
Tumor-Extrinsic Axl Expression Shapes an Inflammatory Microenvironment Independent of Tumor Cell Promoting Axl Signaling in Hepatocellular Carcinoma

 , , and
, , and 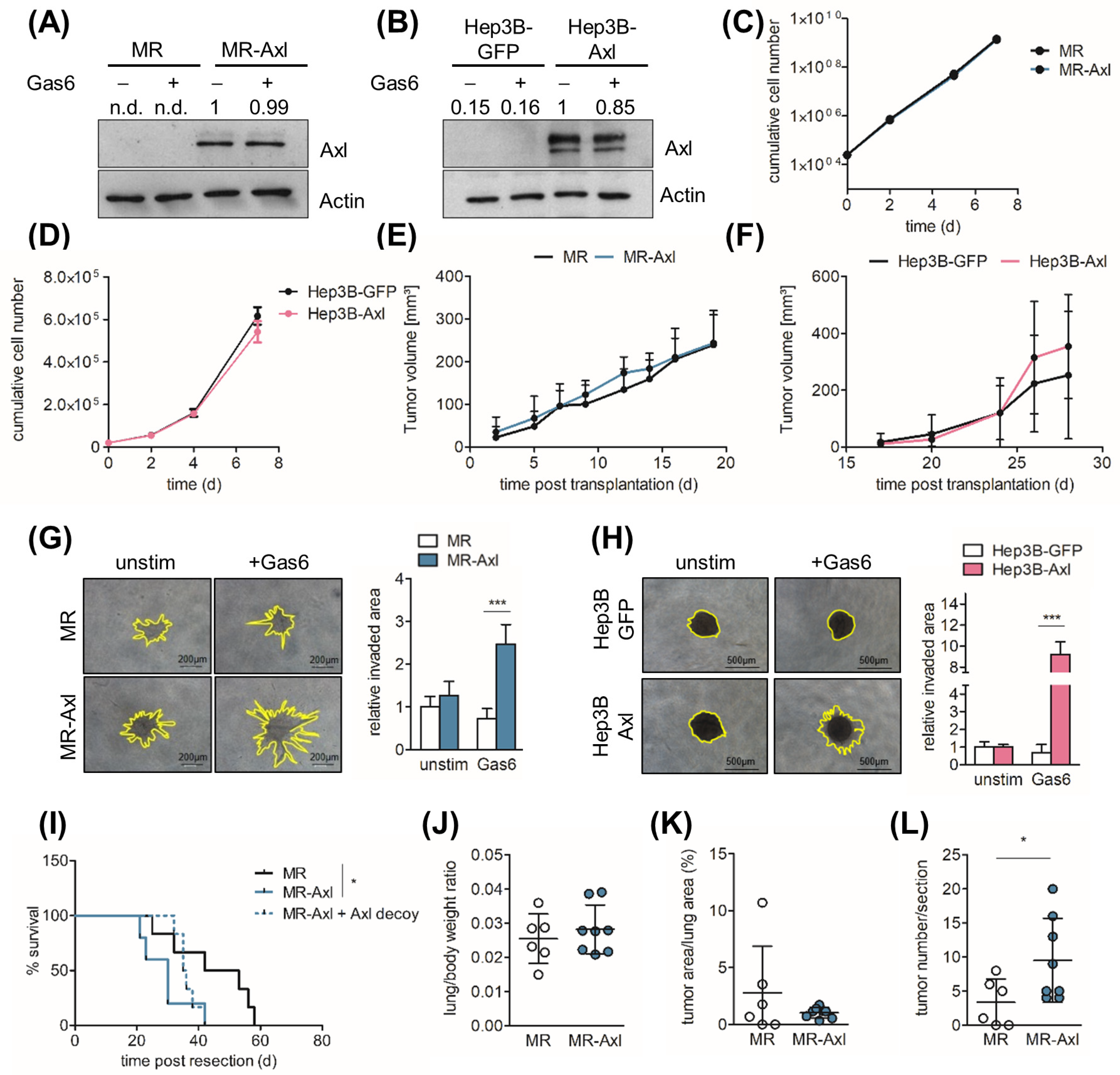{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
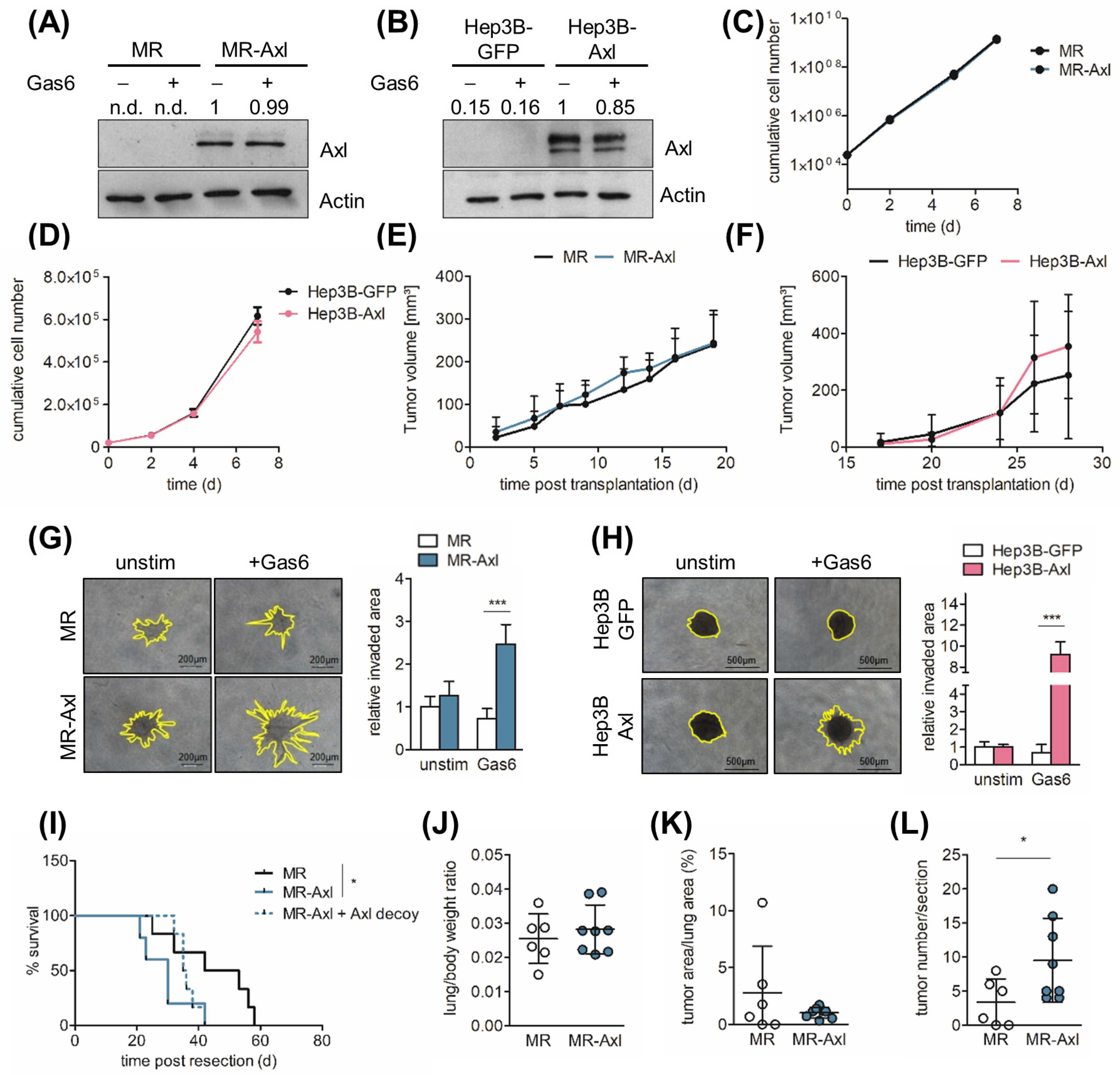
2.1. Axl Augments Invasive Behavior of HCC Cells
2.2. Axl Promotes Metastasis In Vivo
2.3. Axl Is Associated with Partial EMT
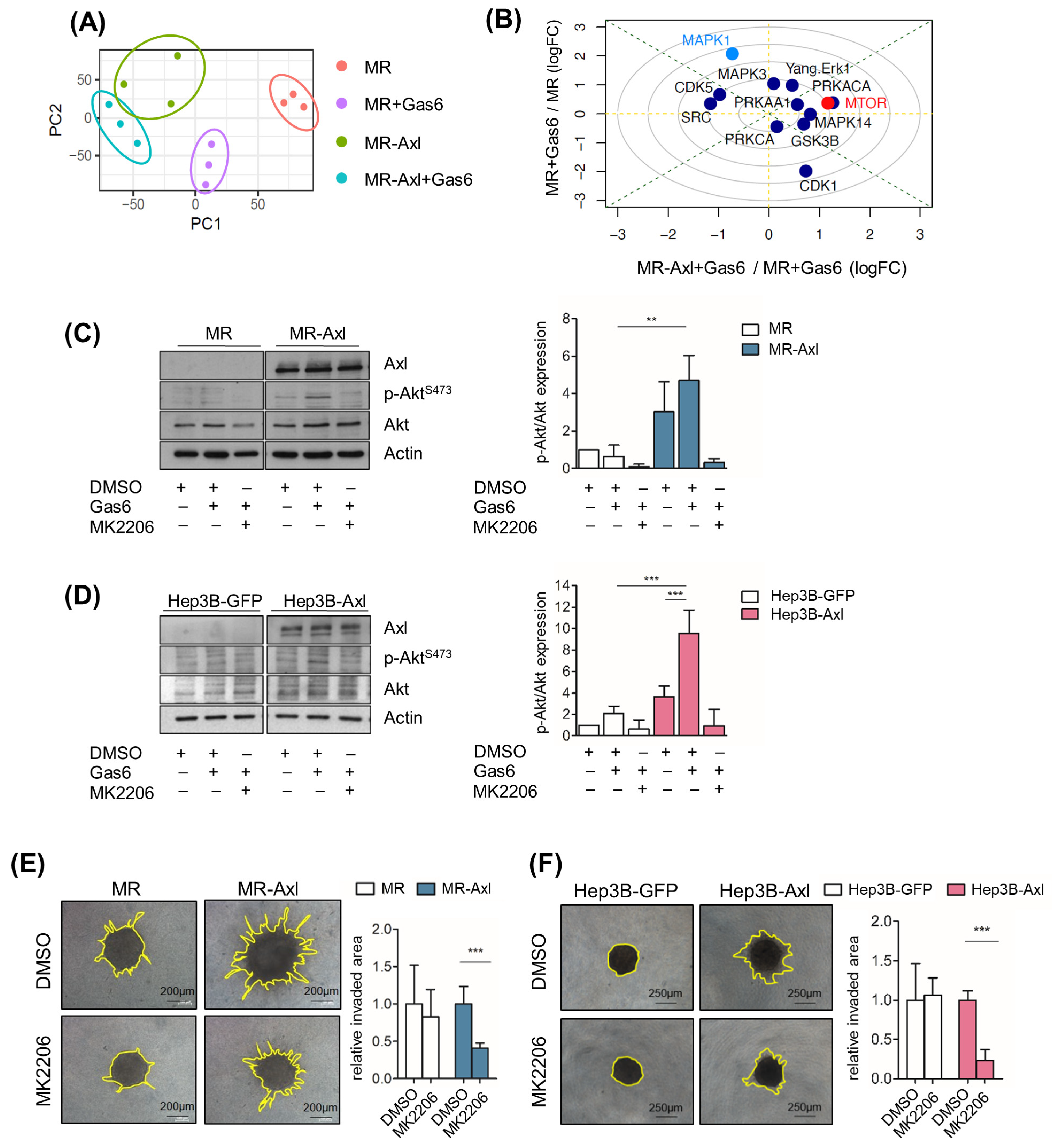
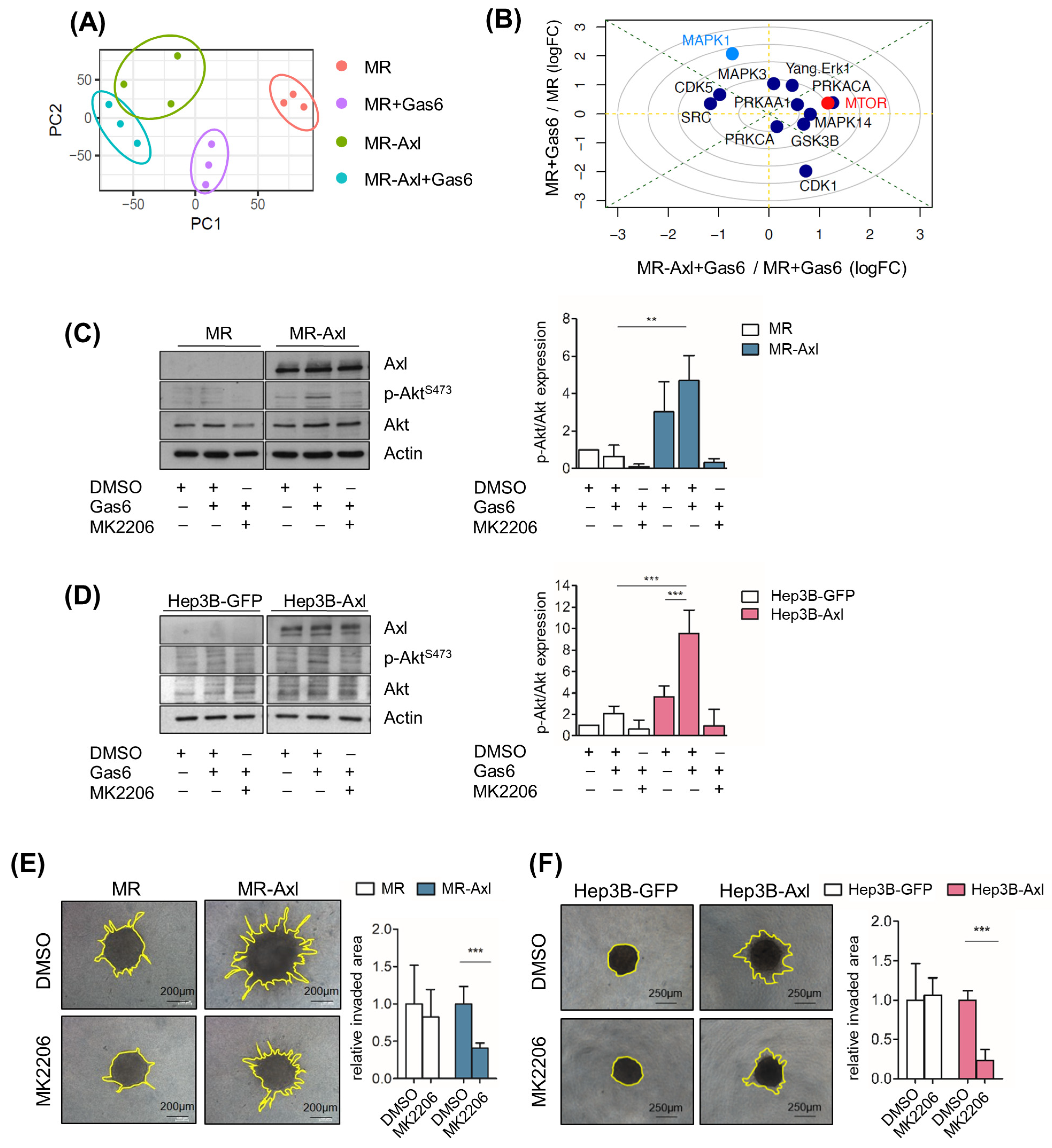
2.4. Gas6/Axl/Akt Signaling Mediates Invasion of Liver Cancer Cells
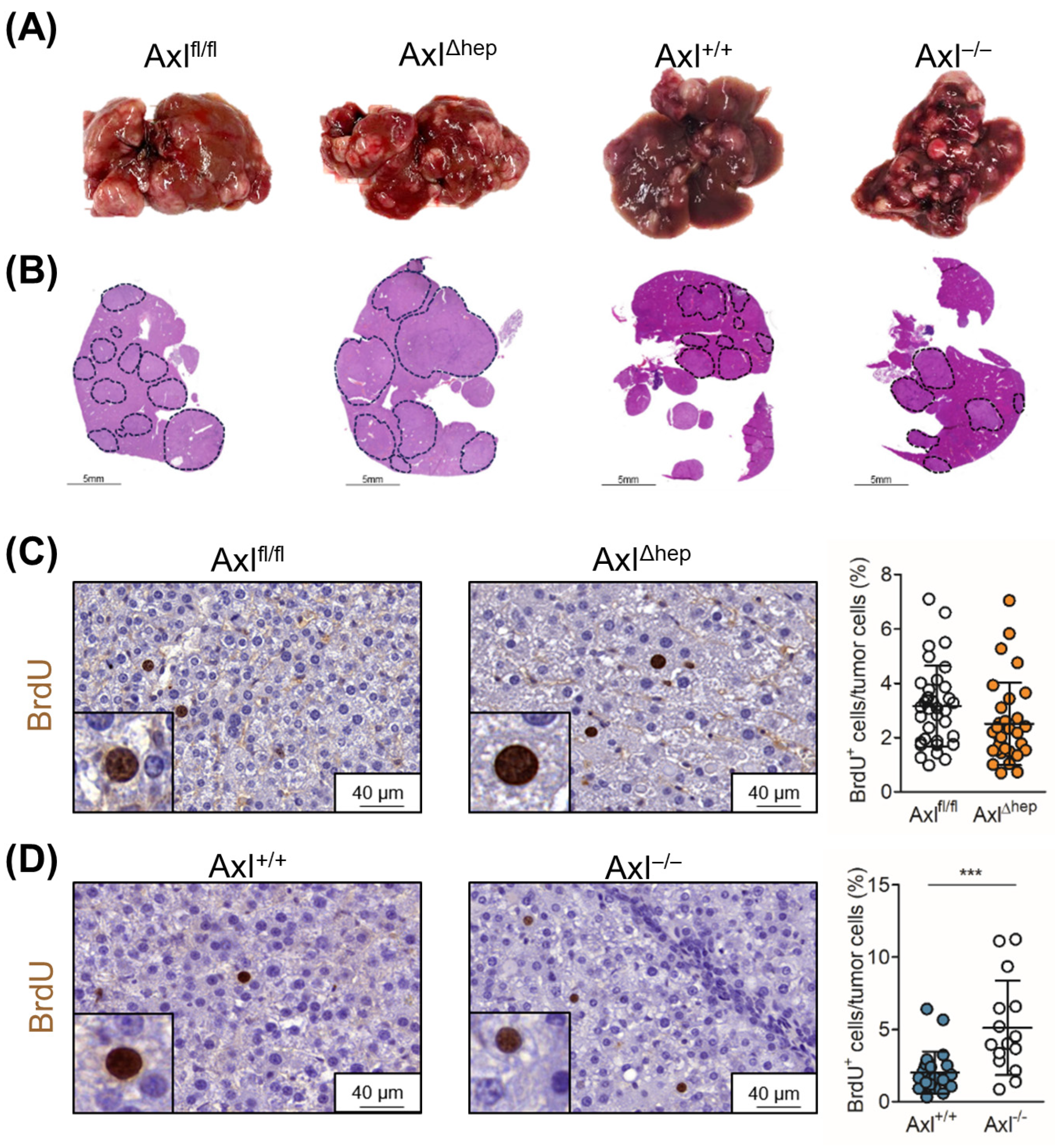
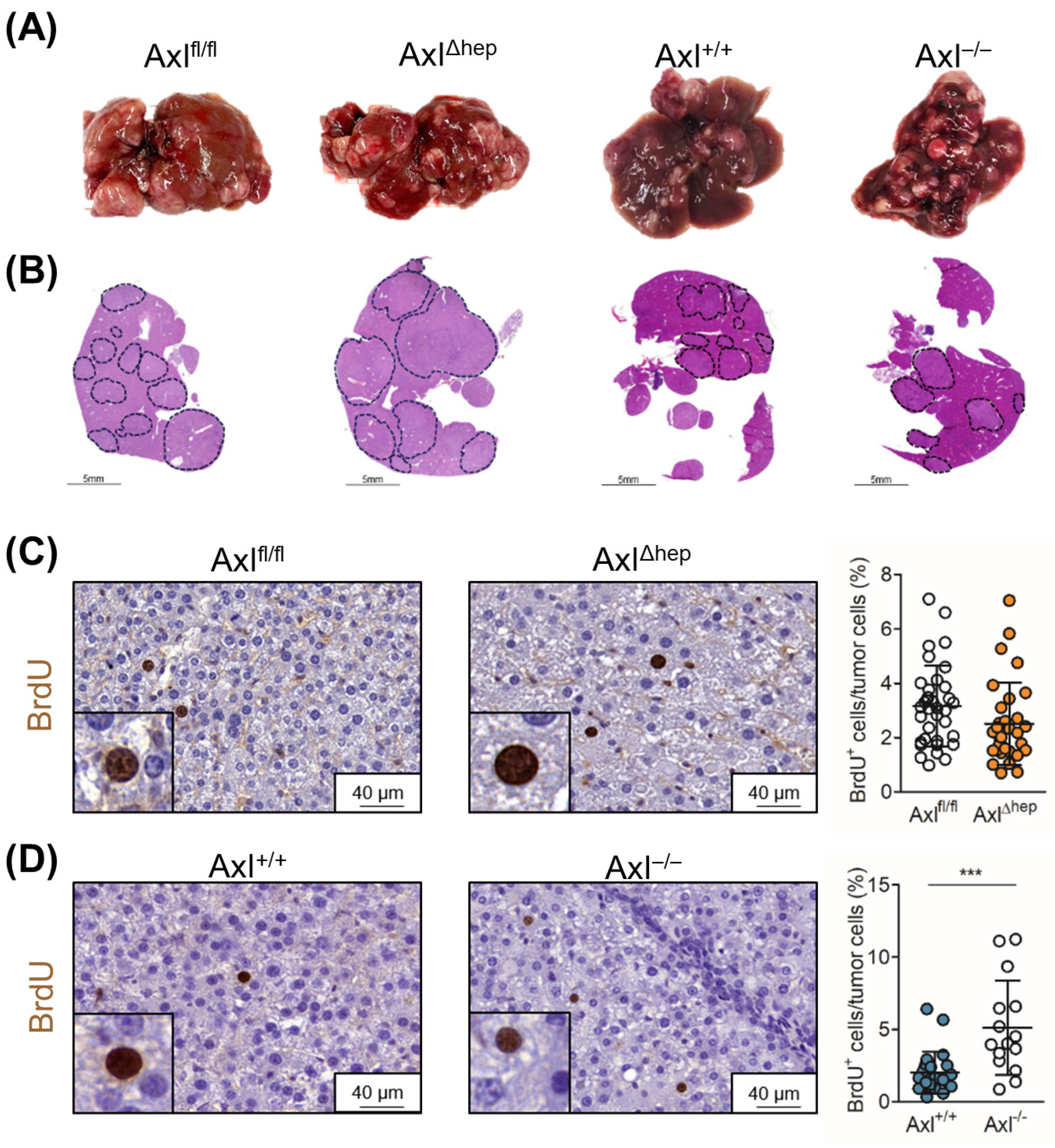
2.5. Tumor Cell-Extrinsic Axl Signaling Promotes Proliferation of Neoplastic Hepatocytes
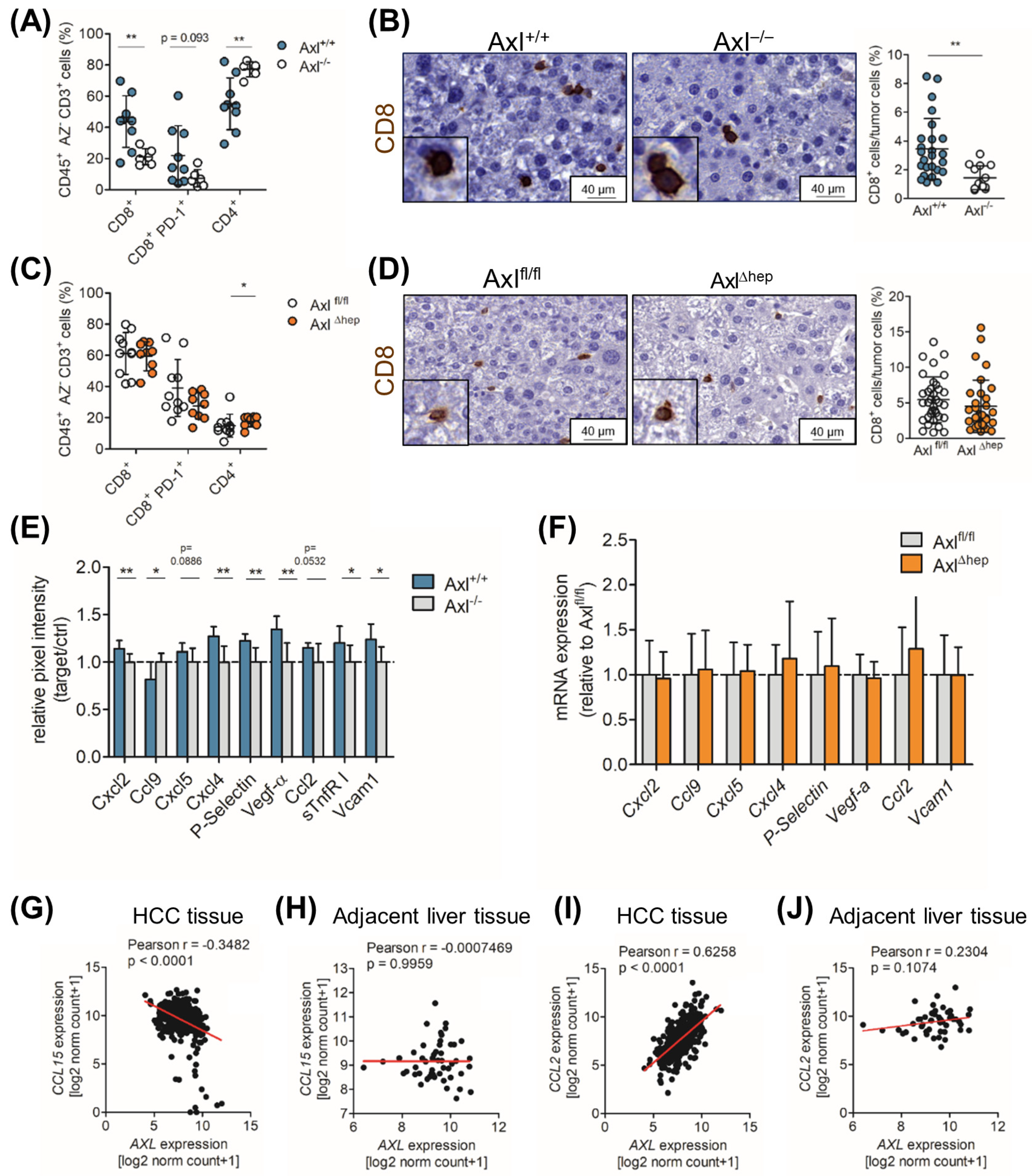
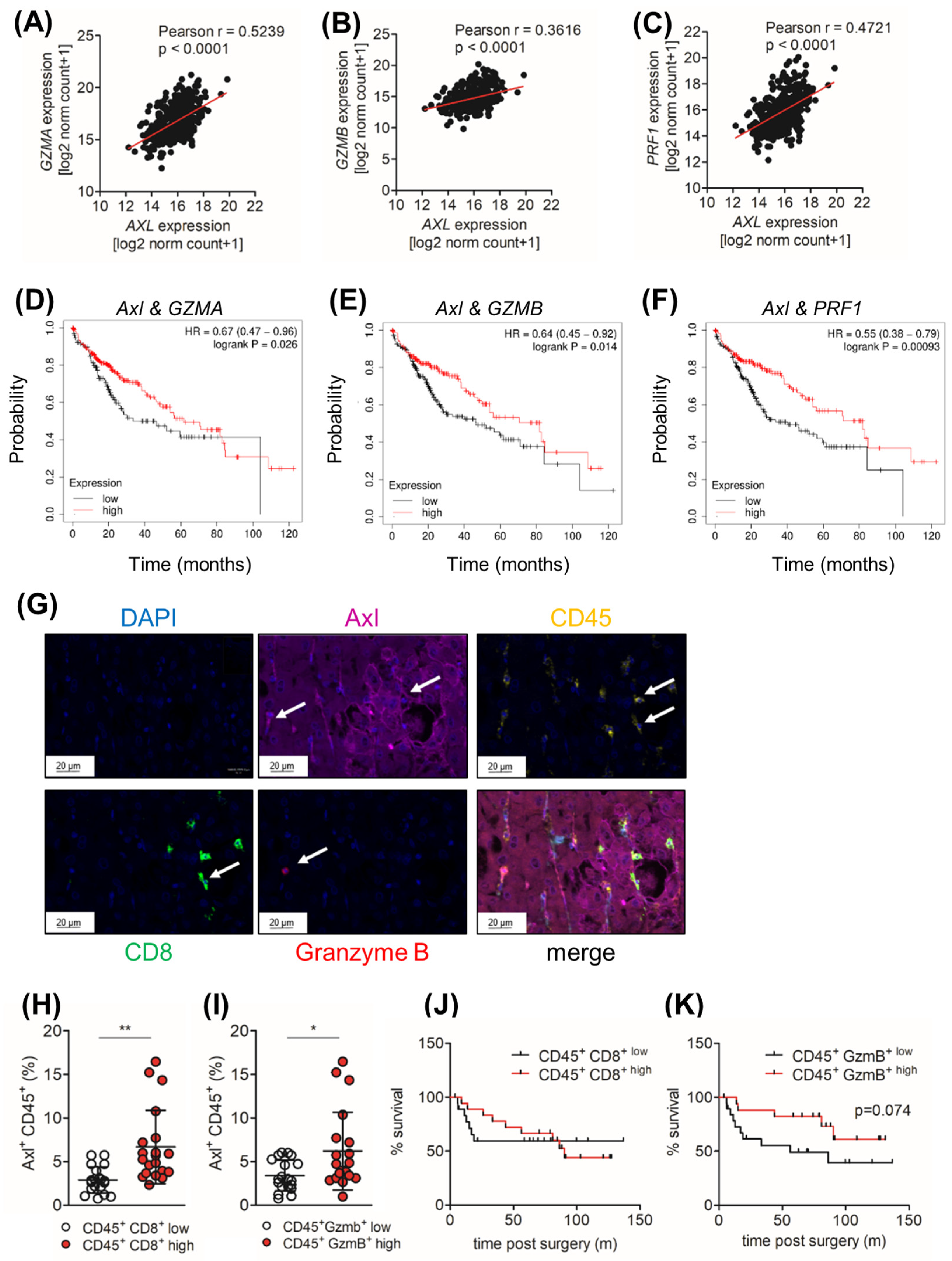
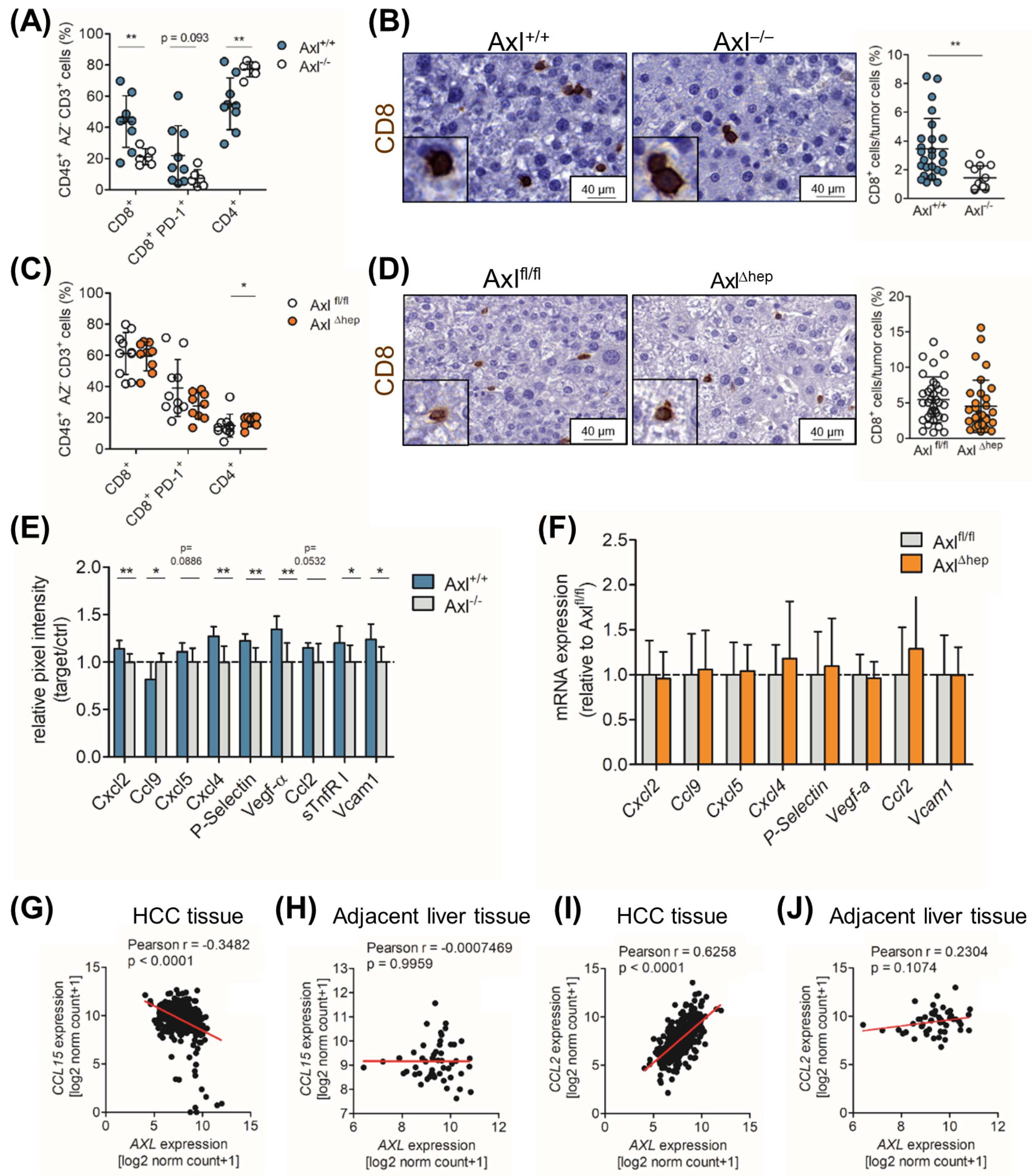
2.6. Axl Stimulates Infiltration of Cytotoxic Immune Cells
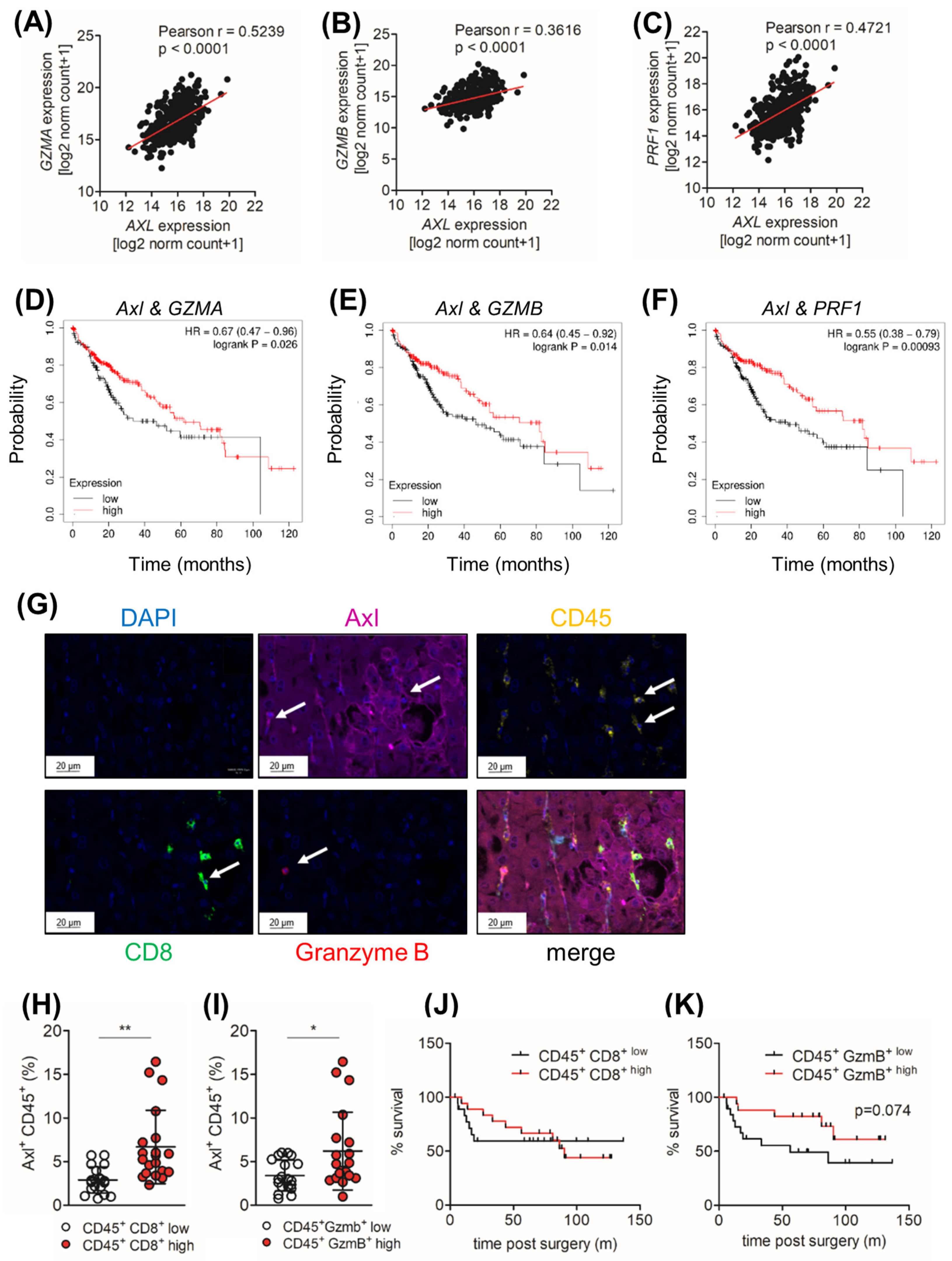
2.7. Ccl9/Ccl15 Negatively Correlates with Axl Expression
2.8. Tumor Cell-Extrinsic Axl Expression Correlates with the Infiltration of CD8+ and Granzyme B+ Cell in HCC Patients
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Patient Samples
