
Error-Corrected Deep Targeted Sequencing of Circulating Cell-Free DNA from Colorectal Cancer Patients for Sensitive Detection of Circulating Tumor DNA

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
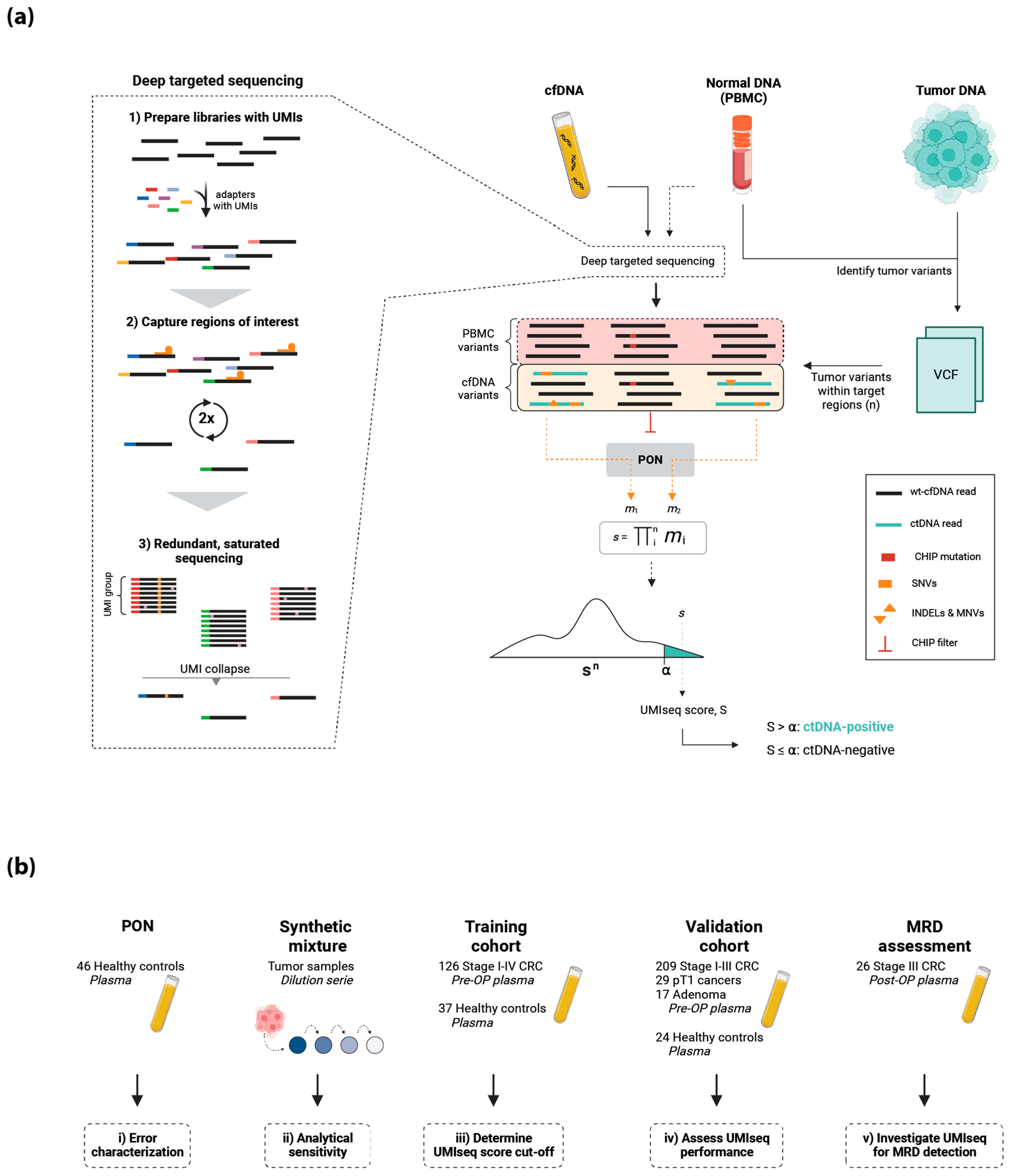
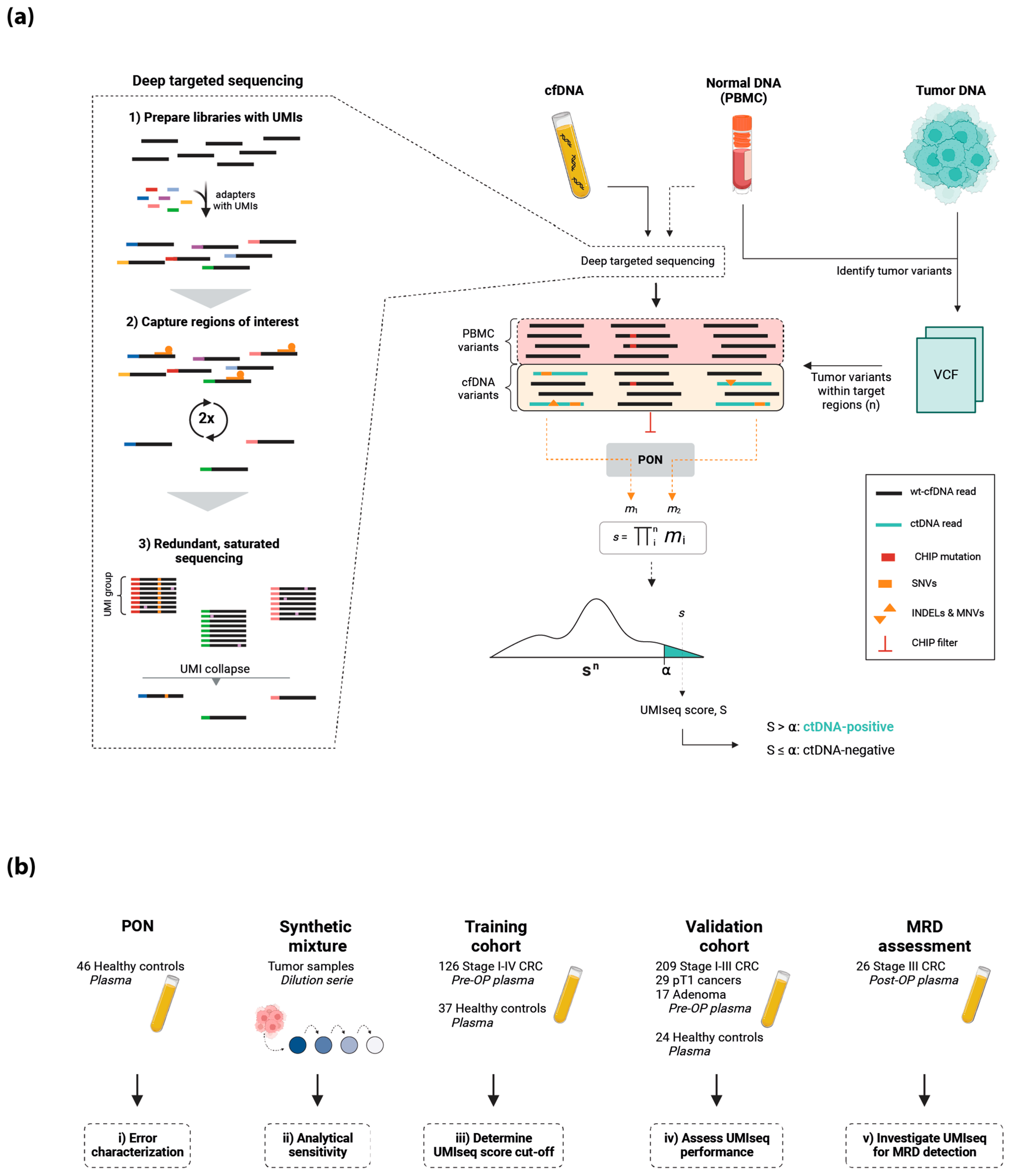
2.1. The UMIseq Method and Study Design
