
Metabolic Considerations in Direct Procurement and Perfusion Protocols with DCD Heart Transplantation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. DCD as a Solution to Cardiac Graft Shortage
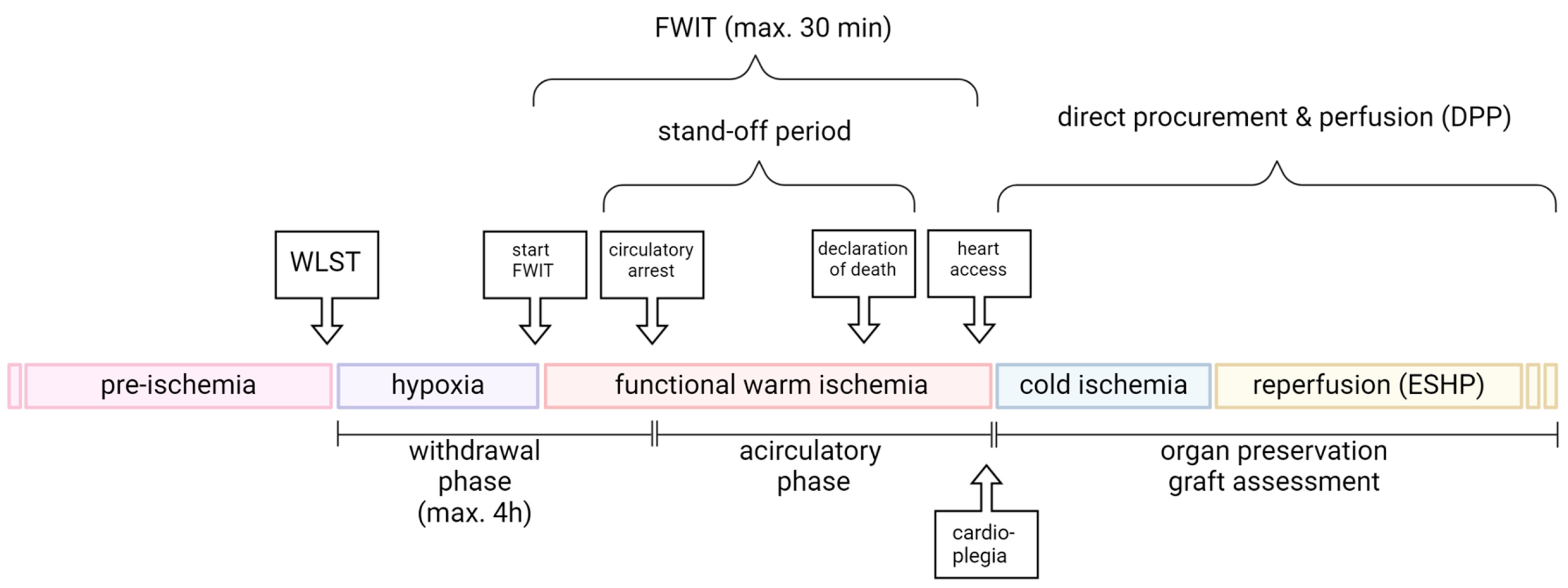
2. Sequence of Events in the DCD Donor
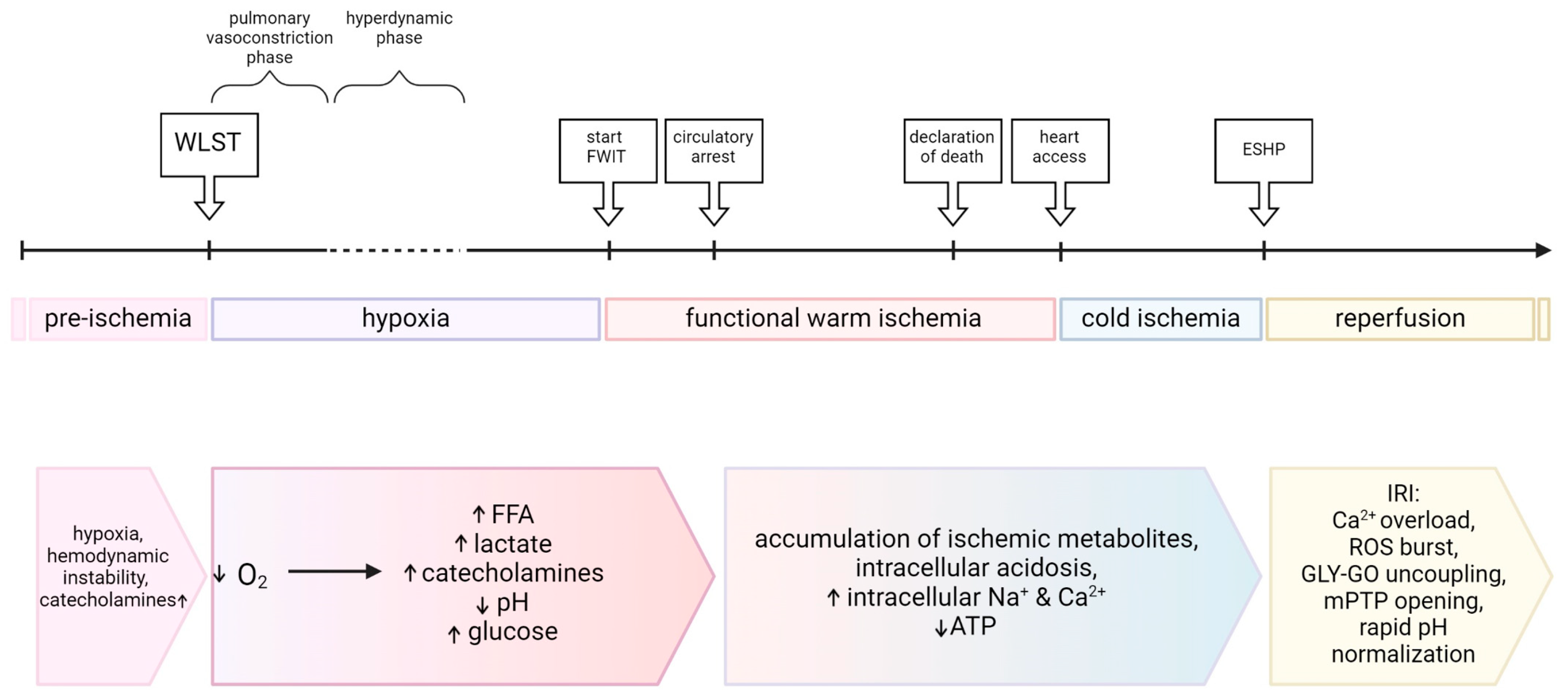
2.1. Withdrawal Phase
2.2. Warm Ischemic Time (WIT)
2.3. Hemodynamic Changes
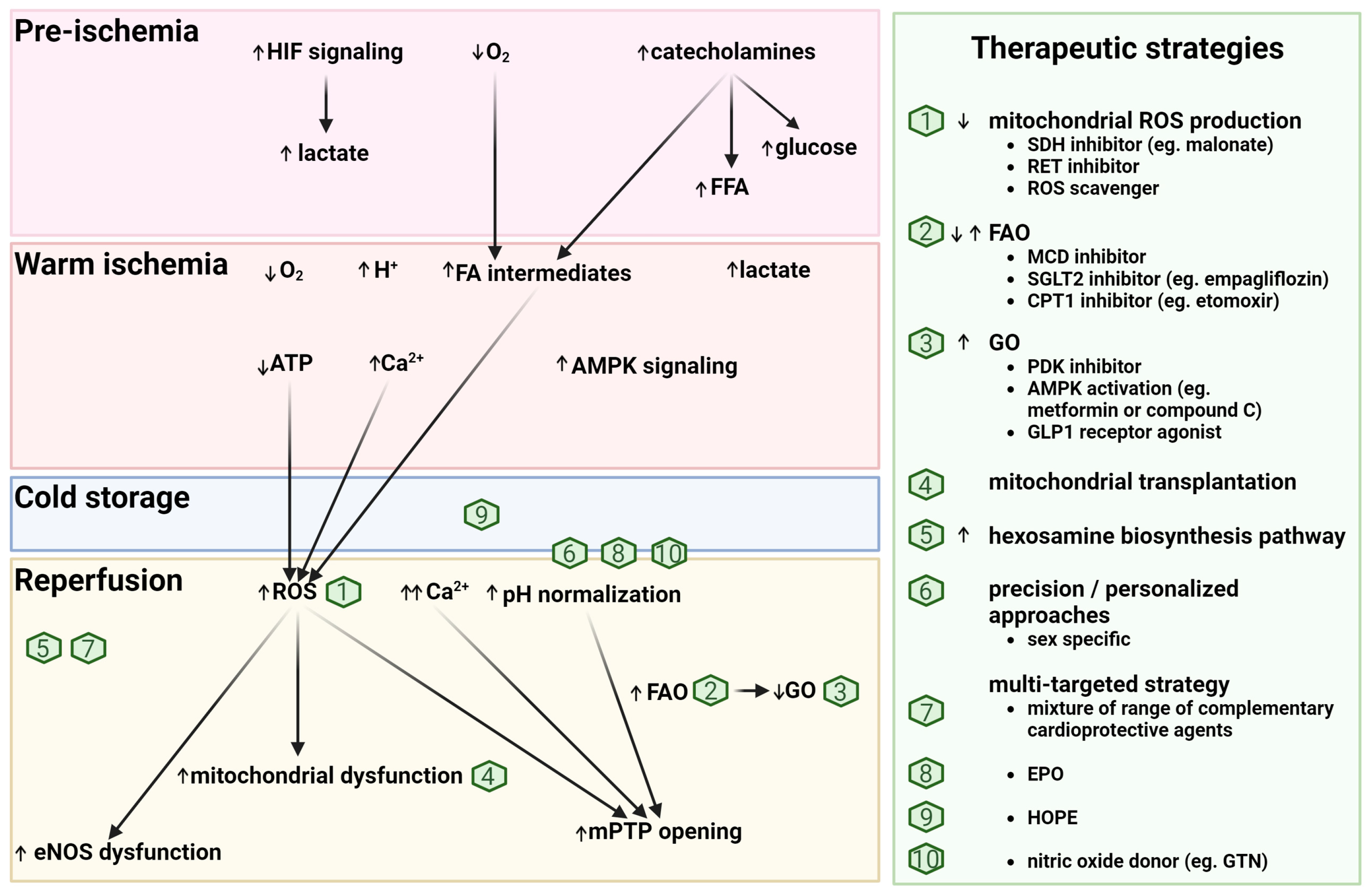
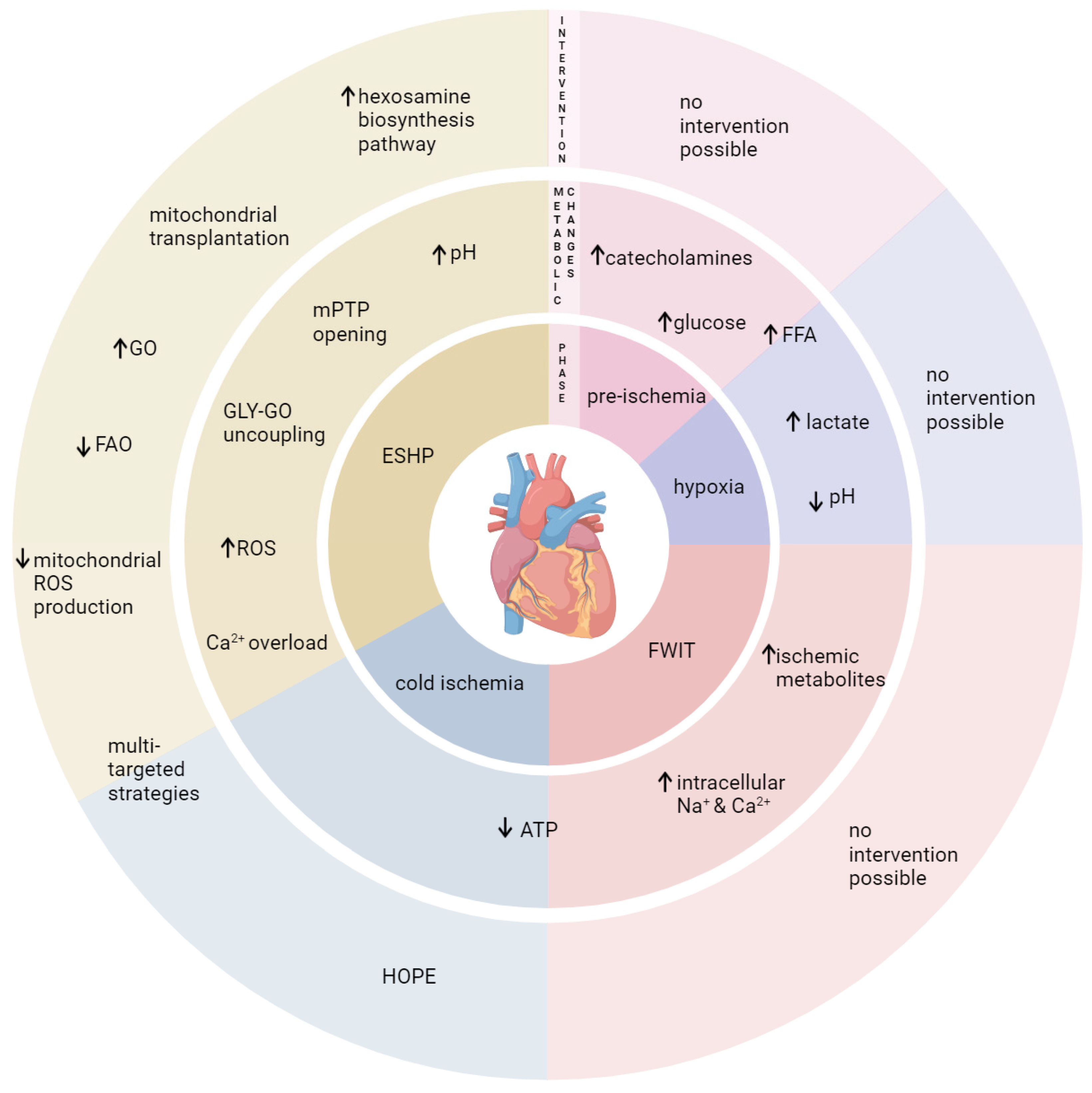
3. Metabolic Changes in the DCD Donor and Their Impacts on the Heart
3.1. Hypoxia
3.2. Catecholamines
3.3. Carbohydrate Metabolism
3.4. Fat Metabolism
3.5. Ketone Metabolism
3.6. Branched-Chain Amino Acid Metabolism
3.7. Cellular Signaling
3.8. Cardiac Status at Procurement
4. Metabolic Considerations during Procurement and Early Reperfusion of Cardiac Grafts
4.1. Cold Ischemia
4.2. Reperfusion Injury
4.3. Challenges and Opportunities
4.4. Potential Metabolic-Based Cardioprotective (Reperfusion) Strategies
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lund, L.H.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Goldfarb, S.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Yusen, R.D.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-Second Official Adult Heart Transplantation Report—2015; Focus Theme: Early Graft Failure. J. Heart Lung Transplant. 2015, 34, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Eurotransplant Annual Report 2020. Available online: https://www.eurotransplant.org/wp-content/uploads/2022/03/ET_AR2020_LR_def.pdf (accessed on 4 April 2024).
- Longnus, S.L.; Mathys, V.; Dornbierer, M.; Dick, F.; Carrel, T.P.; Tevaearai, H.T. Heart Transplantation with Donation after Circulatory Determination of Death. Nat. Rev. Cardiol. 2014, 11, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Dhital, K.K.; Chew, H.C.; Macdonald, P.S. Donation after Circulatory Death Heart Transplantation. Curr. Opin. Organ Transplant. 2017, 22, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Messer, S.; Page, A.; Colah, S.; Axell, R.; Parizkova, B.; Tsui, S.; Large, S. Human Heart Transplantation from Donation after Circulatory-Determined Death Donors Using Normothermic Regional Perfusion and Cold Storage. J. Heart Lung Transplant. 2018, 37, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.; Dhital, K. Heart Transplantation from Donation-after-Circulatory-Death (DCD) Donors: Back to the future―Evolving Trends in Heart Transplantation from DCD Donors. J. Heart Lung Transplant. 2019, 38, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Tchana-Sato, V.; Ledoux, D.; Detry, O.; Hans, G.; Ancion, A.; D’Orio, V.; Massion, P.B.; Amabili, P.; Bruls, S.; Lavigne, J.P.; et al. Successful Clinical Transplantation of Hearts Donated after Circulatory Death Using Normothermic Regional Perfusion. J. Heart Lung Transplant. 2019, 38, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, J. “Donation After Circulatory Death” Heart Transplant Is a US First. JAMA 2020, 323, 111. [Google Scholar] [CrossRef] [PubMed]
- Messer, S.; Cernic, S.; Page, A.; Berman, M.; Kaul, P.; Colah, S.; Ali, J.; Pavlushkov, E.; Baxter, J.; Quigley, R.; et al. A 5-Year Single-Center Early Experience of Heart Transplantation from Donation after Circulatory-Determined Death Donors. J. Heart Lung Transplant. 2020, 39, 1463–1475. [Google Scholar] [CrossRef]
- Schroder, J.N.; Patel, C.B.; DeVore, A.D.; Bryner, B.S.; Casalinova, S.; Shah, A.; Smith, J.W.; Fiedler, A.G.; Daneshmand, M.; Silvestry, S.; et al. Transplantation Outcomes with Donor Hearts after Circulatory Death. N. Engl. J. Med. 2023, 388, 2121–2131. [Google Scholar] [CrossRef]
- Chew, H.C.; Iyer, A.; Connellan, M.; Scheuer, S.; Villanueva, J.; Gao, L.; Hicks, M.; Harkness, M.; Soto, C.; Dinale, A.; et al. Outcomes of Donation After Circulatory Death Heart Transplantation in Australia. J. Am. Coll. Cardiol. 2019, 73, 1447–1459. [Google Scholar] [CrossRef]