4.3. In Silico Analysis
4.4. Proliferation Kinetics
4.5. Invasion Assay
4.6. Quantitative PCR (qPCR)
4.7. Immunofluorescence Analysis of Spheroids
4.8. Phospho-Proteomics by Mass Spectrometry (MS)
4.9. Mass Spectrometry Data Analysis
4.10. Western Blotting
4.11. Cytokine Array
4.12. Animal Experiments
4.13. Immunohistochemistry
4.14. Multiplex IHC
4.15. Flow Cytometry
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Fuhrlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Xu, F.; Dai, C.L. Relationship between epithelial-to-mesenchymal transition and the inflammatory microenvironment of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 203. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Adam-Artigues, A.; Arenas, E.J.; Martinez-Sabadell, A.; Braso-Maristany, F.; Cervera, R.; Tormo, E.; Hernando, C.; Martinez, M.T.; Carbonell-Asins, J.; Simon, S.; et al. Targeting HER2-AXL heterodimerization to overcome resistance to HER2 blockade in breast cancer. Sci. Adv. 2022, 8, eabk2746. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jeng, Y.M.; Chen, Y.L.; Chung, L.; Yuan, R.H. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014, 35, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Golkowski, M.; Lau, H.T.; Chan, M.; Kenerson, H.; Vidadala, V.N.; Shoemaker, A.; Maly, D.J.; Yeung, R.S.; Gujral, T.S.; Ong, S.E. Pharmacoproteomics Identifies Kinase Pathways that Drive the Epithelial-Mesenchymal Transition and Drug Resistance in Hepatocellular Carcinoma. Cell Syst. 2020, 11, 196–207.e197. [Google Scholar] [CrossRef] [PubMed]
- Trojan, J. Cabozantinib for the Treatment of Advanced Hepatocellular Carcinoma: Current Data and Future Perspectives. Drugs 2020, 80, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Giovenzana, C.; Hughes, T.L.; Yu, J.; Trotta, R.; Caligiuri, M.A. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood 2009, 113, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.X.; Kazlauskas, A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012, 31, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef]
- Barcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menendez, A.; Garcia-Ruiz, C.; Sancho-Bru, P.; Mari, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.N.; Herrera, B.; Mikula, M.; Proell, V.; Fuchs, E.; Gotzmann, J.