
Chloride/Multiple Anion Exchanger SLC26A Family: Systemic Roles of SLC26A4 in Various Organs



Abstract
1. Introduction
2. Multiple Physiological Functions of SLC26A4
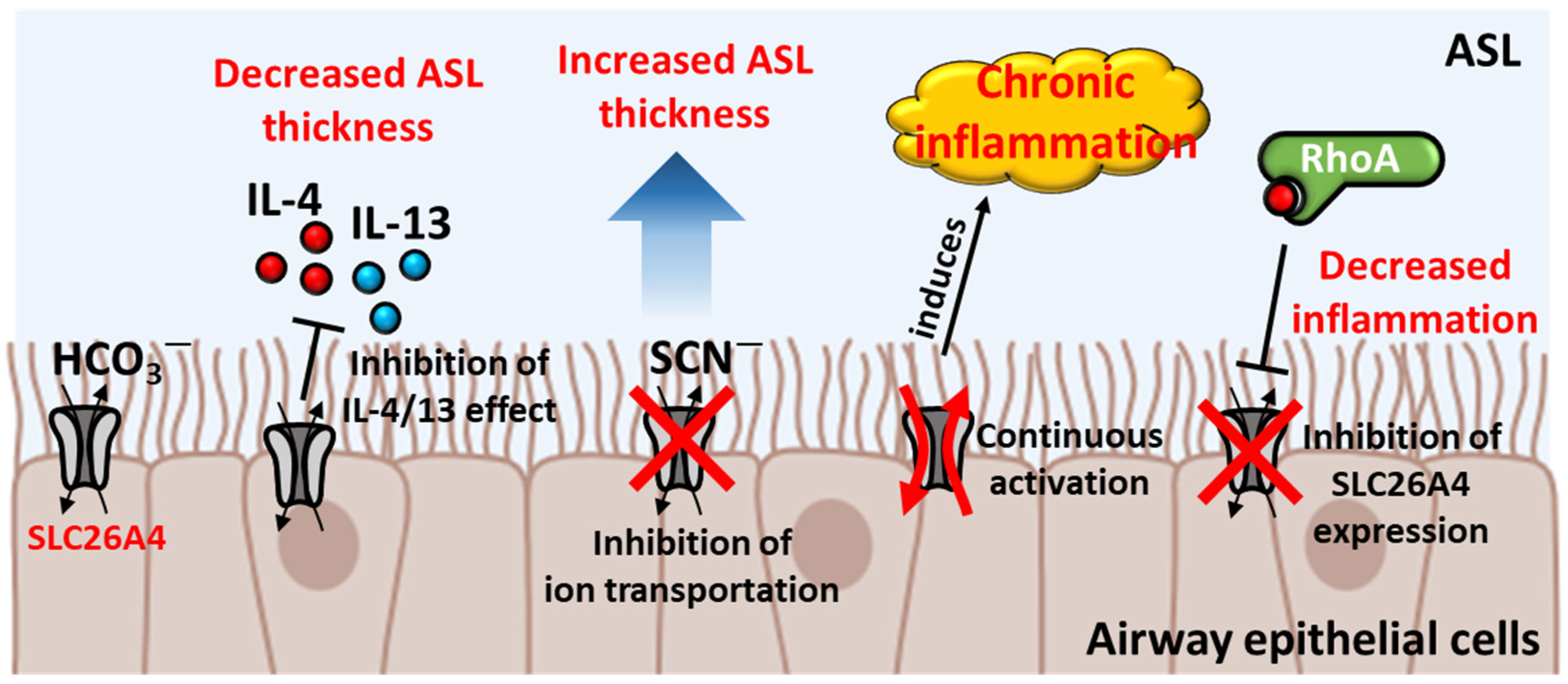
2.1. Protection of SLC26A4 in Airway Epithelium
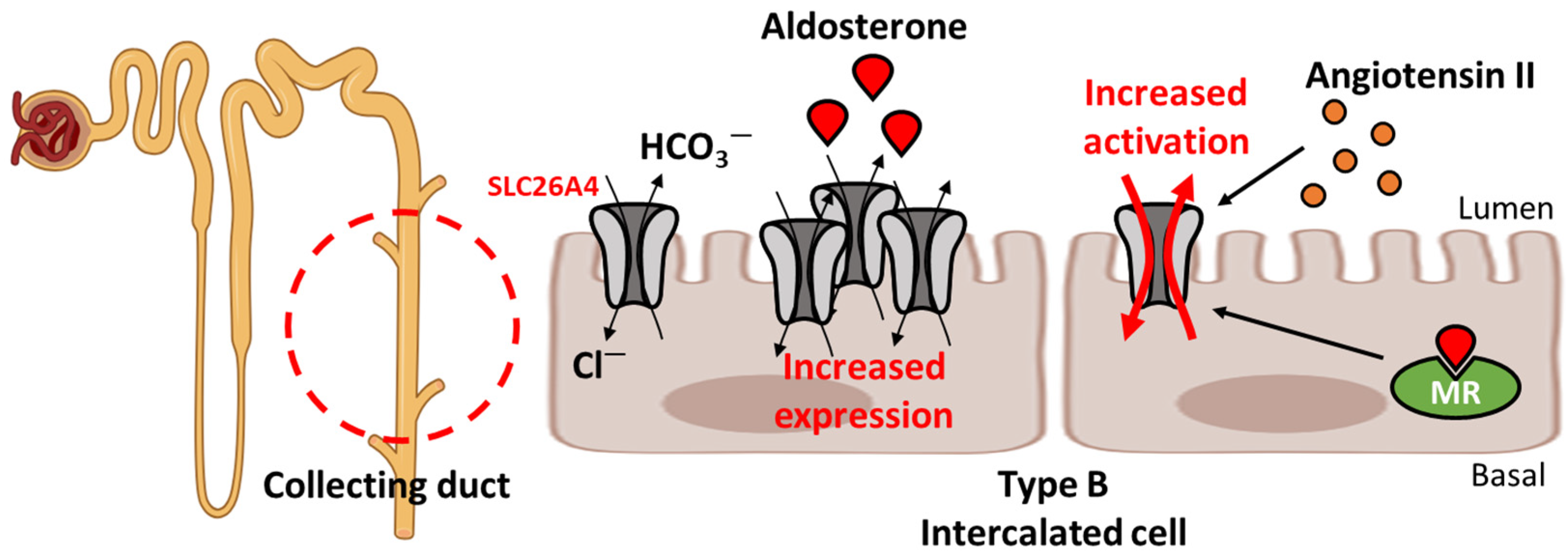
2.2. Regulation of Blood Pressure
2.3. Involvement in Hormone Regulation
2.4. Other Tissues and Potential Negative Regulators of Tumors
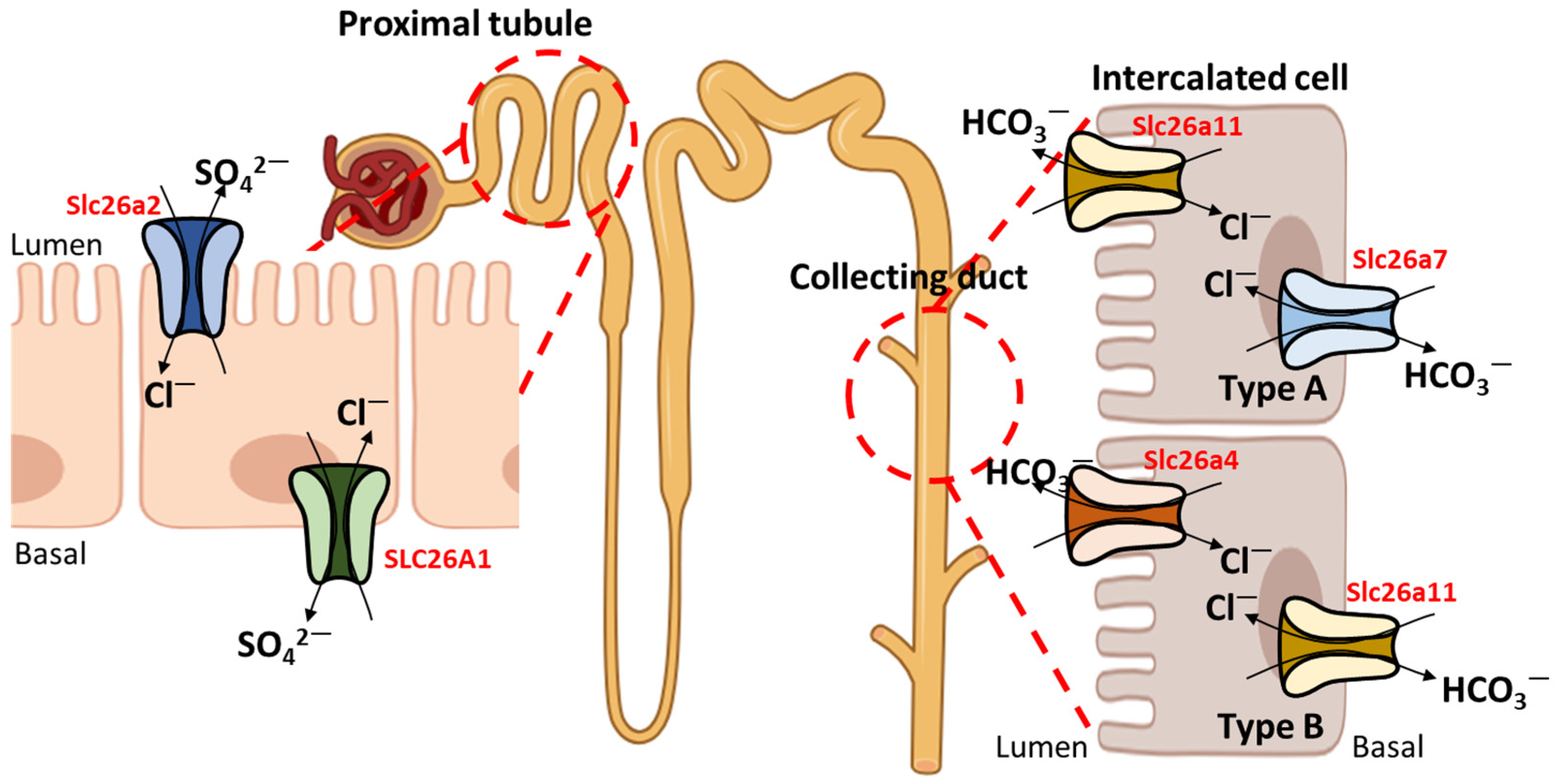
3. Relationship between SLC26A4 and Other Ion Transporters
4. Role of Other SLC26A Transporters with SLC26A4
5. Therapeutic Approaches
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alper, S.L.; Sharma, A.K. The SLC26 gene family of anion transporters and channels. Mol. Asp. Med. 2013, 34, 494–515. [Google Scholar] [CrossRef]
- Schnedler, N.; Burckhardt, G.; Burckhardt, B.C. Glyoxylate is a substrate of the sulfate-oxalate exchanger, sat-1, and increases its expression in HepG2 cells. J. Hepatol. 2011, 54, 513–520. [Google Scholar] [CrossRef]
- Xie, Q.; Welch, R.; Mercado, A.; Romero, M.F.; Mount, D.B. Molecular characterization of the murine Slc26a6 anion exchanger: Functional comparison with Slc26a1. Am. J. Physiol. Ren. Physiol. 2002, 283, F826–F838. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, J.F.; Akhavein, A.; Salas, M.J.; Shmukler, B.E.; Karniski, L.P.; Vandorpe, D.H.; Alper, S.L. Regulated transport of sulfate and oxalate by SLC26A2/DTDST. Am. J. Physiol. Cell Physiol. 2010, 298, C1363–C1375. [Google Scholar] [CrossRef]
- Chernova, M.N.; Jiang, L.; Shmukler, B.E.; Schweinfest, C.W.; Blanco, P.; Freedman, S.D.; Stewart, A.K.; Alper, S.L. Acute regulation of the SLC26A3 congenital chloride diarrhoea anion exchanger (DRA) expressed in Xenopus oocytes. J. Physiol. 2003, 549, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Y.H.; Chi, Z.P.; Huang, R.; Huang, H.; Liu, G.; Zhang, Y.; Yang, H.; Lin, J.; Yang, T.; et al. The Handling of Oxalate in the Body and the Origin of Oxalate in Calcium Oxalate Stones. Urol. Int. 2020, 104, 167–176. [Google Scholar] [CrossRef]
- Reimold, F.R.; Heneghan, J.F.; Stewart, A.K.; Zelikovic, I.; Vandorpe, D.H.; Shmukler, B.E.; Alper, S.L. Pendrin function and regulation in Xenopus oocytes. Cell. Physiol. Biochem. 2011, 28, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Shcheynikov, N.; Yang, D.; Wang, Y.; Zeng, W.; Karniski, L.P.; So, I.; Wall, S.M.; Muallem, S. The Slc26a4 transporter functions as an electroneutral Cl−/I−/HCO3− exchanger: Role of Slc26a4 and Slc26a6 in I− and HCO3− secretion and in regulation of CFTR in the parotid duct. J. Physiol. 2008, 586, 3813–3824. [Google Scholar] [CrossRef]
- Schaechinger, T.J.; Oliver, D. Nonmammalian orthologs of prestin (SLC26A5) are electrogenic divalent/chloride anion exchangers. Proc. Natl. Acad. Sci. USA 2007, 104, 7693–7698. [Google Scholar] [CrossRef]
- Bai, J.P.; Surguchev, A.; Montoya, S.; Aronson, P.S.; Santos-Sacchi, J.; Navaratnam, D. Prestin’s anion transport and voltage-sensing capabilities are independent. Biophys. J. 2009, 96, 3179–3186. [Google Scholar] [CrossRef]
- Alvarez, B.V.; Kieller, D.M.; Quon, A.L.; Markovich, D.; Casey, J.R. Slc26a6: A cardiac chloride-hydroxyl exchanger and predominant chloride-bicarbonate exchanger of the mouse heart. J. Physiol. 2004, 561, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Chernova, M.N.; Jiang, L.; Friedman, D.J.; Darman, R.B.; Lohi, H.; Kere, J.; Vandorpe, D.H.; Alper, S.L. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: Differences in anion selectivity, regulation, and electrogenicity. J. Biol. Chem. 2005, 280, 8564–8580. [Google Scholar] [CrossRef]
- Petrovic, S.; Barone, S.; Xu, J.; Conforti, L.; Ma, L.; Kujala, M.; Kere, J.; Soleimani, M. SLC26A7: A basolateral Cl−/HCO3− exchanger specific to intercalated cells of the outer medullary collecting duct. Am. J. Physiol. Ren. Physiol. 2004, 286, F161–F169. [Google Scholar] [CrossRef] [PubMed]
- Kosiek, O.; Busque, S.M.; Foller, M.; Shcheynikov, N.; Kirchhoff, P.; Bleich, M.; Muallem, S.; Geibel, J.P. SLC26A7 can function as a chloride-loading mechanism in parietal cells. Pflug. Arch. 2007, 454, 989–998. [Google Scholar] [CrossRef]
- Lohi, H.; Kujala, M.; Makela, S.; Lehtonen, E.; Kestila, M.; Saarialho-Kere, U.; Markovich, D.; Kere, J. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J. Biol. Chem. 2002, 277, 14246–14254. [Google Scholar] [CrossRef] [PubMed]
- Toure, A.; Morin, L.; Pineau, C.; Becq, F.; Dorseuil, O.; Gacon, G. Tat1, a novel sulfate transporter specifically expressed in human male germ cells and potentially linked to rhogtpase signaling. J. Biol. Chem. 2001, 276, 20309–20315. [Google Scholar] [CrossRef]
- Xu, J.; Henriksnas, J.; Barone, S.; Witte, D.; Shull, G.E.; Forte, J.G.; Holm, L.; Soleimani, M. SLC26A9 is expressed in gastric surface epithelial cells, mediates Cl−/HCO3− exchange, and is inhibited by NH4+. Am. J. Physiol. Cell Physiol. 2005, 289, C493–C505. [Google Scholar] [CrossRef] [PubMed]