- Messer, S.; Page, A.; Axell, R.; Berman, M.; Hernández-Sánchez, J.; Colah, S.; Parizkova, B.; Valchanov, K.; Dunning, J.; Pavlushkov, E.; et al. Outcome after Heart Transplantation from Donation after Circulatory-Determined Death Donors. J. Heart Lung Transplant. 2017, 36, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Large, S.; Tsui, S.; Messer, S. Clinical and Ethical Challenges in Heart Transplantation from Donation after Circulatory Determined Death Donors. Curr. Opin. Organ Transplant. 2017, 22, 251–259. [Google Scholar] [CrossRef]
- Vandenbriele, C.; Brouckaert, J.; Hans, G.; Tchana-Sato, V.; Vandendriessche, K.; Gunst, J.; Ancion, A.; Van Cleemput, J.; Ledoux, D.; Rex, S.; et al. The Role of Transesophageal Echocardiography in Guiding Heart Donation after Circulatory Death. Clin. Transplant. 2022, 36, e14783. [Google Scholar] [CrossRef] [PubMed]
- Dhital, K.K.; Iyer, A.; Connellan, M.; Chew, H.C.; Gao, L.; Doyle, A.; Hicks, M.; Kumarasinghe, G.; Soto, C.; Dinale, A.; et al. Adult Heart Transplantation with Distant Procurement and Ex-Vivo Preservation of Donor Hearts after Circulatory Death: A Case Series. Lancet 2015, 385, 2585–2591. [Google Scholar] [CrossRef]
- Scheuer, S.E.; Jansz, P.C.; Macdonald, P.S. Heart Transplantation Following Donation after Circulatory Death: Expanding the Donor Pool. J. Heart Lung Transplant. 2021, 40, 882–889. [Google Scholar] [CrossRef]
- Thuong, M.; Ruiz, A.; Evrard, P.; Kuiper, M.; Boffa, C.; Akhtar, M.Z.; Neuberger, J.; Ploeg, R. New Classification of Donation after Circulatory Death Donors Definitions and Terminology. Transpl. Int. 2016, 29, 749–759. [Google Scholar] [CrossRef]
- García Sáez, D.; Bowles, C.T.; Mohite, P.N.; Zych, B.; Maunz, O.; Popov, A.F.; Hurtado, A.; Raj, B.; Rahman-Haley, S.; Banner, N.; et al. Heart Transplantation after Donor Circulatory Death in Patients Bridged to Transplant with Implantable Left Ventricular Assist Devices. J. Heart Lung Transplant. 2016, 35, 1255–1260. [Google Scholar] [CrossRef]
- White, C.W.; Messer, S.J.; Large, S.R.; Conway, J.; Kim, D.H.; Kutsogiannis, D.J.; Nagendran, J.; Freed, D.H. Transplantation of Hearts Donated after Circulatory Death. Front. Cardiovasc. Med. 2018, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Detry, O.; Le Dinh, H.; Noterdaeme, T.; De Roover, A.; Honoré, P.; Squifflet, J.-P.; Meurisse, M. Categories of Donation After Cardiocirculatory Death. Transplant. Proc. 2012, 44, 1189–1195. [Google Scholar] [CrossRef]
- Messer, S.; Page, A.; Berman, M.; Tsui, S.; Large, S. Prolonged Donation Withdrawal Ischaemic Time (DWIT) Does Not Impact on DCD Heart Transplant Outcomes. J. Heart Lung Transplant. 2018, 37, S14–S15. [Google Scholar] [CrossRef]
- White, C.W.; Lillico, R.; Sandha, J.; Hasanally, D.; Wang, F.; Ambrose, E.; Müller, A.; Rachid, O.; Li, Y.; Xiang, B.; et al. Physiologic Changes in the Heart Following Cessation of Mechanical Ventilation in a Porcine Model of Donation After Circulatory Death: Implications for Cardiac Transplantation: Physiologic Response to Donor Extubation. Am. J. Transplant. 2016, 16, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Chew, H.C.; Gao, L.; Villanueva, J.; Hicks, M.; Doyle, A.; Kumarasinghe, G.; Jabbour, A.; Jansz, P.C.; Feneley, M.P.; et al. Pathophysiological Trends During Withdrawal of Life Support: Implications for Organ Donation After Circulatory Death. Transplantation 2016, 100, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Kearns, M.J.; Miller, S.D.; Cheung, A.; Bashir, J.; Wong, S.; Seidman, M.A.; Boyd, J.H. A Rodent Model of Cardiac Donation After Circulatory Death and Novel Biomarkers of Cardiac Viability During Ex Vivo Heart Perfusion. Transplantation 2017, 101, e231–e239. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Méndez-Carmona, N.; Wyss, R.K.; Joachimbauer, A.; Casoni, D.; Carrel, T.; Longnus, S. Comparison of Experimental Rat Models in Donation After Circulatory Death (DCD): In-Situ vs. Ex-Situ Ischemia. Front. Cardiovasc. Med. 2021, 7, 596883. [Google Scholar] [CrossRef] [PubMed]