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis. Carcinogenesis 2005, 26, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, T.; Hoffman, N.J.; Xiao, D.; James, D.E.; Humphrey, S.J.; Yang, P. PhosR enables processing and functional analysis of phosphoproteomic data. Cell Rep. 2021, 34, 108771. [Google Scholar] [CrossRef]
- Yang, P.; Patrick, E.; Tan, S.X.; Fazakerley, D.J.; Burchfield, J.; Gribben, C.; Prior, M.J.; James, D.E.; Hwa Yang, Y. Direction pathway analysis of large-scale proteomics data reveals novel features of the insulin action pathway. Bioinformatics 2014, 30, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-beta and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2019, 69, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeshan, S.; Prasad, M.; Badarni, M.; Ben-Lulu, T.; Liju, V.B.; Mathukkada, S.; Saunders, C.; Shnerb, A.B.; Zorea, J.; Yegodayev, K.M.; et al. Mutated HRAS Activates YAP1-AXL Signaling to Drive Metastasis of Head and Neck Cancer. Cancer Res. 2023, 83, 1031–1047. [Google Scholar] [CrossRef]
- Hedrich, V.; Breitenecker, K.; Ortmayr, G.; Pupp, F.; Huber, H.; Chen, D.; Sahoo, S.; Jolly, M.K.; Mikulits, W. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers 2023, 15, 2415. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e1624. [Google Scholar] [CrossRef] [PubMed]