- Vincourt, J.B.; Jullien, D.; Amalric, F.; Girard, J.P. Molecular and functional characterization of SLC26A11, a sodium-independent sulfate transporter from high endothelial venules. FASEB J. 2003, 17, 890–892. [Google Scholar] [CrossRef]
- Stewart, A.K.; Shmukler, B.E.; Vandorpe, D.H.; Reimold, F.; Heneghan, J.F.; Nakakuki, M.; Akhavein, A.; Ko, S.; Ishiguro, H.; Alper, S.L. SLC26 anion exchangers of guinea pig pancreatic duct: Molecular cloning and functional characterization. Am. J. Physiol. Cell Physiol. 2011, 301, C289–C303. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Huang, H.; Chen, Y.; Wang, F.; Hao, A.; Zhan, W.; Mao, Q.; Hu, Y.; Han, L.; et al. Asymmetric pendrin homodimer reveals its molecular mechanism as anion exchanger. Nat. Commun. 2023, 14, 3012. [Google Scholar] [CrossRef]
- Izuhara, K.; Suzuki, S.; Ogawa, M.; Nunomura, S.; Nanri, Y.; Mitamura, Y.; Yoshihara, T. The Significance of Hypothiocyanite Production via the Pendrin/DUOX/Peroxidase Pathway in the Pathogenesis of Asthma. Oxidative Med. Cell. Longev. 2017, 2017, 1054801. [Google Scholar] [CrossRef]
- Sharma, A.K.; Krieger, T.; Rigby, A.C.; Zelikovic, I.; Alper, S.L. Human SLC26A4/Pendrin STAS domain is a nucleotide-binding protein: Refolding and characterization for structural studies. Biochem. Biophys. Rep. 2016, 8, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, V.C.; Kraiem, Z.; Beck, J.C.; Nishimura, D.; Stone, E.M.; Salameh, M.; Sadeh, O.; Glaser, B. Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organification. Nat. Genet. 1996, 12, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Friis, I.J.; Aaberg, K.; Edholm, B. Causes of hearing loss and implantation age in a cohort of Danish pediatric cochlear implant recipients. Int. J. Pediatr. Otorhinolaryngol. 2023, 171, 111640. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.M.; Ko, H.C.; Tsou, Y.T.; Lin, Y.H.; Lin, J.L.; Chen, C.K.; Chen, P.L.; Wu, C.C. Long-Term Cochlear Implant Outcomes in Children with GJB2 and SLC26A4 Mutations. PLoS ONE 2015, 10, e0138575. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Cucci, R.A.; Prasad, S.; Green, G.E.; Edeal, J.B.; Galer, C.E.; Karniski, L.P.; Sheffield, V.C.; Smith, R.J. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum. Mutat. 2001, 17, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Dossena, S.; Rodighiero, S.; Vezzoli, V.; Nofziger, C.; Salvioni, E.; Boccazzi, M.; Grabmayer, E.; Botta, G.; Meyer, G.; Fugazzola, L.; et al. Functional characterization of wild-type and mutated pendrin (SLC26A4), the anion transporter involved in Pendred syndrome. J. Mol. Endocrinol. 2009, 43, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Amatngalim, G.D.; Hiemstra, P.S. Airway Epithelial Cell Function and Respiratory Host Defense in Chronic Obstructive Pulmonary Disease. Chin. Med. J. 2018, 131, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.F.; Sin, D.D. The airway epithelium: More than just a structural barrier. Ther. Adv. Respir. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef]
- Widdicombe, J.H. Regulation of the depth and composition of airway surface liquid. J. Anat. 2002, 201, 313–318. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [PubMed]
- Widdicombe, J.H.; Wine, J.J. Airway Gland Structure and Function. Physiol. Rev. 2015, 95, 1241–1319. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, S.; Scantamburlo, G.; Dossena, S.; Paulmichl, M.; Nofziger, C. Interleukin-Mediated Pendrin Transcriptional Regulation in Airway and Esophageal Epithelia. Int. J. Mol. Sci. 2019, 20, 731. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Yoo, J.E.; Namkung, W.; Cho, H.J.; Kim, K.; Kang, J.W.; Yoon, J.H.; Choi, J.Y. Thick airway surface liquid volume and weak mucin expression in pendrin-deficient human airway epithelia. Physiol. Rep. 2015, 3, e12480. [Google Scholar] [CrossRef] [PubMed]
- Haggie, P.M.; Phuan, P.W.; Tan, J.A.; Zlock, L.; Finkbeiner, W.E.; Verkman, A.S. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J. 2016, 30, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Scantamburlo, G.; Vanoni, S.; Dossena, S.; Soyal, S.M.; Bernardinelli, E.; Civello, D.A.; Patsch, W.; Paulmichl, M.; Nofziger, C. Interleukin-4 Induces CpG Site-Specific Demethylation of the Pendrin Promoter in Primary Human Bronchial Epithelial Cells. Cell. Physiol. Biochem. 2017, 41, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Ranieri, M.; Di Mise, A.; Dossena, S.; Pellegrino, T.; Furia, E.; Nofziger, C.; Debellis, L.; Paulmichl, M.; Valenti, G.; et al. Interleukin-13 increases pendrin abundance to the cell surface in bronchial NCI-H292 cells via Rho/actin signaling. Pflug. Arch. 2017, 469, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ogawa, M.; Ohta, S.; Nunomura, S.; Nanri, Y.; Shiraishi, H.; Mitamura, Y.; Yoshihara, T.; Lee, J.J.; Izuhara, K. Induction of Airway Allergic Inflammation by Hypothiocyanite via Epithelial Cells. J. Biol. Chem. 2016, 291, 27219–27227. [Google Scholar] [CrossRef]
