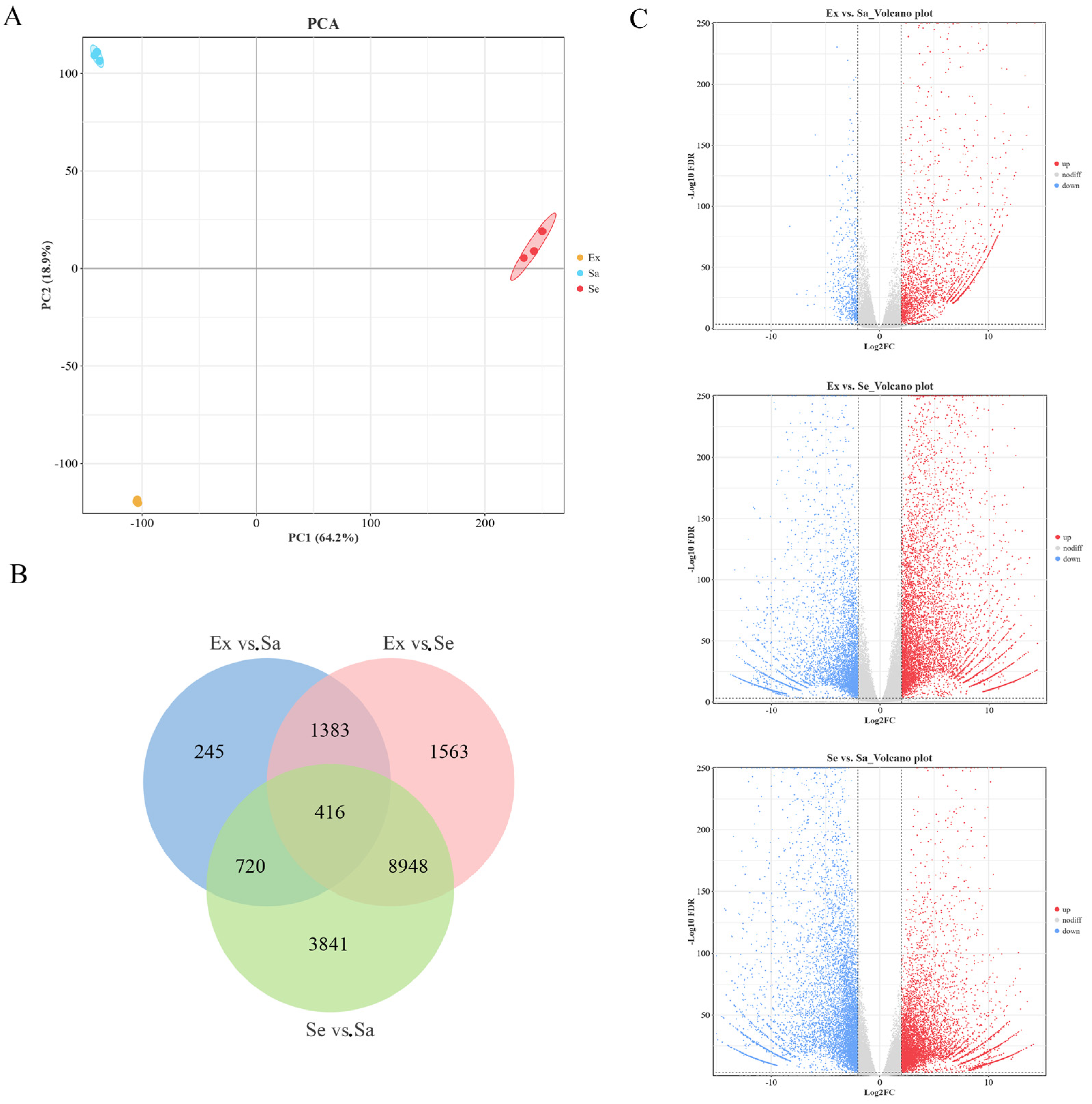
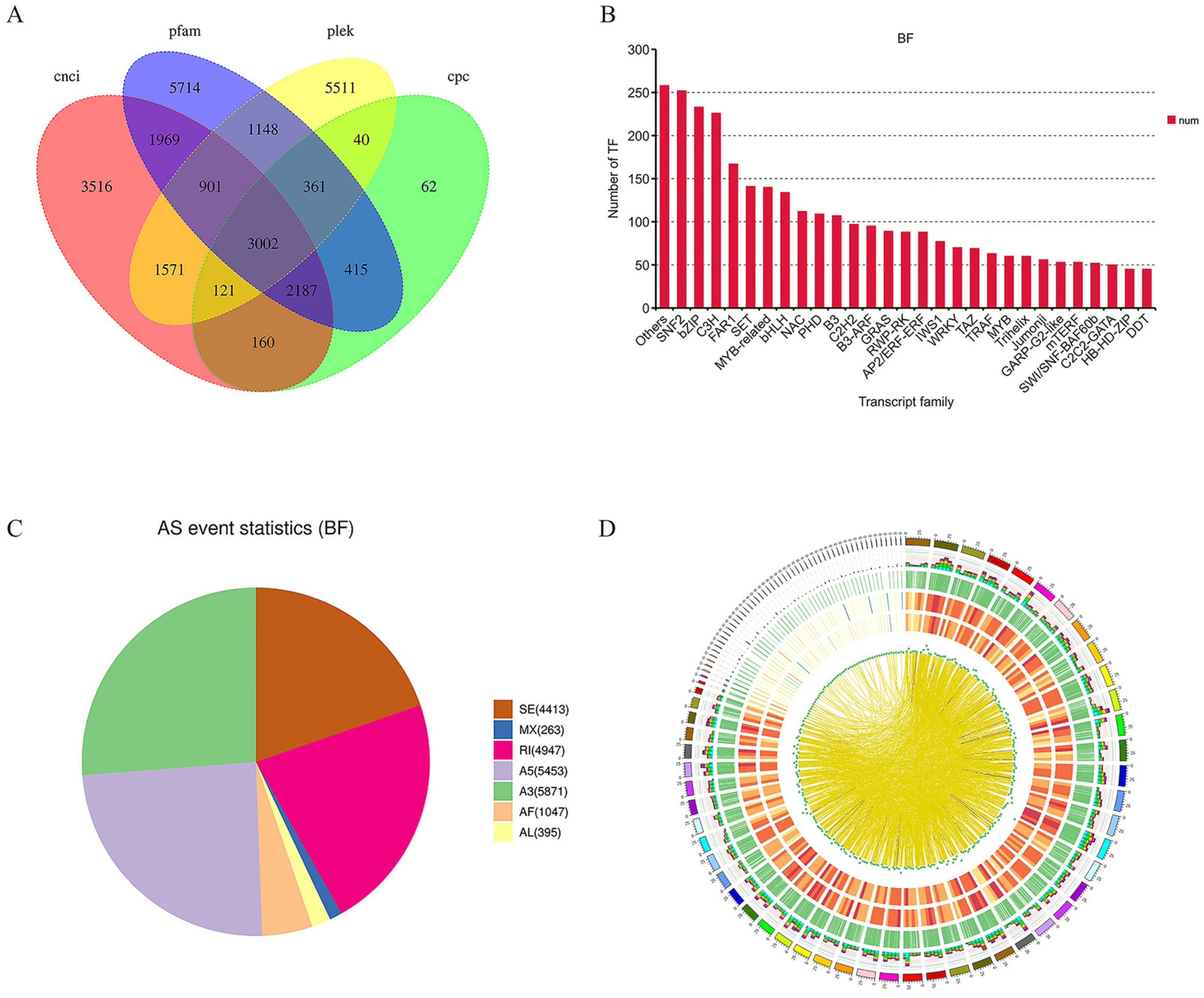
2.3. Full-Length Transcriptome Analysis of Mature Fruit
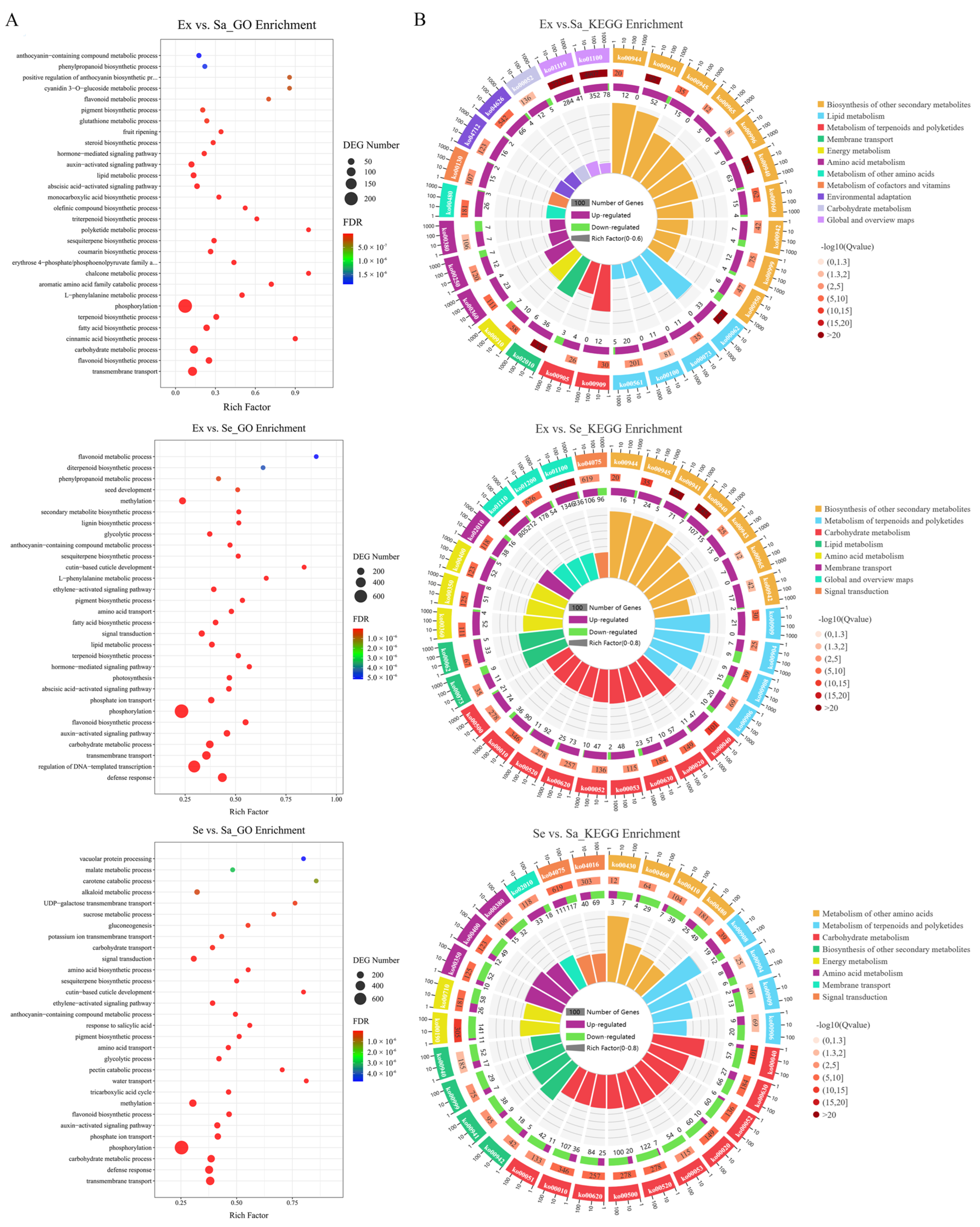
2.4. Identification and Functional Enrichment Analysis of DEGs
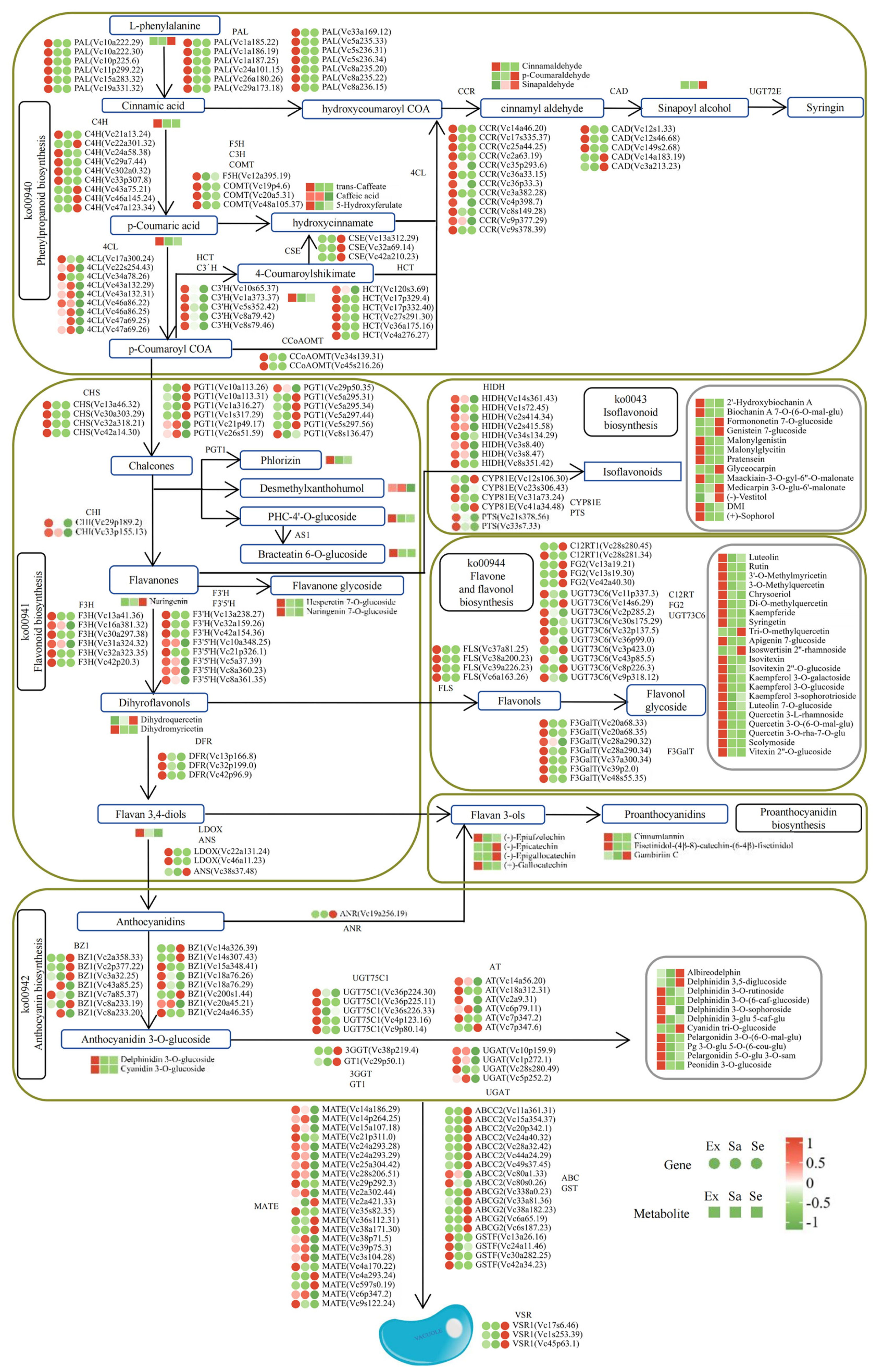
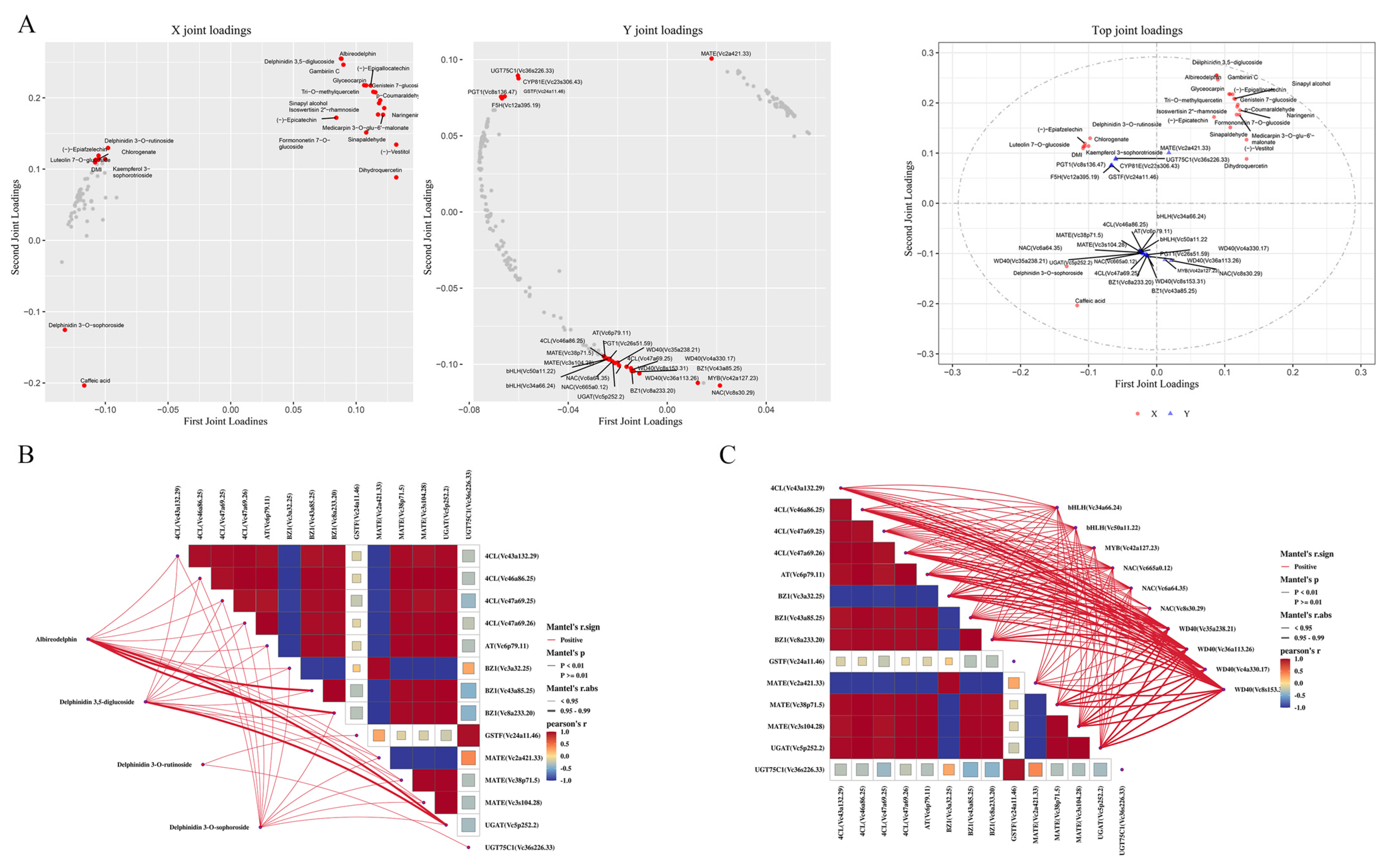
2.5. Integrated Transcriptome and Metabolome Analysis
2.6. Candidate Hub Genes and Metabolites Related to Flavonoid Biosynthesis
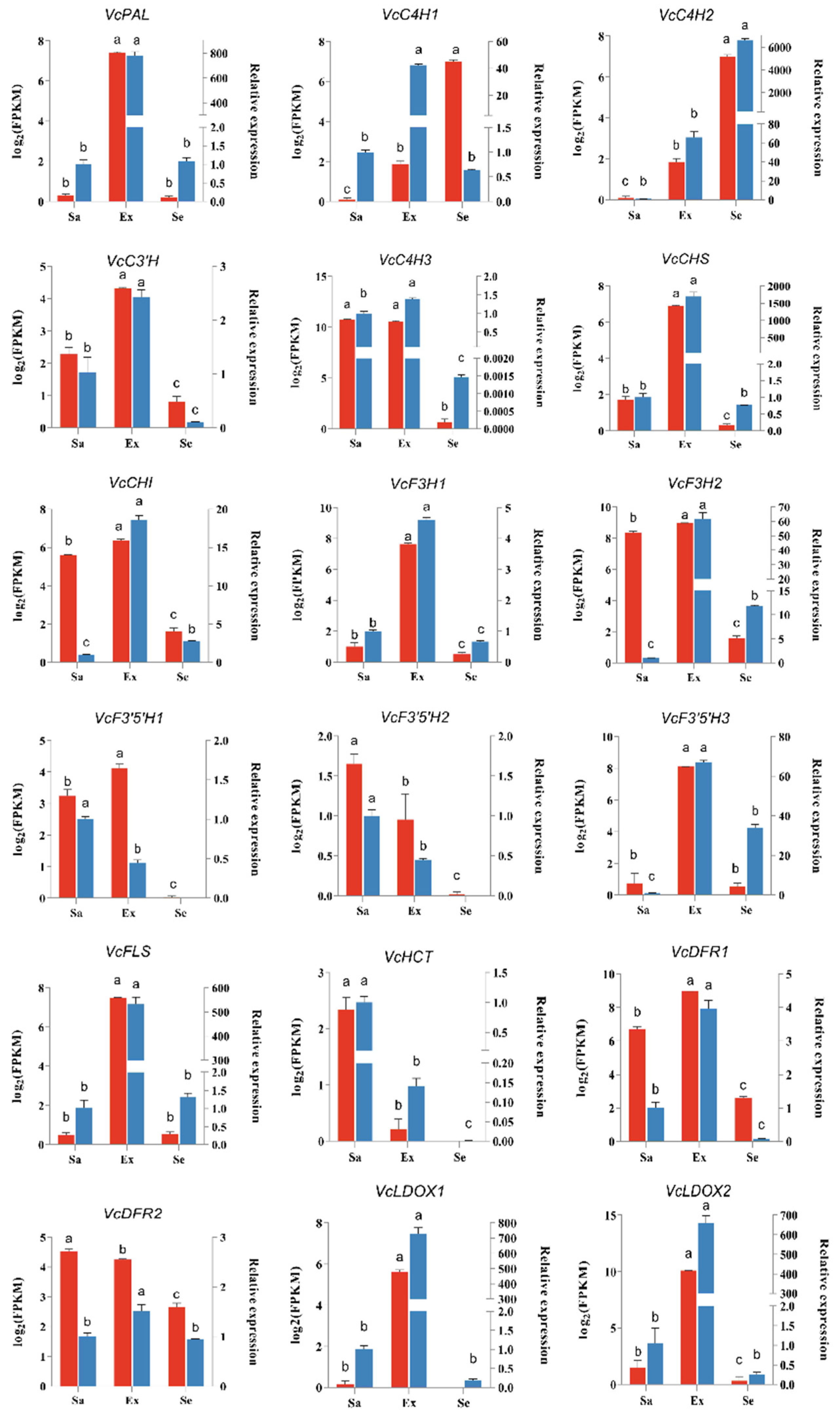
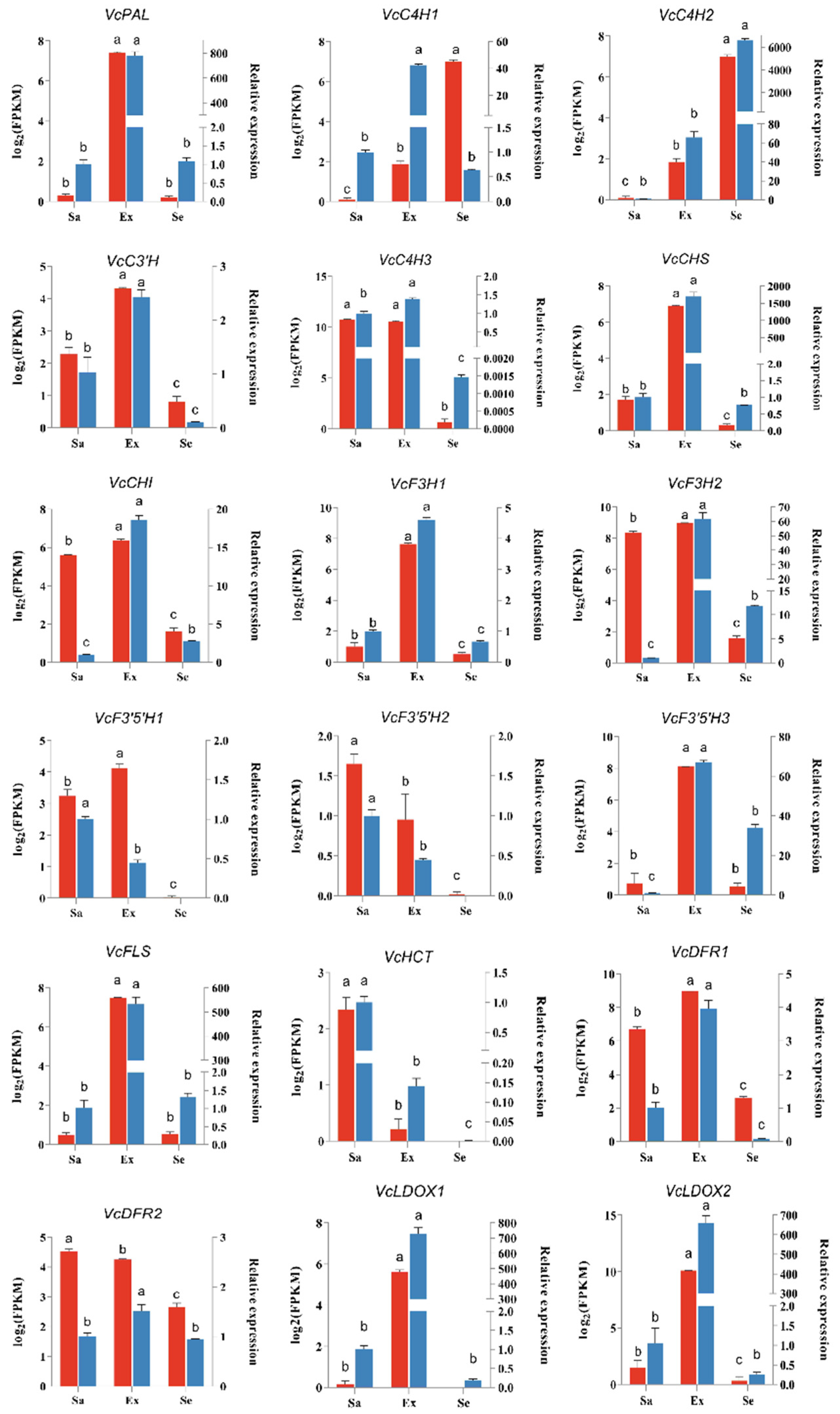
2.7. Quantitative Real-Time PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Metabolite Sequencing and Analysis
4.3. PacBio Iso-Seq and RNA-Seq Library Construction and DEG Analysis
4.4. Gene Functional Annotation
4.5. Integrated Metabolomic and Transcriptomic Analyses
4.6. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Retamales, J.B.; Hancock, J.F. Blueberry taxonomy and breeding. In Blueberries, 2nd ed.; CABI: Wallington, NJ, USA, 2018; pp. 18–60. [Google Scholar]
- Rodriguez-Saona, C.; Vincent, C.; Isaacs, R. Blueberry IPM: Past Successes and Future Challenges. Annu. Rev. Entomol. 2019, 64, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Balandrano, D.D.; Chai, Z.; Beta, T.; Feng, J.; Huang, W. Blueberry anthocyanins: An updated review on approaches to enhancing their bioavailability. Trends Food Sci. Technol. 2021, 118, 808–821. [Google Scholar] [CrossRef]
- Felgus-Lavefve, L.; Howard, L.; Adams, S.H.; Baum, J.I. The Effects of Blueberry Phytochemicals on Cell Models of Inflammation and Oxidative Stress. Adv. Nutr. Int. Rev. J. 2021, 13, 1279–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, B.; Dong, K.; Li, J.; Li, Y.; Sun, H. Identification and quantification of anthocyanins of 62 blueberry cultivars via UPLC-MS. Biotechnol. Biotechnol. Equip. 2022, 36, 587–597. [Google Scholar] [CrossRef]