- Obeid, N.R.; Rojas, A.; Reoma, J.L.; Hall, C.M.; Cook, K.E.; Bartlett, R.H.; Punch, J.D. Organ Donation After Cardiac Determination of Death (DCD): A Swine Model. ASAIO J. 2009, 55, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A.; Steenbergen, C. Myocardial Ischemia Revisited. The Osmolar Load, Membrane Damage, and Reperfusion. J. Mol. Cell. Cardiol. 1986, 18, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A. The Cell Biology of Acute Myocardial Ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A.; Hill, M.L.; Mayer, S.E. Total Ischemia in Dog Hearts, in Vitro. Circ. Res. 1981, 49, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A. Lethal Myocardial Ischemic Injury. Am. J. Pathol. 1981, 102, 241. [Google Scholar]
- Schaefer, S.; Ramasamy, R. Glycogen Utilization and Ischemic Injury in the Isolated Rat Heart. Cardiovasc. Res. 1997, 35, 90–98. [Google Scholar] [CrossRef]
- Knutson, A.K.; Williams, A.L.; Boisvert, W.A.; Shohet, R.V. HIF in the Heart: Development, Metabolism, Ischemia, and Atherosclerosis. J. Clin. Investig. 2021, 131, e137557. [Google Scholar] [CrossRef] [PubMed]
- Verges, S.; Chacaroun, S.; Godin-Ribuot, D.; Baillieul, S. Hypoxic Conditioning as a New Therapeutic Modality. Front. Pediatr. 2015, 3, 58. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting Fatty Acid and Carbohydrate Oxidation—A Novel Therapeutic Intervention in the Ischemic and Failing Heart. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1333–1350. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Lameris, T.W.; de Zeeuw, S.; Alberts, G.; Boomsma, F.; Duncker, D.J.; Verdouw, P.D.; Veld, A.J.M.I.; van den Meiracker, A.H. Time Course and Mechanism of Myocardial Catecholamine Release During Transient Ischemia In Vivo. Circulation 2000, 101, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.A.; See Hoe, L.E.; Heather, L.C.; Molenaar, P.; Suen, J.Y.; Peart, J.; McGiffin, D.; Fraser, J.F. Peritransplant Cardiometabolic and Mitochondrial Function: The Missing Piece in Donor Heart Dysfunction and Graft Failure. Transplantation 2021, 105, 496–508. [Google Scholar] [CrossRef]
- Shimada, Y.; Yamamoto, F.; Yamamoto, H.; Oka, T. Effect of Preischemic Catecholamine Treatment on Ischemia-Reperfusion Injury of the Myocardium: Subtype, Dose, and Temperature Dependency. Jpn. Circ. J. 1998, 62, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Niederberger, P.; Farine, E.; Arnold, M.; Wyss, R.K.; Sanz, M.N.; Méndez-Carmona, N.; Gahl, B.; Fiedler, G.M.; Carrel, T.P.; Tevaearai Stahel, H.T.; et al. High Pre-Ischemic Fatty Acid Levels Decrease Cardiac Recovery in an Isolated Rat Heart Model of Donation after Circulatory Death. Metabolism 2017, 71, 107–117. [Google Scholar] [CrossRef]
- Schömig, A.; Strasser, R.; Richardt, G. Release and Effects of Catecholamines in Myocardial Ischemia. In Pathophysiology of Severe Ischemic Myocardial Injury. Developments in Cardiovascular Medicine; Springer: Dordrecht, The Netherlands, 1990; Volume 104, pp. 381–412. ISBN 978-0-7923-0459-3. [Google Scholar]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy Substrate Metabolism and Mitochondrial Oxidative Stress in Cardiac Ischemia/Reperfusion Injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Hubacher, V.; Egle, M.; Graf, S.; Arnold, M.; Segiser, A.; Sanz, M.N.; Casoni, D.; Garcia Casalta, L.; Nettelbeck, K.; Mihalj, M.; et al. Open- versus Closed-Chest Pig Models of Donation after Circulatory Death. 2024; manuscript in preparation. [Google Scholar]