- Do, D.C.; Zhang, Y.; Tu, W.; Hu, X.; Xiao, X.; Chen, J.; Hao, H.; Liu, Z.; Li, J.; Huang, S.K.; et al. Type II alveolar epithelial cell-specific loss of RhoA exacerbates allergic airway inflammation through SLC26A4. JCI Insight 2021, 6, e148147. [Google Scholar] [CrossRef]
- Jia, C.E.; Jiang, D.Y.; Dai, H.P.; Xiao, F.; Wang, C. Pendrin, an anion exchanger on lung epithelial cells, could be a novel target for lipopolysaccharide-induced acute lung injury mice. Am. J. Transl. Res. 2016, 8, 981–992. [Google Scholar] [PubMed]
- Lee, E.H.; Shin, M.H.; Gi, M.; Park, J.; Song, D.; Hyun, Y.M.; Ryu, J.H.; Seong, J.K.; Jeon, Y.; Han, G.; et al. Inhibition of Pendrin by a small molecule reduces Lipopolysaccharide-induced acute Lung Injury. Theranostics 2020, 10, 9913–9922. [Google Scholar] [CrossRef] [PubMed]
- Guyton, A.C. Roles of the kidneys and fluid volumes in arterial pressure regulation and hypertension. Chin. J. Physiol. 1989, 32, 49–57. [Google Scholar] [PubMed]
- An, C.; Yang, L.; Han, T.; Song, H.; Li, Z.; Zhang, J.; Zhang, K. Kidney ion handling genes and their interaction in blood pressure control. Biosci. Rep. 2022, 42, BSR20220977. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Ehrlich, B.E. Ion channels in renal disease. Chem. Rev. 2012, 112, 6353–6372. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, E.H.J.; Castenmiller, C.; Peters, D.J.M.; Arjona, F.J.; Bindels, R.J.M.; Hoenderop, J.G.J. Sensing of tubular flow and renal electrolyte transport. Nat. Rev. Nephrol. 2020, 16, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.M.; Verlander, J.W.; Romero, C.A. The Renal Physiology of Pendrin-Positive Intercalated Cells. Physiol. Rev. 2020, 100, 1119–1147. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kwon, T.H.; Frische, S.; Kim, J.; Tisher, C.C.; Madsen, K.M.; Nielsen, S. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am. J. Physiol. Ren. Physiol. 2002, 283, F744–F754. [Google Scholar] [CrossRef] [PubMed]
- Royaux, I.E.; Wall, S.M.; Karniski, L.P.; Everett, L.A.; Suzuki, K.; Knepper, M.A.; Green, E.D. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc. Natl. Acad. Sci. USA 2001, 98, 4221–4226. [Google Scholar] [CrossRef]
- Wall, S.M.; Hassell, K.A.; Royaux, I.E.; Green, E.D.; Chang, J.Y.; Shipley, G.L.; Verlander, J.W. Localization of pendrin in mouse kidney. Am. J. Physiol. Ren. Physiol. 2003, 284, F229–F241. [Google Scholar] [CrossRef]
- Wall, S.M. Renal intercalated cells and blood pressure regulation. Kidney Res. Clin. Pract. 2017, 36, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.M. The role of pendrin in blood pressure regulation. Am. J. Physiol. Ren. Physiol. 2016, 310, F193–F203. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Yoo, T.H.; Yoo, J.E.; Seo, Y.J.; Jung, J.; Choi, J.Y. Resistance to hypertension and high Cl− excretion in humans with SLC26A4 mutations. Clin. Genet. 2017, 91, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Trepiccione, F.; Soukaseum, C.; Baudrie, V.; Kumai, Y.; Teulon, J.; Villoutreix, B.; Corniere, N.; Wangemann, P.; Griffith, A.J.; Byung Choi, Y.; et al. Acute genetic ablation of pendrin lowers blood pressure in mice. Nephrol. Dial. Transpl. 2017, 32, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S. Role of Pendrin in the Pathophysiology of Aldosterone-Induced Hypertension. Am. J. Hypertens. 2019, 32, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.; Verlander, J.W.; Wang, Y.; Romero, C.A.; Yue, Q.; Chen, C.; Thumova, M.; Eaton, D.C.; Lazo-Fernandez, Y.; Wall, S.M. Aldosterone Regulates Pendrin and Epithelial Sodium Channel Activity through Intercalated Cell Mineralocorticoid Receptor-Dependent and -Independent Mechanisms over a Wide Range in Serum Potassium. J. Am. Soc. Nephrol. 2020, 31, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Ayuzawa, N.; Nishimoto, M.; Ueda, K.; Hirohama, D.; Kawarazaki, W.; Shimosawa, T.; Marumo, T.; Fujita, T. Two Mineralocorticoid Receptor-Mediated Mechanisms of Pendrin Activation in Distal Nephrons. J. Am. Soc. Nephrol. 2020, 31, 748–764. [Google Scholar] [CrossRef] [PubMed]
- Hirohama, D.; Ayuzawa, N.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Watanabe, A.; Shimosawa, T.; Marumo, T.; Shibata, S.; Fujita, T. Aldosterone Is Essential for Angiotensin II-Induced Upregulation of Pendrin. J. Am. Soc. Nephrol. 2018, 29, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Nanami, M.; Pham, T.D.; Kim, Y.H.; Yang, B.; Sutliff, R.L.; Staub, O.; Klein, J.D.; Lopez-Cayuqueo, K.I.; Chambrey, R.; Park, A.Y.; et al. The Role of Intercalated Cell Nedd4-2 in BP Regulation, Ion Transport, and Transporter Expression. J. Am. Soc. Nephrol. 2018, 29, 1706–1719. [Google Scholar] [CrossRef]