- Cho, M.J.; Howard, L.R.; Prior, R.L.; Clark, J.R. Flavonol glycosides and antioxidant capacity of various blackberry and blueberry genotypes determined by high-performance liquid chromatography/mass spectrometry. J. Sci. Food Agric. 2005, 85, 2149–2158. [Google Scholar] [CrossRef]
- Vrhovsek, U.; Masuero, D.; Palmieri, L.; Mattivi, F. Identification and quantification of flavonol glycosides in cultivated blueberry cultivars. J. Food Compos. Anal. 2011, 25, 9–16. [Google Scholar] [CrossRef]
- Rodriguez-Mateos, A.; Cifuentes-Gomez, T.; Tabatabaee, S.; Lecras, C.; Spencer, J.P.E. Procyanidin, Anthocyanin, and Chlorogenic Acid Contents of Highbush and Lowbush Blueberries. J. Agric. Food Chem. 2012, 60, 5772–5778. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, L.; Liu, X.; Hasan, K.F.; Li, H.; Zhou, S.; Zhang, Q.; Zhou, Y. Effect of thermosonication treatment on blueberry juice quality: Total phenolics, flavonoids, anthocyanin, and antioxidant activity. LWT 2021, 150, 112021. [Google Scholar] [CrossRef]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: Structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Luan, A.; Zhang, W.; Yang, M.; Zhong, Z.; Wu, J.; He, Y.; He, J. Unveiling the molecular mechanism involving anthocyanins in pineapple peel discoloration during fruit maturation. Food Chem. 2023, 412, 135482. [Google Scholar] [CrossRef]
- Ju, Y.; Wang, W.; Yue, X.; Xue, W.; Zhang, Y.; Fang, Y. Integrated metabolomic and transcriptomic analysis reveals the mechanism underlying the accumulation of anthocyanins and other flavonoids in the flesh and skin of teinturier grapes. Plant Physiol. Biochem. 2023, 197, 107667. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Zhu, C.; Qiu, D.; Mao, G.; Mueller-Roeber, B.; Zeng, J. Integrated transcriptomic and metabolomic analyses reveal key genes controlling flavonoid biosynthesis in Citrus grandis ‘Tomentosa’ fruits. Plant Physiol. Biochem. 