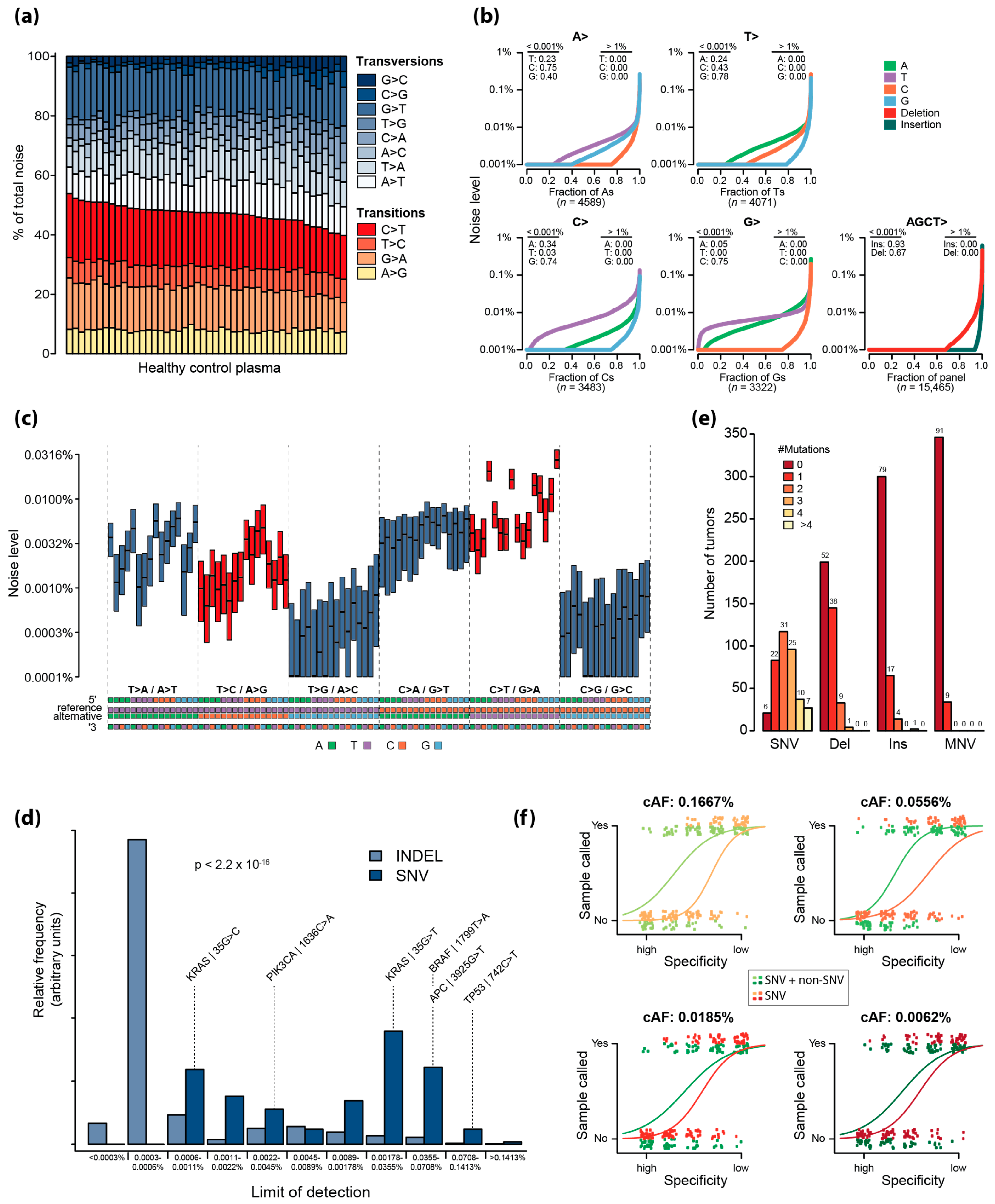
2.2. Error Characterization of UMIseq
2.3. Assessment of the Theoretical Limit of Detection