- Vissenberg, R.; Manders, V.D.; Mastenbroek, S.; Fliers, E.; Afink, G.B.; Ris-Stalpers, C.; Goddijn, M.; Bisschop, P.H. Pathophysiological aspects of thyroid hormone disorders/thyroid peroxidase autoantibodies and reproduction. Hum. Reprod. Update 2015, 21, 378–387. [Google Scholar] [CrossRef]
- Salazar, P.; Cisternas, P.; Martinez, M.; Inestrosa, N.C. Hypothyroidism and Cognitive Disorders during Development and Adulthood: Implications in the Central Nervous System. Mol. Neurobiol. 2019, 56, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Markou, K.; Georgopoulos, N.; Kyriazopoulou, V.; Vagenakis, A.G. Iodine-Induced hypothyroidism. Thyroid 2001, 11, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.C.; Kopp, P.A. Pendrin and anoctamin as mediators of apical iodide efflux in thyroid cells. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Bizhanova, A.; Kopp, P. Minireview: The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology 2009, 150, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Royaux, I.E.; Suzuki, K.; Mori, A.; Katoh, R.; Everett, L.A.; Kohn, L.D.; Green, E.D. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 2000, 141, 839–845. [Google Scholar] [CrossRef]
- Pesce, L.; Bizhanova, A.; Caraballo, J.C.; Westphal, W.; Butti, M.L.; Comellas, A.; Kopp, P. TSH regulates pendrin membrane abundance and enhances iodide efflux in thyroid cells. Endocrinology 2012, 153, 512–521. [Google Scholar] [CrossRef]
- Raad, H.; Eskalli, Z.; Corvilain, B.; Miot, F.; De Deken, X. Thyroid hydrogen peroxide production is enhanced by the Th2 cytokines, IL-4 and IL-13, through increased expression of the dual oxidase 2 and its maturation factor DUOXA2. Free Radic. Biol. Med. 2013, 56, 216–225. [Google Scholar] [CrossRef]
- Eskalli, Z.; Achouri, Y.; Hahn, S.; Many, M.C.; Craps, J.; Refetoff, S.; Liao, X.H.; Dumont, J.E.; Van Sande, J.; Corvilain, B.; et al. Overexpression of Interleukin-4 in the Thyroid of Transgenic Mice Upregulates the Expression of Duox1 and the Anion Transporter Pendrin. Thyroid 2016, 26, 1499–1512. [Google Scholar] [CrossRef]
- Merakchi, K.; Djerbib, S.; Soleimani, M.; Dumont, J.E.; Miot, F.; De Deken, X. Murine Thyroid IL-4 Expression Worsens Hypothyroidism on Iodine Restriction and Mitigates Graves Disease Development. Endocrinology 2022, 163, bqac107. [Google Scholar] [CrossRef]
- Calcaterra, V.; Lamberti, R.; Viggiano, C.; Gatto, S.; Spaccini, L.; Lista, G.; Zuccotti, G. Neonatal Dyshormonogenetic Goiter with Hypothyroidism Associated with Novel Mutations in Thyroglobulin and SLC26A4 Gene. Pediatr. Rep. 2021, 13, 210–215. [Google Scholar] [CrossRef]
- Mukherjee, S.; Guha, M.; Adhikary, B.; Bankura, B.; Mitra, P.; Chowdhury, S.; Das, M. Genetic Alterations in Pendrin (SLC26A4) Gene in Adult Hypothyroid Patients. Horm. Metab. Res. 2017, 49, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Farebrother, J.; Zimmermann, M.B.; Andersson, M. Excess iodine intake: Sources, assessment, and effects on thyroid function. Ann. N. Y. Acad. Sci. 2019, 1446, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Lin, C.H.; Yang, L.H.; Li, W.G.; Zhang, J.W.; Zheng, W.W.; Wang, X.; Qian, J.; Huang, J.L.; Lei, Y.X. The Effect on Sodium/Iodide Symporter and Pendrin in Thyroid Colloid Retention Developed by Excess Iodide Intake. Biol. Trace Elem. Res. 2016, 172, 193–200. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Do, B.H.; Kitamura, T.; Ohkubo, J.I.; Wakasugi, T.; Ohbuchi, T.; Suzuki, H. Expression of Cl− channels/transporters in nasal polyps. Eur. Arch. Otorhinolaryngol. 2020, 277, 2263–2270. [Google Scholar] [CrossRef]
- Seshadri, S.; Lu, X.; Purkey, M.R.; Homma, T.; Choi, A.W.; Carter, R.; Suh, L.; Norton, J.; Harris, K.E.; Conley, D.B.; et al. Increased expression of the epithelial anion transporter pendrin/SLC26A4 in nasal polyps of patients with chronic rhinosinusitis. J. Allergy Clin. Immunol. 2015, 136, 1548–1558.e7. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ikeda, S.; Asamori, T.; Honda, K.; Kawashima, Y.; Kitamura, K.; Suzuki, K.; Tsutsumi, T. Increased expression of pendrin in eosinophilic chronic rhinosinusitis with nasal polyps. Braz. J. Otorhinolaryngol. 2019, 85, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Helguera, O.; Anguiano, B.; Delgado, G.; Aceves, C. Uptake and antiproliferative effect of molecular iodine in the MCF-7 breast cancer cell line. Endocr. Relat. Cancer 2006, 13, 1147–1158. [Google Scholar] [CrossRef][Green Version]
- Garcia-Solis, P.; Alfaro, Y.; Anguiano, B.; Delgado, G.; Guzman, R.C.; Nandi, S.; Diaz-Munoz, M.; Vazquez-Martinez, O.; Aceves, C. Inhibition of N-methyl-N-nitrosourea-induced mammary carcinogenesis by molecular iodine (I2) but not by iodide (I−) treatment Evidence that I2 prevents cancer promotion. Mol. Cell. Endocrinol. 2005, 236, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Arczewska, K.D.; Godlewska, M.; Krasuska, W.; Lyczkowska, A.; Kiedrowski, M.; Czarnocka, B. Expression of pendrin and NIS iodide transporters in human breast tumor and peri-tumoral tissue. Arch. Med. Sci. 2022, 18, 1041–1050. [Google Scholar] [CrossRef]