2023, 196, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Liu, Z.; Wang, L.; Lin-Wang, K.; Zhu, J.; Bi, Z.; Sun, C.; Zhang, J.; Bai, J. Integrative analysis of metabolome and transcriptome reveals a dynamic regulatory network of potato tuber pigmentation. iScience 2023, 26, 105903. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, Y.; Li, B.; Tan, H.; Li, D.; Li, L.; Liu, X.; Han, J.; Meng, X. Comparative transcriptome analysis of genes involved in anthocyanin synthesis in blueberry. Plant Physiol. Biochem. 2018, 127, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Colle, M.; Leisner, C.P.; Wai, C.M.; Ou, S.; Bird, K.A.; Wang, J.; Wisecaver, J.H.; Yocca, A.E.; Alger, E.I.; Tang, H.; et al. Haplotype-phased genome and evolution of phytonutrient pathways of tetraploid blueberry. GigaScience 2019, 8, giz012. [Google Scholar] [CrossRef]
- Mengist, M.F.; Grace, M.H.; Xiong, J.; Kay, C.D.; Bassil, N.; Hummer, K.; Ferruzzi, M.G.; Lila, M.A.; Iorizzo, M. Diversity in Metabolites and Fruit Quality Traits in Blueberry Enables Ploidy and Species Differentiation and Establishes a Strategy for Future Genetic Studies. Front. Plant Sci. 2020, 11, 370. [Google Scholar] [CrossRef]
- Guo, X.; Shakeel, M.; Wang, D.; Qu, C.; Yang, S.; Ahmad, S.; Song, Z. Metabolome and transcriptome profiling unveil the mechanisms of light-induced anthocyanin synthesis in rabbiteye blueberry (Vaccinium ashei: Reade). BMC Plant Biol. 2022, 22, 223. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, L.; Määttä, K.; Pirttilä, A.M.; Törrönen, R.; Kärenlampi, S.; Hohtola, A. Expression of Genes Involved in Anthocyanin Biosynthesis in Relation to Anthocyanin, Proanthocyanidin, and Flavonol Levels during Bilberry Fruit Development. Plant Physiol. 2002, 130, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, D.J.; Espley, R.V.; Deng, C.H.; Dare, A.P.; Günther, C.S.; Jaakola, L.; Karppinen, K.; Boase, M.R.; Wang, L.; Luo, H.; et al. The Coordinated Action of MYB Activators and Repressors Controls Proanthocyanidin and Anthocyanin Biosynthesis in Vaccinium. Front. Plant Sci. 2022, 13, 910155. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, F.; Wang, B.; Wu, H.; Wu, J.; Liu, J.; Sun, Y.; Cheng, C.; Qiu, D. Identification, characterization and expression analysis of anthocyanin biosynthesis-related bHLH genes in blueberry (Vaccinium corymbosum L.). Int. J. Mol. Sci. 2021, 22, 13274. [Google Scholar] [CrossRef]
- Zhao, M.; Li, J.; Zhu, L.; Chang, P.; Li, L.; Zhang, L. Identification and characterization of MYB-bHLH-WD40 regulatory complex members controlling anthocyanidin biosynthesis in blueberry fruits development. Genes 2019, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2007, 53, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.J.; Zuo, H.; Xu, Q. Genomic insights into citrus domestication and its important agronomic traits. Plant Commun. 2020, 2, 100138. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jiang, S.; Zhang, T.; Xu, H.; Fang, H.; Zhang, J.; Su, M.; Wang, Y.; Zhang, Z.; Wang, N.; et al. Apple NAC transcription factor MdNAC52 regulates biosynthesis of anthocyanin and proanthocyanidin through MdMYB9 and MdMYB11. Plant Sci. 2019, 289, 110286. [Google Scholar] [CrossRef] [PubMed]
- An, J.-P.; Zhang, X.-W.; Bi, S.-Q.; You, C.-X.; Wang, X.-F.; Hao, Y.-J. The ERF transcription factor MdERF38 promotes drought stress-induced anthocyanin biosynthesis in apple. Plant J. 2019, 101, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Liu, Y.; Luo, Y.; Cui, Y.; Lu, C.; Li, H.; Huang, H.; Dai, S. Functional analysis of the ScAG and ScAGL11 MADS-box transcription factors for anthocyanin biosynthesis and bicolour pattern formation in Senecio cruentus ray florets. Hortic. Res. 2022, 9, uhac071. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, X.; Li, B.; Zhao, X.; Shen, Y.; Yuan, Z. Genome-wide identification and characterization of bZIP gene family and cloning of candidate genes for anthocyanin biosynthesis in pomegranate (Punica granatum). BMC Plant Biol. 2022, 22, 170. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Nambeesan, S.U. Full-length fruit transcriptomes of southern highbush (Vaccinium sp.) and rabbiteye (V. virgatum Ait.) blueberry. BMC Genom. 2022, 23, 733. [Google Scholar] [CrossRef]