- van der Vusse, G.J.; de Groot, M.J.M. Interrelationship between Lactate and Cardiac Fatty Acid Metabolism. Mol. Cell. Biochem. 1992, 116, 11–17. [Google Scholar] [CrossRef]
- Arnold, M.; Segiser, A.; Graf, S.; Méndez-Carmona, N.; Sanz, M.N.; Wyss, R.K.; Kalbermatter, N.; Keller, N.; Carrel, T.; Longnus, S. Pre-Ischemic Lactate Levels Affect Post-Ischemic Recovery in an Isolated Rat Heart Model of Donation After Circulatory Death (DCD). Front. Cardiovasc. Med. 2021, 8, 669205. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Vanoverschelde, J.-L.J.; Taegtmeyer, H. Glucose for the Heart. Circulation 1999, 99, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Willis, M.S.; Berthiaume, J.M. Influence of Ischemia-Reperfusion Injury on Cardiac Metabolism. In The Scientist’s Guide to Cardiac Metabolism; Elsevier: London, UK, 2016; pp. 155–167. [Google Scholar]
- Doenst, T.; Bugger, H.; Schwarzer, M.; Faerber, G.; Borger, M.A.; Mohr, F.W. Three Good Reasons for Heart Surgeons to Understand Cardiac Metabolism. Eur. J. Cardio-Thorac. Surg. 2008, 33, 862–871. [Google Scholar] [CrossRef] [PubMed]
- King, L.M.; Opie, L.H. Glucose Delivery Is a Major Determinant of Glucose Utilisation in the Ischemic Myocardium with a Residual Coronary Flow. Cardiovasc. Res. 1998, 39, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Eric Botker, H.; Dambrova, M.; et al. Cardiac Metabolism as a Driver and Therapeutic Target of Myocardial Infarction. J. Cell. Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Collins-Nakai, R.; Olley, P.M.; Montague, T.J.; McNeil, G.; Gayle, M.; Penkoske, P.; Finegan, B.A. Plasma Fatty Acid Levels in Infants and Adults after Myocardial Ischemia. Am. Heart J. 1994, 128, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Oliver, M.F. Fatty Acids and the Risk of Death during Acute Myocardial Ischaemia. Clin. Sci. 2015, 128, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Marina Prendes, M.G.; Hermann, R.; Torresin, M.E.; Souto, P.; Tallis, S.; Savino, E.A.; Varela, A. Involvement of Energetic Metabolism in the Effects of Ischemic Postconditioning on the Ischemic-Reperfused Heart of Fed and Fasted Rats. J. Physiol. Sci. 2011, 61, 303–312. [Google Scholar] [CrossRef]
- Doenst, T.; Guthrie, P.H.; Chemnitius, J.-M.; Zech, R.; Taegtmeyer, H. Fasting, Lactate, and Insulin Improve Ischemia Tolerance in Rat Heart: A Comparison with Ischemic Preconditioning. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H1607–H1615. [Google Scholar] [CrossRef]
- Schneider, C.A.; Taegtmeyer, H. Fasting in Vivo Delays Myocardial Cell Damage after Brief Periods of Ischemia in the Isolated Working Rat Heart. Circ. Res. 1991, 68, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, C.; Papageorgiou, I.; Tardy, I.; Lerch, R. Effect of Nutritional State on Substrate Metabolism and Contractile Function in Postischemic Rat Myocardium. Am. J. Physiol.-Heart Circ. Physiol. 1996, 271, H2060–H2070. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Makrecka, M.; Kuka, J.; Makarova, E.; Vilskersts, R.; Cirule, H.; Sevostjanovs, E.; Grinberga, S.; Pugovics, O.; Dambrova, M. The Heart Is Better Protected against Myocardial Infarction in the Fed State Compared to the Fasted State. Metabolism 2014, 63, 127–136. [Google Scholar] [CrossRef] [PubMed]
- van Hoorn, D.E.C.; Boelens, P.G.; van Middelaar-Voskuilen, M.C.; Nijveldt, R.J.; Prins, H.; Bouritius, H.; Hofman, Z.; M’rabet, L.; van Leeuwen, P.A.M.; van Norren, K. Preoperative Feeding Preserves Heart Function and Decreases Oxidative Injury in Rats. Nutrition 2005, 21, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Makrecka-Kuka, M.; Volska, K.; Kuka, J.; Makarova, E.; Antone, U.; Sevostjanovs, E.; Vilskersts, R.; Strods, A.; Tars, K.; et al. Long-Chain Acylcarnitines Determine Ischaemia/Reperfusion-Induced Damage in Heart Mitochondria. Biochem. J. 2016, 473, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid Metabolism and Signaling in Cardiac Lipotoxicity. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R. Evolving Concepts of Myocardial Energy Metabolism: More Than Just Fats and Carbohydrates. Circ. Res. 2016, 119, 1173–1176. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Lopaschuk, G.D. CrossTalk Proposal: Ketone Bodies Are an Important Metabolic Fuel for the Heart. J. Physiol. 2022, 600, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Brunner, M.P.; Shah, S.H.; Craig, D.M.; Stevens, R.D.; Muehlbauer, M.J.; Bain, J.R.; Newgard, C.B.; Kraus, W.E.; Granger, C.B.; Sketch, M.H.; et al. Effect of Heparin Administration on Metabolomic Profiles in Samples Obtained During Cardiac Catheterization. Circ. Cardiovasc. Genet. 2011, 4, 695–700. [Google Scholar] [CrossRef]
- Bahnsen, M.; Burrin, J.M.; Johnston, D.G.; Pernet, A.; Walker, M.; Alberti, K.G. Mechanisms of Catecholamine Effects on Ketogenesis. Am. J. Physiol.-Endocrinol. Metab. 1984, 247, E173–E180. [Google Scholar] [CrossRef]
- Krentz, A.J.; Freedman, D.; Greene, R.; McKinley, M.; Boyle, P.J.; Schade, D.S. Differential Effects of Physiological versus Pathophysiological Plasma Concentrations of Epinephrine and Norepinephrine on Ketone Body Metabolism and Hepatic Portal Blood Flow in Man. Metabolism 1996, 45, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R.; Taegtmeyer, H. Changes in Citric Acid Cycle Flux and Anaplerosis Antedate the Functional Decline in Isolated Rat Hearts Utilizing Acetoacetate. J. Clin. Investig. 1991, 87, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.D.; Elkins, N.; Reiss, O.K.; Chan, L.; Shapiro, J.I. Effects of Acetate on Energy Metabolism and Function in the Isolated Perfused Rat Heart. Kidney Int. 1997, 52, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Hua, Y.; He, L.; He, J.; Chen, Y.; Yang, J.; Mahmoud, I.; Zeng, F.; Zeng, X.; Benavides, G.A.; et al. β-Hydroxybutyrate Administered at Reperfusion Reduces Infarct Size and Preserves Cardiac Function by Improving Mitochondrial Function through Autophagy in Male Mice. J. Mol. Cell. Cardiol. 2024, 186, 31–44. [Google Scholar] [CrossRef] [PubMed]
- McGarrah, R.W.; White, P.J. Branched-Chain Amino Acids in Cardiovascular Disease. Nat. Rev. Cardiol 2023, 20, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G. The Role of Fatty Acid Oxidation in Cardiac Ischemia and Reperfusion. Adv. Stud. Med. 2004, 4, S803–S807. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, F.; Schotola, H.; Popov, A.F.; Sohns, C.; Schuenemann, J.; Friedrich, M.; Coskun, K.O.; von Lewinski, D.; Hinz, J.; Bauer, M.; et al. AMPK—Activated Protein Kinase and Its Role in Energy Metabolism of the Heart. Curr. Cardiol. Rev. 2010, 6, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R.B. The Role of AMPK in Cardiomyocyte Health and Survival. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. In AMP-Activated Protein Kinase; Cordero, M.D., Viollet, B., Eds.; Experientia Supplementum; Springer International Publishing: Cham, Switzerland, 2016; Volume 107, pp. 23–43. ISBN 978-3-319-43587-9. [Google Scholar]
- Ferguson, B.S.; Rogatzki, M.J.; Goodwin, M.L.; Kane, D.A.; Rightmire, Z.; Gladden, L.B. Lactate Metabolism: Historical Context, Prior Misinterpretations, and Current Understanding. Eur. J. Appl. Physiol. 2018, 118, 691–728. [Google Scholar] [CrossRef]
- Nalbandian, M.; Takeda, M. Lactate as a Signaling Molecule That Regulates Exercise-Induced Adaptations. Biology 2016, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically Derived Lactate Stimulates Revascularization and Tissue Repair via Redox Mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Dunn, R.B.; Griggs, D.M. Transmural Gradients in Ventricular Tissue Metabolites Produced by Stopping Coronary Blood Flow in the Dog. Circ. Res. 1975, 37, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Murry, C.E.; Steenbergen, C.; Reimer, K.A. Development of Cell Injury in Sustained Acute Ischemia. Circulation 1990, 82, II2-12. [Google Scholar] [PubMed]