- Luo, C.; Liu, Z.; Gan, Y.; Gao, X.; Zu, X.; Zhang, Y.; Ye, W.; Cai, Y. SLC26A4 correlates with homologous recombination deficiency and patient prognosis in prostate cancer. J. Transl. Med. 2022, 20, 313. [Google Scholar] [CrossRef] [PubMed]
- Makhlouf, A.M.; Chitikova, Z.; Pusztaszeri, M.; Berczy, M.; Delucinge-Vivier, C.; Triponez, F.; Meyer, P.; Philippe, J.; Dibner, C. Identification of CHEK1, SLC26A4, c-KIT, TPO and TG as new biomarkers for human follicular thyroid carcinoma. Oncotarget 2016, 7, 45776–45788. [Google Scholar] [CrossRef] [PubMed]
- Zane, M.; Agostini, M.; Enzo, M.V.; Casal Ide, E.; Del Bianco, P.; Torresan, F.; Merante Boschin, I.; Pennelli, G.; Saccani, A.; Rubello, D.; et al. Circulating cell-free DNA, SLC5A8 and SLC26A4 hypermethylation, BRAFV600E: A non-invasive tool panel for early detection of thyroid cancer. Biomed. Pharmacother. 2013, 67, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Rauluseviciute, I.; Drablos, F.; Rye, M.B. DNA hypermethylation associated with upregulated gene expression in prostate cancer demonstrates the diversity of epigenetic regulation. BMC Med. Genom. 2020, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Dhar, G.A.; Saha, S.; Mitra, P.; Nag Chaudhuri, R. DNA methylation and regulation of gene expression: Guardian of our health. Nucleus 2021, 64, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Dossena, S. Functional interplay between CFTR and pendrin: Physiological and pathophysiological relevance. Front. Biosci. (Landmark Ed.) 2022, 27, 75. [Google Scholar] [CrossRef] [PubMed]
- Bajko, J.; Duguid, M.; Altmann, S.; Hurlbut, G.D.; Kaczmarek, J.S. Pendrin stimulates a chloride absorption pathway to increase CFTR-mediated chloride secretion from Cystic Fibrosis airway epithelia. FASEB BioAdv. 2020, 2, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Pranke, I.; Villeret, B.; Cottart, C.H.; et al. Airway surface liquid acidification initiates host defense abnormalities in Cystic Fibrosis. Sci. Rep. 2019, 9, 6516. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Huang, J.; Billet, A.; Abu-Arish, A.; Goepp, J.; Matthes, E.; Tewfik, M.A.; Frenkiel, S.; Hanrahan, J.W. Pendrin Mediates Bicarbonate Secretion and Enhances Cystic Fibrosis Transmembrane Conductance Regulator Function in Airway Surface Epithelia. Am. J. Respir. Cell Mol. Biol. 2019, 60, 705–716. [Google Scholar] [CrossRef]
- Cheng, S.H.; Fang, S.L.; Zabner, J.; Marshall, J.; Piraino, S.; Schiavi, S.C.; Jefferson, D.M.; Welsh, M.J.; Smith, A.E. Functional activation of the cystic fibrosis trafficking mutant delta F508-CFTR by overexpression. Am. J. Physiol. 1995, 268, L615–L624. [Google Scholar] [CrossRef]
- Park, H.; Shin, D.H.; Sim, J.R.; Aum, S.; Lee, M.G. IRE1α kinase-mediated unconventional protein secretion rescues misfolded CFTR and pendrin. Sci. Adv. 2020, 6, eaax9914. [Google Scholar] [CrossRef]
- Park, H.; Seo, S.K.; Sim, J.R.; Hwang, S.J.; Kim, Y.J.; Shin, D.H.; Jang, D.G.; Noh, S.H.; Park, P.G.; Ko, S.H.; et al. TMED3 Complex Mediates ER Stress-Associated Secretion of CFTR, Pendrin, and SARS-CoV-2 Spike. Adv. Sci. 2022, 9, e2105320. [Google Scholar] [CrossRef]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef]
- Jung, J.; Kim, J.; Roh, S.H.; Jun, I.; Sampson, R.D.; Gee, H.Y.; Choi, J.Y.; Lee, M.G. The HSP70 co-chaperone DNAJC14 targets misfolded pendrin for unconventional protein secretion. Nat. Commun. 2016, 7, 11386. [Google Scholar] [CrossRef]
- Berg, P.; Svendsen, S.L.; Sorensen, M.V.; Larsen, C.K.; Andersen, J.F.; Jensen-Fangel, S.; Jeppesen, M.; Schreiber, R.; Cabrita, I.; Kunzelmann, K.; et al. Impaired Renal HCO3− Excretion in Cystic Fibrosis. J. Am. Soc. Nephrol. 2020, 31, 1711–1727. [Google Scholar] [CrossRef]
- Berg, P.; Svendsen, S.L.; Hoang, T.T.L.; Praetorius, H.A.; Sorensen, M.V.; Leipziger, J. Impaired renal HCO3− secretion in CFTR deficient mice causes metabolic alkalosis during chronic base-loading. Acta Physiol. 2021, 231, e13591. [Google Scholar] [CrossRef]
- Varasteh Kia, M.; Barone, S.; McDonough, A.A.; Zahedi, K.; Xu, J.; Soleimani, M. Downregulation of the Cl−/HCO3− Exchanger Pendrin in Kidneys of Mice with Cystic Fibrosis: Role in the Pathogenesis of Metabolic Alkalosis. Cell. Physiol. Biochem. 2018, 45, 1551–1565. [Google Scholar] [CrossRef]
- Baum, M. Developmental changes in proximal tubule NaCl transport. Pediatr. Nephrol. 2008, 23, 185–194. [Google Scholar] [CrossRef]
- Wang, X.; Armando, I.; Upadhyay, K.; Pascua, A.; Jose, P.A. The regulation of proximal tubular salt transport in hypertension: An update. Curr. Opin. Nephrol. Hypertens. 2009, 18, 412–420. [Google Scholar] [CrossRef]