- Lam, P.Y.; Wang, L.; Lo, C.; Zhu, F.-Y. Alternative Splicing and Its Roles in Plant Metabolism. Int. J. Mol. Sci. 2022, 23, 7355. [Google Scholar] [CrossRef]
- Lim, S.-H.; Kim, D.-H.; Jung, J.-A.; Lee, J.-Y. Alternative Splicing of the Basic Helix–Loop–Helix Transcription Factor Gene CmbHLH2 Affects Anthocyanin Biosynthesis in Ray Florets of Chrysanthemum (Chrysanthemum morifolium). Front. Plant Sci. 2021, 12, 669315. [Google Scholar] [CrossRef] [PubMed]
- Albert, N.W.; Iorizzo, M.; Mengist, M.F.; Montanari, S.; Zalapa, J.; Maule, A.; Edger, P.P.; Yocca, A.E.; Platts, A.E.; Pucker, B.; et al. Vaccinium as a comparative system for understanding of complex flavonoid accumulation profiles and regulation in fruit. Plant Physiol. 2023, 192, 1696–1710. [Google Scholar] [CrossRef] [PubMed]
- Rha, C.-S.; Jeong, H.W.; Park, S.; Lee, S.; Jung, Y.S.; Kim, D.-O. Antioxidative, Anti-Inflammatory, and Anticancer Effects of Purified Flavonol Glycosides and Aglycones in Green Tea. Antioxidants 2019, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Walle, T. Methylated Flavonoids Have Greatly Improved Intestinal Absorption and Metabolic Stability. Drug Metab. Dispos. 2006, 34, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Wang, S.; Li, J.; Bacha, S.A.S.; Xu, G. Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway in blueberry (Vaccinium spp.). Front. Plant Sci. 2023, 14, 1082245. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-P.; Xu, J.-G. Profiles of Carotenoids, Anthocyanins, Phenolics, and Antioxidant Activity of Selected Color Waxy Corn Grains during Maturation. J. Agric. Food Chem. 2011, 59, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef] [PubMed]