- Kroger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [PubMed]
- Zdzalik-Bielecka, D.; Poswiata, A.; Kozik, K.; Jastrzebski, K.; Schink, K.O.; Brewinska-Olchowik, M.; Piwocka, K.; Stenmark, H.; Miaczynska, M. The GAS6-AXL signaling pathway triggers actin remodeling that drives membrane ruffling, macropinocytosis, and cancer-cell invasion. Proc. Natl. Acad. Sci. USA 2021, 118, e2024596118. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.G.; Fingar, D.C. Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, Y.; Fujino, N.; Miyauchi, E.; Saito, R.; Fujishima, F.; Itakura, K.; Kyogoku, Y.; Okutomo, K.; Yamada, M.; Okazaki, T.; et al. Axl kinase drives immune checkpoint and chemokine signalling pathways in lung adenocarcinomas. Mol. Cancer 2019, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, Y.; Zhang, D.; Ma, J. Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget 2017, 8, 89761–89774. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Koga, H.; Suzuki, H.; Iwamoto, H.; Sakaue, T.; Masuda, A.; Nakamura, T.; Akiba, J.; Yano, H.; Torimura, T.; et al. Anti-PD-L1 antibodies promote cellular proliferation by activating the PD-L1-AXL signal relay in liver cancer cells. Hepatol. Int. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Schmid, E.T.; Pang, I.K.; Carrera Silva, E.A.; Bosurgi, L.; Miner, J.J.; Diamond, M.S.; Iwasaki, A.; Rothlin, C.V. AXL receptor tyrosine kinase is required for T cell priming and antiviral immunity. eLife 2016, 5, e12414. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, J.; Zhang, W.; Zhang, N.; Guo, H. Identification of serum CCL15 in hepatocellular carcinoma. Br. J. Cancer 2013, 108, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Zhang, Z.; Zheng, B.H.; Shi, Y.; Duan, M.; Ma, L.J.; Wang, Z.C.; Dong, L.Q.; Dong, P.P.; Shi, J.Y.; et al. CCL15 Recruits Suppressive Monocytes to Facilitate Immune Escape and Disease Progression in Hepatocellular Carcinoma. Hepatology 2019, 69, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Bosurgi, L.; Bernink, J.H.; Delgado Cuevas, V.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Huser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Banaszek, A.; Bumm, T.G.P.; Nowotny, B.; Geis, M.; Jacob, K.; Wolfl, M.; Trebing, J.; Kucka, K.; Kouhestani, D.; Gogishvili, T.; et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun. 2019, 10, 5387. [Google Scholar] [CrossRef]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Sachsenmeier, K.F.; Yang, C.; Hansen, A.; Filderman, J.; Mulgrew, K.; Wu, H.; Dall’Acqua, W.F. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 2017, 7, 40098. [Google Scholar] [CrossRef] [PubMed]
- Mikula, M.; Fuchs, E.; Huber, H.; Beug, H.; Schulte-Hermann, R.; Mikulits, W. Immortalized p19ARF null hepatocytes restore liver injury and generate hepatic progenitors after transplantation. Hepatology 2004, 39, 628–634. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Nagy, A.; Gyorffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [PubMed]
- Osswald, A.; Hedrich, V.; Sommergruber, W. 3D-3 Tumor Models in Drug Discovery for Analysis of Immune Cell Infiltration. Methods Mol. Biol. 2019, 1953, 151–162. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Petz, M.; Them, N.; Huber, H.; Beug, H.; Mikulits, W. La enhances IRES-mediated translation of laminin B1 during malignant epithelial to mesenchymal transition. Nucleic Acids Res. 2012, 40, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breitenecker, K.; Heiden, D.; Demmer, T.; Weber, G.; Primorac, A.