- Pech, V.; Pham, T.D.; Hong, S.; Weinstein, A.M.; Spencer, K.B.; Duke, B.J.; Walp, E.; Kim, Y.H.; Sutliff, R.L.; Bao, H.F.; et al. Pendrin modulates ENaC function by changing luminal HCO3−. J. Am. Soc. Nephrol. 2010, 21, 1928–1941. [Google Scholar] [CrossRef]
- Pech, V.; Wall, S.M.; Nanami, M.; Bao, H.F.; Kim, Y.H.; Lazo-Fernandez, Y.; Yue, Q.; Pham, T.D.; Eaton, D.C.; Verlander, J.W. Pendrin gene ablation alters ENaC subcellular distribution and open probability. Am. J. Physiol. Ren. Physiol. 2015, 309, F154–F163. [Google Scholar] [CrossRef]
- Patel-Chamberlin, M.; Varasteh Kia, M.; Xu, J.; Barone, S.; Zahedi, K.; Soleimani, M. The Role of Epithelial Sodium Channel ENaC and the Apical Cl−/HCO3− Exchanger Pendrin in Compensatory Salt Reabsorption in the Setting of Na-Cl Cotransporter (NCC) Inactivation. PLoS ONE 2016, 11, e0150918. [Google Scholar] [CrossRef]
- Relman, A.S.; Schwartz, W.B. The nephropathy of potassium depletion; a clinical and pathological entity. N. Engl. J. Med. 1956, 255, 195–203. [Google Scholar] [CrossRef]
- Smith, S.G.; Lasater, T.E. A diabetes insipidus-like condition produced in dogs by a potassium deficient diet. Proc. Soc. Exp. Biol. Med. 1950, 74, 427–431. [Google Scholar] [CrossRef]
- Xu, N.; Hirohama, D.; Ishizawa, K.; Chang, W.X.; Shimosawa, T.; Fujita, T.; Uchida, S.; Shibata, S. Hypokalemia and Pendrin Induction by Aldosterone. Hypertension 2017, 69, 855–862. [Google Scholar] [CrossRef]
- Boyd-Shiwarski, C.R.; Weaver, C.J.; Beacham, R.T.; Shiwarski, D.J.; Connolly, K.A.; Nkashama, L.J.; Mutchler, S.M.; Griffiths, S.E.; Knoell, S.A.; Sebastiani, R.S.; et al. Effects of extreme potassium stress on blood pressure and renal tubular sodium transport. Am. J. Physiol. Ren. Physiol. 2020, 318, F1341–F1356. [Google Scholar] [CrossRef]
- Regeer, R.R.; Markovich, D. A dileucine motif targets the sulfate anion transporter sat-1 to the basolateral membrane in renal cell lines. Am. J. Physiol. Cell Physiol. 2004, 287, C365–C372. [Google Scholar] [CrossRef][Green Version]
- Chapman, J.M.; Karniski, L.P. Protein localization of SLC26A2 (DTDST) in rat kidney. Histochem. Cell Biol. 2010, 133, 541–547. [Google Scholar] [CrossRef]
- Xu, J.; Barone, S.; Li, H.; Holiday, S.; Zahedi, K.; Soleimani, M. Slc26a11, a chloride transporter, localizes with the vacuolar H+-ATPase of A-intercalated cells of the kidney. Kidney Int. 2011, 80, 926–937. [Google Scholar] [CrossRef]
- Aronson, P.S. Essential roles of CFEX-mediated Cl−-oxalate exchange in proximal tubule NaCl transport and prevention of urolithiasis. Kidney Int. 2006, 70, 1207–1213. [Google Scholar] [CrossRef]
- Petrovic, S.; Barone, S.; Wang, Z.; McDonough, A.A.; Amlal, H.; Soleimani, M. Slc26a6 (PAT1) deletion downregulates the apical Na+/H+ exchanger in the straight segment of the proximal tubule. Am. J. Nephrol. 2008, 28, 330–338. [Google Scholar] [CrossRef]
- Barone, S.; Amlal, H.; Kujala, M.; Xu, J.; Karet, F.; Blanchard, A.; Kere, J.; Soleimani, M. Regulation of the basolateral chloride/base exchangers AE1 and SLC26A7 in the kidney collecting duct in potassium depletion. Nephrol. Dial. Transpl. 2007, 22, 3462–3470. [Google Scholar] [CrossRef]
- Xu, J.; Worrell, R.T.; Li, H.C.; Barone, S.L.; Petrovic, S.; Amlal, H.; Soleimani, M. Chloride/bicarbonate exchanger SLC26A7 is localized in endosomes in medullary collecting duct cells and is targeted to the basolateral membrane in hypertonicity and potassium depletion. J. Am. Soc. Nephrol. 2006, 17, 956–967. [Google Scholar] [CrossRef]
- Xu, J.; Song, P.; Nakamura, S.; Miller, M.; Barone, S.; Alper, S.L.; Riederer, B.; Bonhagen, J.; Arend, L.J.; Amlal, H.; et al. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J. Biol. Chem. 2009, 284, 29470–29479. [Google Scholar] [CrossRef]
- Cangul, H.; Liao, X.H.; Schoenmakers, E.; Kero, J.; Barone, S.; Srichomkwun, P.; Iwayama, H.; Serra, E.G.; Saglam, H.; Eren, E.; et al. Homozygous loss-of-function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI Insight 2018, 3, e99631. [Google Scholar] [CrossRef]
- Tanimura, Y.; Kiriya, M.; Kawashima, A.; Mori, H.; Luo, Y.; Kondo, T.; Suzuki, K. Regulation of solute carrier family 26 member 7 (Slc26a7) by thyroid stimulating hormone in thyrocytes. Endocr. J. 2021, 68, 691–699. [Google Scholar] [CrossRef]
- Jo, S.; Centeio, R.; Park, J.; Ousingsawat, J.; Jeon, D.K.; Talbi, K.; Schreiber, R.; Ryu, K.; Kahlenberg, K.; Somoza, V.; et al. The SLC26A9 inhibitor S9-A13 provides no evidence for a role of SLC26A9 in airway chloride secretion but suggests a contribution to regulation of ASL pH and gastric proton secretion. FASEB J. 2022, 36, e22534. [Google Scholar] [CrossRef]