- Khodzhaieva, R.S.; Gladkov, E.S.; Kyrychenko, A.; Roshal, A.D. Progress and Achievements in Glycosylation of Flavonoids. Front. Chem. 2021, 9, 637994. [Google Scholar] [CrossRef] [PubMed]
- Hofer, B. Recent developments in the enzymatic O-glycosylation of flavonoids. Appl. Microbiol. Biotechnol. 2016, 100, 4269–4281. [Google Scholar] [CrossRef]
- Wen, L.; Jiang, Y.; Yang, J.; Zhao, Y.; Tian, M.; Yang, B. Structure, bioactivity, and synthesis of methylated flavonoids. Ann. N. Y. Acad. Sci. 2017, 1398, 120–129. [Google Scholar] [CrossRef]
- Chebil, L.; Humeau, C.; Falcimaigne, A.; Engasser, J.-M.; Ghoul, M. Enzymatic acylation of flavonoids. Process Biochem. 2006, 41, 2237–2251. [Google Scholar] [CrossRef]
- Mackon, E.; Mackon, G.C.J.D.E.; Yao, Y.; Guo, Y.; Ma, Y.; Dai, X.; Jandan, T.H.; Liu, P. Integrative HPLC profiling and transcriptome analysis revealed insights into anthocyanin accumulation and key genes at three developmental stages of black rice (Oryza sativa L.) caryopsis. Front. Plant Sci. 2023, 14, 1211326. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Giusti, M.M.; Stoner, G.D.; Schwartz, S.J. Screening for anthocyanins using high-performance liquid chromatography coupled to electrospray ionization tandem mass spectrometry with precursor-ion analysis, product-ion analysis, common-neutral-loss analysis, and selected reaction monitoring. J. Chromatogr. A 2005, 1091, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Mazza, G. Quantitation and Distribution of Simple and Acylated Anthocyanins and Other Phenolics in Blueberries. J. Food Sci. 1994, 59, 1057–1059. [Google Scholar] [CrossRef]
- Xu, C.; Min, J. Structure and function of WD40 domain proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Chen, L.; Cui, Y.; Yao, Y.; An, L.; Bai, Y.; Li, X.; Yao, X.; Wu, K. Genome-wide identification of WD40 transcription factors and their regulation of the MYB-bHLH-WD40 (MBW) complex related to anthocyanin synthesis in Qingke (Hordeum vulgare L. var. nudum Hook. f.). BMC Genom. 