- Page, A.; Messer, S.; Large, S.R. Heart Transplantation from Donation after Circulatory Determined Death. Ann. Cardiothorac. Surg. 2018, 7, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Smail, H.; Garcia-Saez, D.; Stock, U.; Ahmed-Hassan, H.; Bowles, C.; Zych, B.; Mohite, P.N.; Maunz, O.; Simon, A.R. Direct Heart Procurement After Donation After Circulatory Death With Ex Situ Reperfusion. Ann. Thorac. Surg. 2018, 106, e211–e214. [Google Scholar] [CrossRef] [PubMed]
- Parolari, A.; Rubini, P.; Cannata, A.; Bonati, L.; Alamanni, F.; Tremoli, E.; Biglioli, P. Endothelial Damage during Myocardial Preservation and Storage. Ann. Thorac. Surg. 2002, 73, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Docherty, J.C.; Rendell, J.C.T.; Clanachan, A.S.; Lopaschuk, G.D. High Levels of Fatty Acids Delay the Recovery of Intracellular pH and Cardiac Efficiency Inpost-Ischemic Hearts by Inhibiting Glucose Oxidation. J. Am. Coll. Cardiol. 2002, 39, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Ussher, J.R.; Wang, W.; Gandhi, M.; Keung, W.; Samokhvalov, V.; Oka, T.; Wagg, C.S.; Jaswal, J.S.; Harris, R.A.; Clanachan, A.S.; et al. Stimulation of Glucose Oxidation Protects against Acute Myocardial Infarction and Reperfusion Injury. Cardiovasc. Res. 2012, 94, 359–369. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Switching Metabolic Genes to Build a Better Heart. Circulation 2002, 106, 2043–2045. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Clanachan, A.S.; Schulz, R.; Lopaschuk, G.D. Cardiac Efficiency Is Improved After Ischemia by Altering Both the Source and Fate of Protons. Circ. Res. 1996, 79, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Mallet, R.T.; Sun, J.; Knott, E.M.; Sharma, A.B.; Olivencia-Yurvati, A.H. Metabolic Cardioprotection by Pyruvate: Recent Progress. Exp. Biol. Med. 2005, 230, 435–443. [Google Scholar] [CrossRef]
- Smolenski, R.T.; Amrani, M.; Jayakumar, J.; Jagodzinski, P.; Gray, C.C.; Goodwin, A.T.; Sammut, I.A.; Yacoub, M.H. Pyruvate/Dichloroacetate Supply during Reperfusion Accelerates Recovery of Cardiac Energetics and Improves Mechanical Function Following Cardioplegic Arrest. Eur. J. Cardio-Thorac. Surg. 2001, 19, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.B.; Cheng, J.-F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl Coenzyme A Decarboxylase Inhibition Protects the Ischemic Heart by Inhibiting Fatty Acid Oxidation and Stimulating Glucose Oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Barr, R.; Thomas, P.D.; Dyck, J.R.B. Beneficial Effects of Trimetazidine in Ex Vivo Working Ischemic Hearts Are Due to a Stimulation of Glucose Oxidation Secondary to Inhibition of Long-Chain 3-Ketoacyl Coenzyme A Thiolase. Circ. Res. 2003, 93, e33–e37. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; McNeil, G.F.; McVeigh, J.J. Glucose Oxidation Is Stimulated in Reperfused Ischemic Hearts with the Carnitine Palmitoyltransferase 1 Inhibitor, Etomoxir. Mol. Cell. Biochem. 1989, 88, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Functional Coupling between Glycolysis and Sarcoplasmic Reticulum Ca2+ Transport. Circ. Res. 1995, 77, 88–97. [Google Scholar] [CrossRef]
- Fillmore, N.; Lopaschuk, G.D. Impact of Fatty Acid Oxidation on Cardiac Efficiency. Heart Metab. 2011, 53, 33–37. [Google Scholar]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Niederberger, P.; Farine, E.; Raillard, M.; Dornbierer, M.; Freed, D.H.; Large, S.R.; Chew, H.C.; MacDonald, P.S.; Messer, S.J.; White, C.W.; et al. Heart Transplantation with Donation after Circulatory Death: What Have We Learned From Preclinical Studies? Circ. Heart Fail. 2019, 12, e005517. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.L.; Howlett, S.E. Age and Ovariectomy Abolish Beneficial Effects of Female Sex on Rat Ventricular Myocytes Exposed to Simulated Ischemia and Reperfusion. PLoS ONE 2012, 7, e38425. [Google Scholar] [CrossRef] [PubMed]