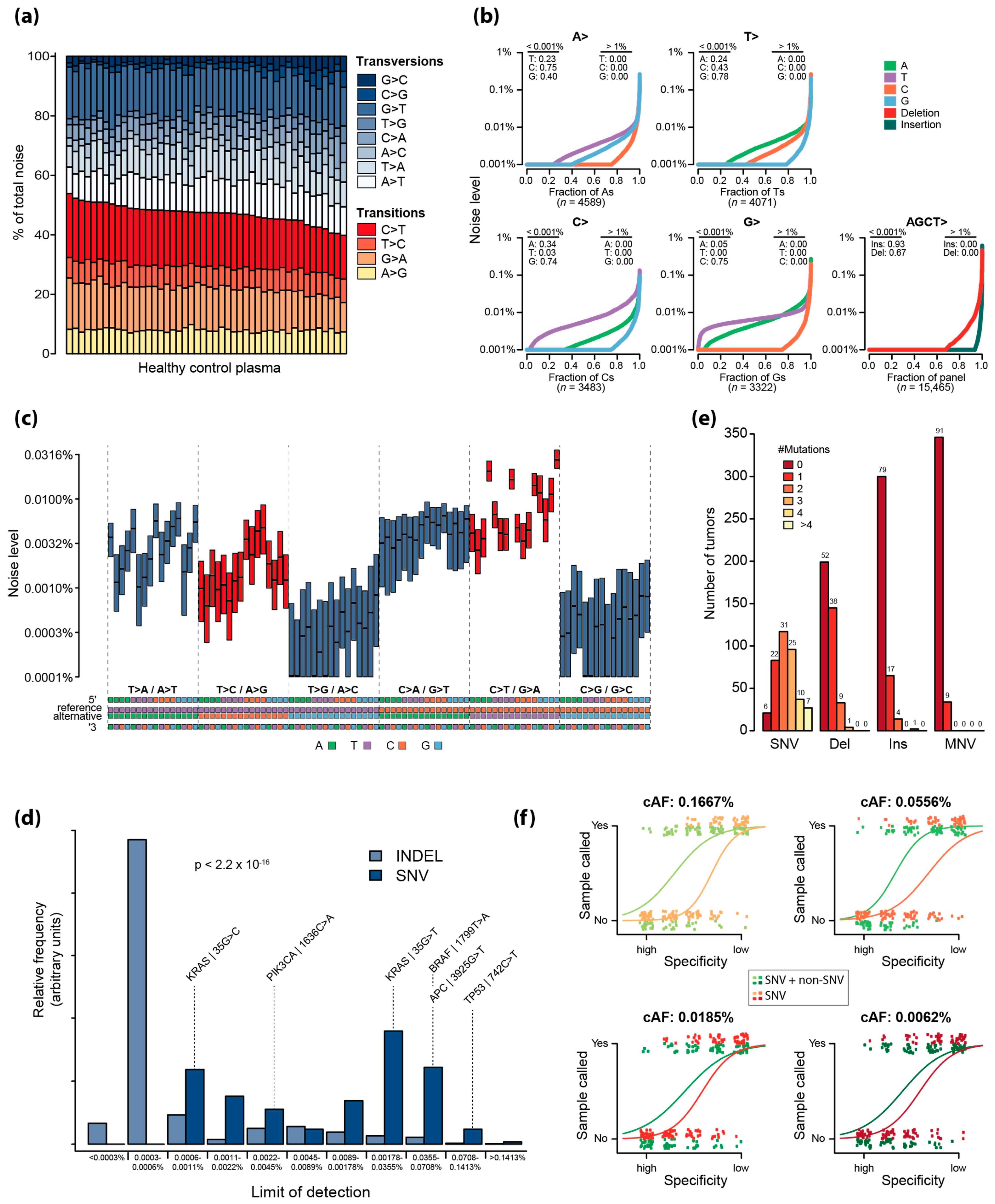
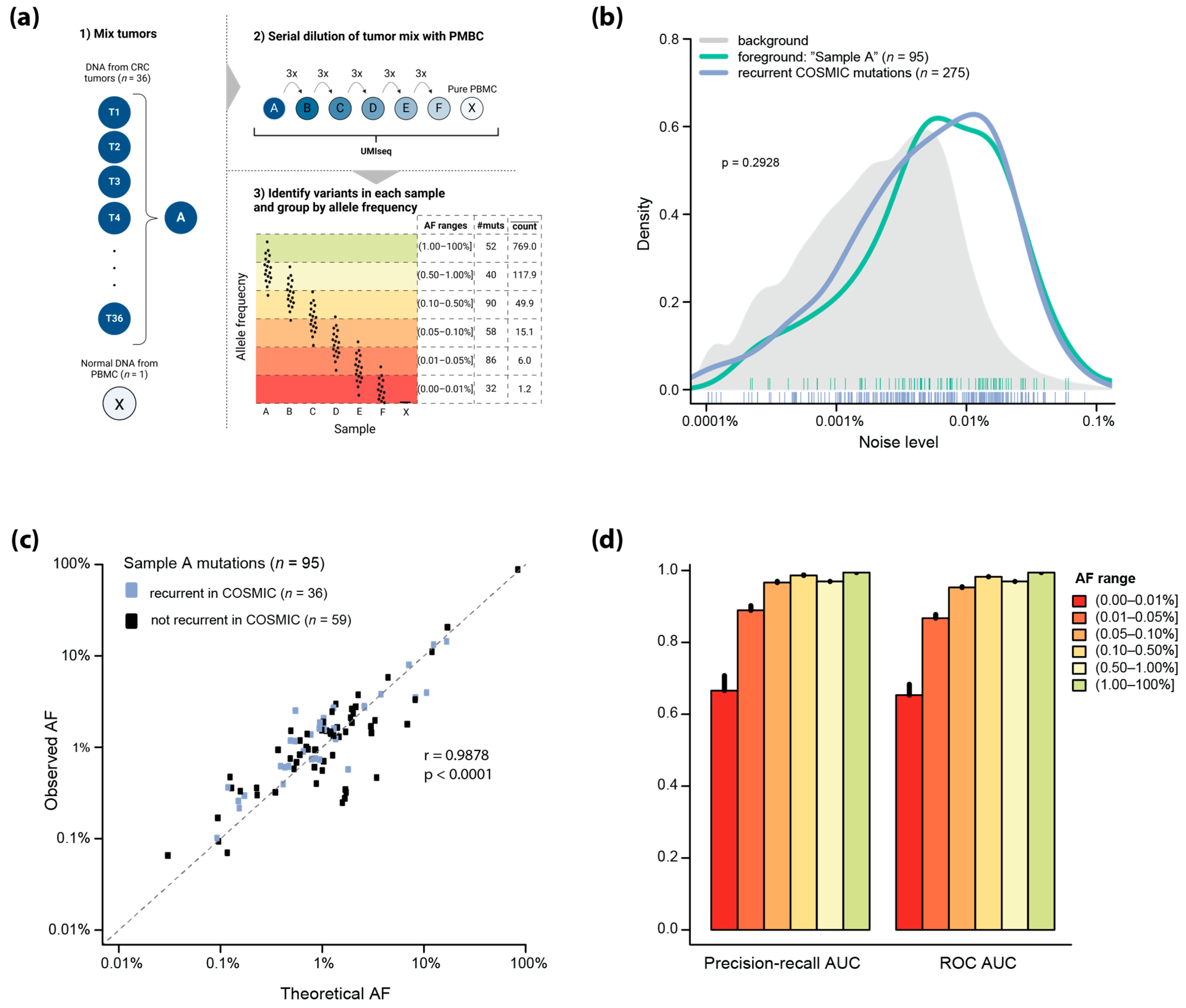
2.4. Analytical