2023, 24, 166. [Google Scholar] [CrossRef] [PubMed]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.P. Genome-wide and constrained ordination-based analyses of EC code data support reclassification of the species of Massilia La Scola et al. 2000 into Telluria Bowman et al. 1993, Mokoshia gen. nov. and Zemynaea gen. nov. Int. J. Syst. Evol. Microbiol. 2023, 73, 005991. [Google Scholar] [CrossRef]
- Chen, S.; Komatsu, S. Plant Proteomic Research 4.0: Frontiers in Stress Resilience. Int. J. Mol. Sci. 2021, 22, 13362. [Google Scholar] [CrossRef]
- García, J.C.; Guadagno, A.; Paytuvi-Gallart, A.; Saera-Vila, A.; Amoroso, C.G.; D’esposito, D.; Andolfo, G.; Cigliano, R.A.; Sanseverino, W.; Ercolano, M.R. PRGdb 4.0: An updated database dedicated to genes involved in plant disease resistance process. Nucleic Acids Res. 2021, 50, D1483–D1490. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-M.; Wang, Z.; Wang, L.; Alejos-Gonzales, F.; Sun, M.-A.; Xie, D.-Y. A Genome-Wide Scenario of Terpene Pathways in Self-pollinated Artemisia annua. Mol. Plant 2015, 8, 1580–1598. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, P.; Sun, J.; Li, B.; Jia, J.; Yuan, J.; Li, X.; Gu, S.; Pang, X. CsMYC2 is involved in the regulation of phenylpropanoid biosynthesis induced by trypsin in cucumber (Cucumis sativus) during storage. Plant Physiol. Biochem. 2023, 196, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Chao, K.; Qi, T.; Wan, Q.; Li, T. Insights into the Flavor Differentiation between Two Wild Edible Boletus Species through Metabolomic and Transcriptomic Analyses. Foods 2023, 12, 2728. [Google Scholar] [CrossRef]
- Deng, Y.; Li, Y.; Sun, H. Selection of reference genes for RT-qPCR normalization in blueberry (Vaccinium corymbosum × an-gustifolium) under various abiotic stresses. FEBS Open Bio 2020, 10, 1418–1435. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PacBio Sequencing | |
|---|---|
| Subreads base (G) | 9.4 |
| Average subreads length (bp) | 1496 |