- Avella, M.; Loriol, C.; Boulukos, K.; Borgese, F.; Ehrenfeld, J. SLC26A9 stimulates CFTR expression and function in human bronchial cell lines. J. Cell. Physiol. 2011, 226, 212–223. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Centeio, R.; Schreiber, R.; Kunzelmann, K. Expression of SLC26A9 in Airways and Its Potential Role in Asthma. Int. J. Mol. Sci. 2022, 23, 2998. [Google Scholar] [CrossRef]
- Anagnostopoulou, P.; Riederer, B.; Duerr, J.; Michel, S.; Binia, A.; Agrawal, R.; Liu, X.; Kalitzki, K.; Xiao, F.; Chen, M.; et al. SLC26A9-mediated chloride secretion prevents mucus obstruction in airway inflammation. J. Clin. Investig. 2012, 122, 3629–3634. [Google Scholar] [CrossRef]
- Amlal, H.; Xu, J.; Barone, S.; Zahedi, K.; Soleimani, M. The chloride channel/transporter Slc26a9 regulates the systemic arterial pressure and renal chloride excretion. J. Mol. Med. 2013, 91, 561–572. [Google Scholar] [CrossRef]
- Bertrand, C.A.; Zhang, R.; Pilewski, J.M.; Frizzell, R.A. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J. Gen. Physiol. 2009, 133, 421–438. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. Differential contribution of SLC26A9 to Cl− conductance in polarized and non-polarized epithelial cells. J. Cell. Physiol. 2012, 227, 2323–2329. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Centeio, R.; Ousingsawat, J.; Talbi, K.; Seidler, U.; Schreiber, R. SLC26A9 in airways and intestine: Secretion or absorption? Channels 2023, 17, 2186434. [Google Scholar] [CrossRef]
- Bertrand, C.A.; Mitra, S.; Mishra, S.K.; Wang, X.; Zhao, Y.; Pilewski, J.M.; Madden, D.R.; Frizzell, R.A. The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L912–L925. [Google Scholar] [CrossRef]
- Gorrieri, G.; Zara, F.; Scudieri, P. SLC26A9 as a Potential Modifier and Therapeutic Target in Cystic Fibrosis Lung Disease. Biomolecules 2022, 12, 202. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Ousingsawat, J.; Kraus, A.; Park, J.H.; Marquardt, T.; Schreiber, R.; Buchholz, B. Pathogenic Relationships in Cystic Fibrosis and Renal Diseases: CFTR, SLC26A9 and Anoctamins. Int. J. Mol. Sci. 2023, 24, 13278. [Google Scholar] [CrossRef]
- Liu, L.; Yamamoto, A.; Yamaguchi, M.; Taniguchi, I.; Nomura, N.; Nakakuki, M.; Kozawa, Y.; Fukuyasu, T.; Higuchi, M.; Niwa, E.; et al. Bicarbonate transport of airway surface epithelia in luminally perfused mice bronchioles. J. Physiol. Sci. 2022, 72, 4. [Google Scholar] [CrossRef]
- Wheat, V.J.; Shumaker, H.; Burnham, C.; Shull, G.E.; Yankaskas, J.R.; Soleimani, M. CFTR induces the expression of DRA along with Cl−/HCO3− exchange activity in tracheal epithelial cells. Am. J. Physiol. Cell Physiol. 2000, 279, C62–C71. [Google Scholar] [CrossRef]
- Kim, B.S.; Yu, M.Y.; Shin, J. Effect of low sodium and high potassium diet on lowering blood pressure and cardiovascular events. Clin. Hypertens. 2024, 30, 2. [Google Scholar] [CrossRef]
- Dror, A.A.; Politi, Y.; Shahin, H.; Lenz, D.R.; Dossena, S.; Nofziger, C.; Fuchs, H.; Hrabe de Angelis, M.; Paulmichl, M.; Weiner, S.; et al. Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J. Biol. Chem. 2010, 285, 21724–21735. [Google Scholar] [CrossRef]
- Tarhuni, M.; Fotso, M.N.; Gonzalez, N.A.; Sanivarapu, R.R.; Osman, U.; Latha Kumar, A.; Sadagopan, A.; Mahmoud, A.; Begg, M.; Hamid, P. Estrogen’s Tissue-Specific Regulation of the SLC26A6 Anion Transporter Reveal a Phenotype of Kidney Stone Disease in Estrogen-Deficient Females: A Systematic Review. Cureus 2023, 15, e45839. [Google Scholar] [CrossRef]
- Ullah, A.; Rumley, A.C.; Peleh, V.; Fernandes, D.; Almomani, E.Y.; Berrini, M.; Lashhab, R.; Touret, N.; Alexander, R.T.; Herrmann, J.M.; et al. SLC26A7 protein is a chloride/bicarbonate exchanger and its abundance is osmolarity- and pH-dependent in renal epithelial cells. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183238. [Google Scholar] [CrossRef]
- Ishii, J.; Suzuki, A.; Kimura, T.; Tateyama, M.; Tanaka, T.; Yazawa, T.; Arimasu, Y.; Chen, I.S.; Aoyama, K.; Kubo, Y.; et al. Congenital goitrous hypothyroidism is caused by dysfunction of the iodide transporter SLC26A7. Commun. Biol. 2019, 2, 270. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Suzuki, A.; Yoshida, A.; Tanaka, T.; Aoyama, K.; Oishi, H.; Hara, Y.; Ogi, T.; Amano, I.; Kameo, S.; et al. The iodide transporter Slc26a7 impacts thyroid function more strongly than Slc26a4 in mice. Sci. Rep. 2022, 12, 11259. [Google Scholar] [CrossRef]
- Bull, S.C.; Doig, A.J. Properties of protein drug target classes. PLoS ONE 2015, 10, e0117955. [Google Scholar] [CrossRef]
- Fujioka, M.; Akiyama, T.; Hosoya, M.; Kikuchi, K.; Fujiki, Y.; Saito, Y.; Yoshihama, K.; Ozawa, H.; Tsukada, K.; Nishio, S.Y.; et al. A phase I/IIa double blind single institute trial of low dose sirolimus for Pendred syndrome/DFNB4. Medicine 2020, 99, e19763. [Google Scholar] [CrossRef]