- Lagranha, C.J.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex Differences in the Phosphorylation of Mitochondrial Proteins Result in Reduced Production of Reactive Oxygen Species and Cardioprotection in Females. Circ. Res. 2010, 106, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Devanathan, S.; Whitehead, T.D.; Fettig, N.; Gropler, R.J.; Nemanich, S.; Shoghi, K.I. Sexual Dimorphism in Myocardial Acylcarnitine and Triglyceride Metabolism. Biol. Sex Differ. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, R.; Wambolt, R.B.; Parsons, H.; Antler, C.; Leong, H.S.; Keller, A.; Dunaway, G.A.; Popov, K.M.; Allard, M.F. Gender and Post-Ischemic Recovery of Hypertrophied Rat Hearts. BMC Cardiovasc. Disord. 2006, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Clausen, C.F.; Morton, J.S.; Lopaschuk, G.D.; Davidge, S.T. Long-Term Effects of Intrauterine Growth Restriction on Cardiac Metabolism and Susceptibility to Ischaemia/Reperfusion. Cardiovasc. Res. 2011, 90, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Oliver, J.; Roca, P.; Garciapalmer, F. Caloric Restriction and Gender Modulate Cardiac Muscle Mitochondrial H2O2 Production and Oxidative Damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.F.; Ronconi, K.S.; Morra, E.A.; Do Val Lima, P.R.; Porto, M.L.; Vassallo, D.V.; Figueiredo, S.G.; Stefanon, I. Sex Differences in the Regulation of Spatially Distinct Cardiac Mitochondrial Subpopulations. Mol. Cell. Biochem. 2016, 419, 41–51. [Google Scholar] [CrossRef]
- Chen, Q.; Akande, O.; Lesnefsky, E.J.; Quader, M. Influence of Sex on Global Myocardial Ischemia Tolerance and Mitochondrial Function in Circulatory Death Donor Hearts. Am. J. Physiol.-Heart Circ. Physiol. 2023, 324, H57–H66. [Google Scholar] [CrossRef]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef]
- Holubarsch, C.J.F.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A Double-Blind Randomized Multicentre Clinical Trial to Evaluate the Efficacy and Safety of Two Doses of Etomoxir in Comparison with Placebo in Patients with Moderate Congestive Heart Failure: The ERGO (Etomoxir for the Recovery of Glucose Oxidation) Study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Truby, L.K.; Kwee, L.C.; Bowles, D.E.; Casalinova, S.; Ilkayeva, O.; Muehlbauer, M.J.; Huebner, J.L.; Holley, C.L.; DeVore, A.D.; Patel, C.B.; et al. Metabolomic Profiling during Ex Situ Normothermic Perfusion before Heart Transplantation Defines Patterns of Substrate Utilization and Correlates with Markers of Allograft Injury. J. Heart Lung Transplant. 2023; in press. [Google Scholar] [CrossRef] [PubMed]
- Hautbergue, T.; Laverdure, F.; Van, S.D.; Vallee, A.; Sanchis-Borja, M.; Decante, B.; Gaillard, M.; Junot, C.; Fenaille, F.; Mercier, O.; et al. Metabolomic Profiling of Cardiac Allografts after Controlled Circulatory Death. J. Heart Lung Transplant. 2023, 42, 870–879. [Google Scholar] [CrossRef]
- Wyss, R.K.; Méndez Carmona, N.; Arnold, M.; Segiser, A.; Mueller, M.; Dutkowski, P.; Carrel, T.P.; Longnus, S.L. Hypothermic, Oxygenated Perfusion (HOPE) Provides Cardioprotection via Succinate Oxidation Prior to Normothermic Perfusion in a Rat Model of Donation after Circulatory Death (DCD). Am. J. Transplant. 2021, 21, 1003–1011. [Google Scholar] [CrossRef]