| FLNC | |
| CCS number | 390,855 |
| FLNC number | 267,687 |
| Mean length (bp) | 2518 |
| After correction by Illumina data | |
| Consensus number | 147,569 |
| Mean length/N50 (bp) | 2738/3176 |
| Mapped to the genome | 138,908 (94.13%) |
| After correction and de-redundancy | |
| High-quality isoform number | 63,425 |
| Isoforms of known genes number | 7584 |
| Novel isoform of known genes number | 49,005 |
| Isoforms of novel genes number | 6836 |
| Novel genes number | 5530 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Liu, X.; Chen, X.; Wang, B.; Dong, M.; Chen, L.; Yang, Z.; Li, Y.; Sun, H. Integrated Untargeted Metabolome, Full-Length Sequencing and Transcriptome Analyses Reveal the Mechanism of Flavonoid Biosynthesis in Blueberry (Vaccinium spp.) Fruit. Int. J. Mol. Sci. 2024, 25, 4137. https://doi.org/10.3390/ijms25084137
Tian Y, Liu X, Chen X, Wang B, Dong M, Chen L, Yang Z, Li Y, Sun H. Integrated Untargeted Metabolome, Full-Length Sequencing and Transcriptome Analyses Reveal the Mechanism of Flavonoid Biosynthesis in Blueberry (Vaccinium spp.) Fruit. International Journal of Molecular Sciences. 2024; 25(8):4137. https://doi.org/10.3390/ijms25084137
Chicago/Turabian StyleTian, Youwen, Xinlei Liu, Xuyang Chen, Bowei Wang, Mei Dong, Li Chen, Zhengsong Yang, Yadong Li, and Haiyue Sun. 2024. "Integrated Untargeted Metabolome, Full-Length Sequencing and Transcriptome Analyses Reveal the Mechanism of Flavonoid Biosynthesis in Blueberry (Vaccinium spp.) Fruit" International Journal of Molecular Sciences 25, no. 8: 4137. https://doi.org/10.3390/ijms25084137
APA StyleTian, Y., Liu, X., Chen, X., Wang, B., Dong, M., Chen, L., Yang, Z., Li, Y., & Sun, H. (2024). Integrated Untargeted Metabolome, Full-Length Sequencing and Transcriptome Analyses Reveal the Mechanism of Flavonoid Biosynthesis in Blueberry (Vaccinium spp.) Fruit. International Journal of Molecular Sciences, 25(8), 4137. https://doi.org/10.3390/ijms25084137