-M.; Hedrich, V.; Ortmayr, G.; Gruenberger, T.; Starlinger, P.; Herndler-Brandstetter, D.; et al. Tumor-Extrinsic Axl Expression Shapes an Inflammatory Microenvironment Independent of Tumor Cell Promoting Axl Signaling in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2024, 25, 4202. https://doi.org/10.3390/ijms25084202
Breitenecker K, Heiden D, Demmer T, Weber G, Primorac A-M, Hedrich V, Ortmayr G, Gruenberger T, Starlinger P, Herndler-Brandstetter D, et al. Tumor-Extrinsic Axl Expression Shapes an Inflammatory Microenvironment Independent of Tumor Cell Promoting Axl Signaling in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2024; 25(8):4202. https://doi.org/10.3390/ijms25084202
Chicago/Turabian StyleBreitenecker, Kristina, Denise Heiden, Tobias Demmer, Gerhard Weber, Ana-Maria Primorac, Viola Hedrich, Gregor Ortmayr, Thomas Gruenberger, Patrick Starlinger, Dietmar Herndler-Brandstetter, and et al. 2024. "Tumor-Extrinsic Axl Expression Shapes an Inflammatory Microenvironment Independent of Tumor Cell Promoting Axl Signaling in Hepatocellular Carcinoma" International Journal of Molecular Sciences 25, no. 8: 4202. https://doi.org/10.3390/ijms25084202