- Egle, M.; Mendez-Carmona, N.; Segiser, A.; Graf, S.; Siepe, M.; Longnus, S. Hypothermic Oxygenated Perfusion Improves Vascular and Contractile Function by Preserving Endothelial Nitric Oxide Production in Cardiac Grafts Obtained with Donation after Circulatory Death. J. Am. Heart Assoc. 2024, 13, e033503. [Google Scholar]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate Dehydrogenase Inhibition with Malonate during Reperfusion Reduces Infarct Size by Preventing Mitochondrial Permeability Transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef]
- Akande, O.; Chen, Q.; Cholyway, R.; Toldo, S.; Lesnefsky, E.J.; Quader, M. Modulation of Mitochondrial Respiration During Early Reperfusion Reduces Cardiac Injury in Donation After Circulatory Death Hearts. J. Cardiovasc. Pharmacol. 2022, 80, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.J.; Kim, S.; Cui, H.X.; Han, K.; Lee, H.K.; Kim, C.-H.; Kang, Y.C.; Zhang, Y.H. Human Platelet Mitochondria Improve the Mitochondrial and Cardiac Function of Donor Heart. Pflug. Arch.-Eur. J. Physiol. 2023, 475, 267–275. [Google Scholar] [CrossRef]
- Guariento, A.; Doulamis, I.P.; Duignan, T.; Kido, T.; Regan, W.L.; Saeed, M.Y.; Hoganson, D.M.; Emani, S.M.; Fynn-Thompson, F.; Matte, G.S.; et al. Mitochondrial Transplantation for Myocardial Protection in Ex-Situ—perfused Hearts Donated after Circulatory Death. J. Heart Lung Transplant. 2020, 39, 1279–1288. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Korzh, S.; Videja, M.; Vilks, K.; Cirule, H.; Kuka, J.; Dambrova, M.; Liepinsh, E. Empagliflozin Protects Cardiac Mitochondrial Fatty Acid Metabolism in a Mouse Model of Diet-Induced Lipid Overload. Cardiovasc. Drugs Ther. 2020, 34, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Paiva, M.A.; Gonçalves, L.M.; Providência, L.A.; Davidson, S.M.; Yellon, D.M.; Mocanu, M.M. Transitory Activation of AMPK at Reperfusion Protects the Ischaemic-Reperfused Rat Myocardium Against Infarction. Cardiovasc. Drugs Ther. 2010, 24, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.P.; Zachara, N.E.; Ngoh, G.A.; Hill, B.G.; Teshima, Y.; Bhatnagar, A.; Hart, G.W.; Marbán, E. Cardioprotection by N -Acetylglucosamine Linkage to Cellular Proteins. Circulation 2008, 117, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, S.; Siraj, M.A.; Chen, W.; Wang, J.; Parker, M.; Nagy, A.; Steve Fan, C.; Runeckles, K.; Li, J.; Kobayashi, J.; et al. Cardioprotective Actions of a Glucagon-like Peptide-1 Receptor Agonist on Hearts Donated After Circulatory Death. J. Am. Heart Assoc. 2023, 12, e027163. [Google Scholar] [CrossRef]
- Andrijevic, D.; Vrselja, Z.; Lysyy, T.; Zhang, S.; Skarica, M.; Spajic, A.; Dellal, D.; Thorn, S.L.; Duckrow, R.B.; Ma, S.; et al. Cellular Recovery after Prolonged Warm Ischaemia of the Whole Body. Nature 2022, 608, 405–412. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, M.; Do, P.; Davidson, S.M.; Large, S.R.; Helmer, A.; Beer, G.; Siepe, M.; Longnus, S.L. Metabolic Considerations in Direct Procurement and Perfusion Protocols with DCD Heart Transplantation. Int. J. Mol. Sci. 2024, 25, 4153. https://doi.org/10.3390/ijms25084153
Arnold M, Do P, Davidson SM, Large SR, Helmer A, Beer G, Siepe M, Longnus SL. Metabolic Considerations in Direct Procurement and Perfusion Protocols with DCD Heart Transplantation. International Journal of Molecular Sciences. 2024; 25(8):4153. https://doi.org/10.3390/ijms25084153
Chicago/Turabian StyleArnold, Maria, Peter Do, Sean M. Davidson, Stephen R. Large, Anja Helmer, Georgia Beer, Matthias Siepe, and Sarah L. Longnus. 2024. "Metabolic Considerations in Direct Procurement and Perfusion Protocols with DCD Heart Transplantation" International Journal of Molecular Sciences 25, no. 8: 4153. https://doi.org/10.3390/ijms25084153
APA StyleArnold, M., Do, P., Davidson, S. M., Large, S. R., Helmer, A., Beer, G., Siepe, M., & Longnus, S. L. (2024). Metabolic Considerations in Direct Procurement and Perfusion Protocols with DCD Heart Transplantation. International Journal of Molecular Sciences, 25(8), 4153. https://doi.org/10.3390/ijms25084153