- Busi, M.; Rosignoli, M.; Castiglione, A.; Minazzi, F.; Trevisi, P.; Aimoni, C.; Calzolari, F.; Granieri, E.; Martini, A. Cochlear Implant Outcomes and Genetic Mutations in Children with Ear and Brain Anomalies. BioMed Res. Int. 2015, 2015, 696281. [Google Scholar] [CrossRef]
- Kim, M.A.; Kim, S.H.; Ryu, N.; Ma, J.H.; Kim, Y.R.; Jung, J.; Hsu, C.J.; Choi, J.Y.; Lee, K.Y.; Wangemann, P.; et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics 2019, 9, 7184–7199. [Google Scholar] [CrossRef]
- Feng, P.; Xu, Z.; Chen, J.; Liu, M.; Zhao, Y.; Wang, D.; Han, L.; Wang, L.; Wan, B.; Xu, X.; et al. Rescue of mis-splicing of a common SLC26A4 mutant associated with sensorineural hearing loss by antisense oligonucleotides. Mol. Ther. Nucleic Acids 2022, 28, 280–292. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, D.; He, Y.; Shu, Y. Advances in gene therapy hold promise for treating hereditary hearing loss. Mol. Ther. 2023, 31, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Park, H.J.; Yoo, S.Y.; Namkung, W.; Jo, M.J.; Koo, S.K.; Park, H.Y.; Lee, W.S.; Kim, K.H.; Lee, M.G. Heterogeneity in the processing defect of SLC26A4 mutants. J. Med. Genet. 2008, 45, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Liantonio, A.; Camerino, G.M.; De Bellis, M.; Camerino, C.; Mele, A.; Giustino, A.; Pierno, S.; De Luca, A.; Tricarico, D.; et al. Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery. Front. Pharmacol. 2016, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Camerino, D.C.; Desaphy, J.F.; Tricarico, D.; Pierno, S.; Liantonio, A. Therapeutic approaches to ion channel diseases. Adv. Genet. 2008, 64, 81–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, J.; Jin, H.; He, S.; Liao, C.; Wang, J.; Tuo, B. Roles of Cl−/HCO3− anion exchanger 2 in the physiology and pathophysiology of the digestive system (Review). Mol. Med. Rep. 2021, 24, 491. [Google Scholar] [CrossRef]
- Ryu, N.; Kim, M.A.; Choi, D.G.; Kim, Y.R.; Sonn, J.K.; Lee, K.Y.; Kim, U.K. CRISPR/Cas9-mediated genome editing of splicing mutation causing congenital hearing loss. Gene 2019, 703, 83–90. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Transporting Ions | Expression | Refs. |
|---|---|---|---|---|
| SLC26A1 | Sat-1 | SO42−, oxalate, glyoxylate | Hepatocyte, renal proximal tubule, intestine | [2,3] |
| SLC26A2 | DTDST | SO42−, oxalate, Cl− | Chondrocyte, renal proximal tubule, intestine, pancreatic duct | [4] |
| SLC26A3 | DRA, CLD | Cl−, HCO3−, oxalate | Enterocyte, sperm, epididymis | [5,6] |
| SLC26A4 | Pendrin | I−, Cl−, HCO3−, SCN− | Cochlear, vestibular epithelial cell, thyrocyte, type B intercalated cell, airway epithelial cell | [7,8] |
| SLC26A5 | Prestin | Cl−, formate, oxalate, SO42− | Cochlear hair cell | [9,10] |
| SLC26A6 | Pat-1, CFEX | Cl−, HCO3−, oxalate, OH−, formate | Enterocyte, pancreatic duct, renal proximal tubule, cardiac myocyte, sperm | [3,11,12] |
| SLC26A7 | SUT2 | Cl−, HCO3−, OH− | Gastric parietal cell, type A intercalated cell, endothelial cell | [13,14] |
| SLC26A8 | TAT1 | Cl−, SO42− | Male germ cell, sperm | [15,16] |
| SLC26A9 | - | Cl−, HCO3− | Airway epithelial cell, gastric parietal cell | [17] |
| SLC26A11 | SUT1, KBAT | Cl−, HCO3−, SO42−, oxalate | Renal intercalated cell, pancreatic duct, endothelial cell, brain | [18,19] |
| Transporters | Expression | Functions | Refs. |
|---|---|---|---|
| Slc26a6 | Mouse bladder | Induction of calcium oxalate stones | [110] |
| Mouse proximal tubule | Decrease in Nhe3 expression | [111] | |
| SLC26A7, Slc26a7 | Mouse renal outer medulla | Increased by high blood pressure | [112,113] |
| Mouse distal renal tubule | Induction of acidosis | [114] | |
| Mouse thyroid | Decrease in thyroid hormone | [115] | |
| FRTL-5 | Translocated by thyroid stimulating hormone | [116] | |
| SLC26A9, Slc26a9 | Mouse airway surface liquid | Induction of acidification | [117] |
| CFBE41o | Increase in CFTR current | [118] | |
| Human Asthmatic airway | Overexpressed in cells | [119,120] | |
| Mouse kidney medullary collecting duct | Increase in arterial pressure | [121] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.; Hong, J.H. Chloride/Multiple Anion Exchanger SLC26A Family: Systemic Roles of SLC26A4 in Various Organs. Int. J. Mol. Sci. 2024, 25, 4190. https://doi.org/10.3390/ijms25084190
Lee D, Hong JH. Chloride/Multiple Anion Exchanger SLC26A Family: Systemic Roles of SLC26A4 in Various Organs. International Journal of Molecular Sciences. 2024; 25(8):4190. https://doi.org/10.3390/ijms25084190
Chicago/Turabian StyleLee, Dongun, and Jeong Hee Hong. 2024. "Chloride/Multiple Anion Exchanger SLC26A Family: Systemic Roles of SLC26A4 in Various Organs" International Journal of Molecular Sciences 25, no. 8: 4190. https://doi.org/10.3390/ijms25084190
APA StyleLee, D., & Hong, J. H. (2024). Chloride/Multiple Anion Exchanger SLC26A Family: Systemic Roles of SLC26A4 in Various Organs. International Journal of Molecular Sciences, 25(8), 4190. https://doi.org/10.3390/ijms25084190