Sensitivity of UMIseq
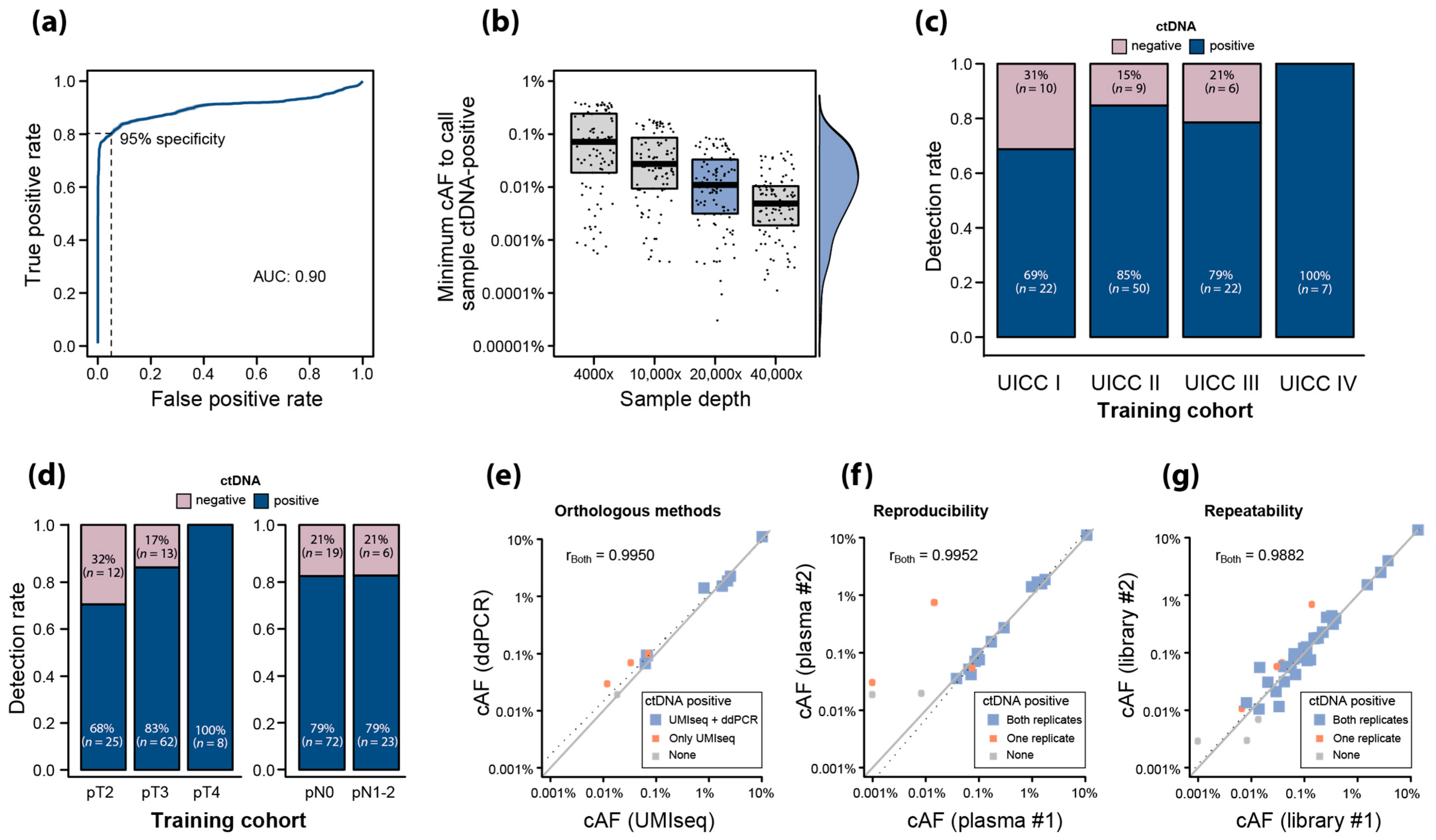
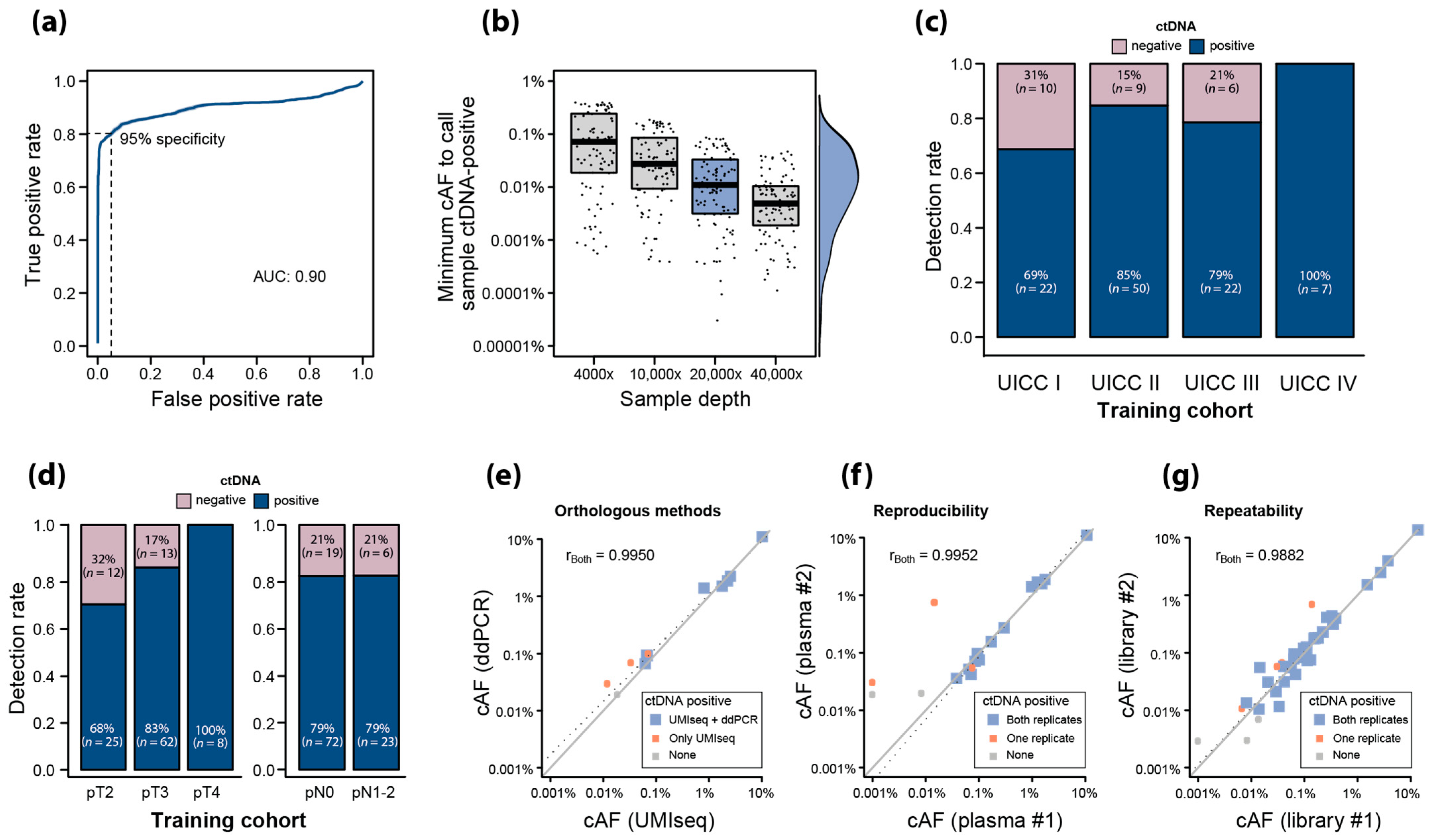
2.5. Training of UMIseq for ctDNA Detection Using Pre-OP Plasma
2.6. Analytical Robustness of ctDNA Detection Using UMIseq
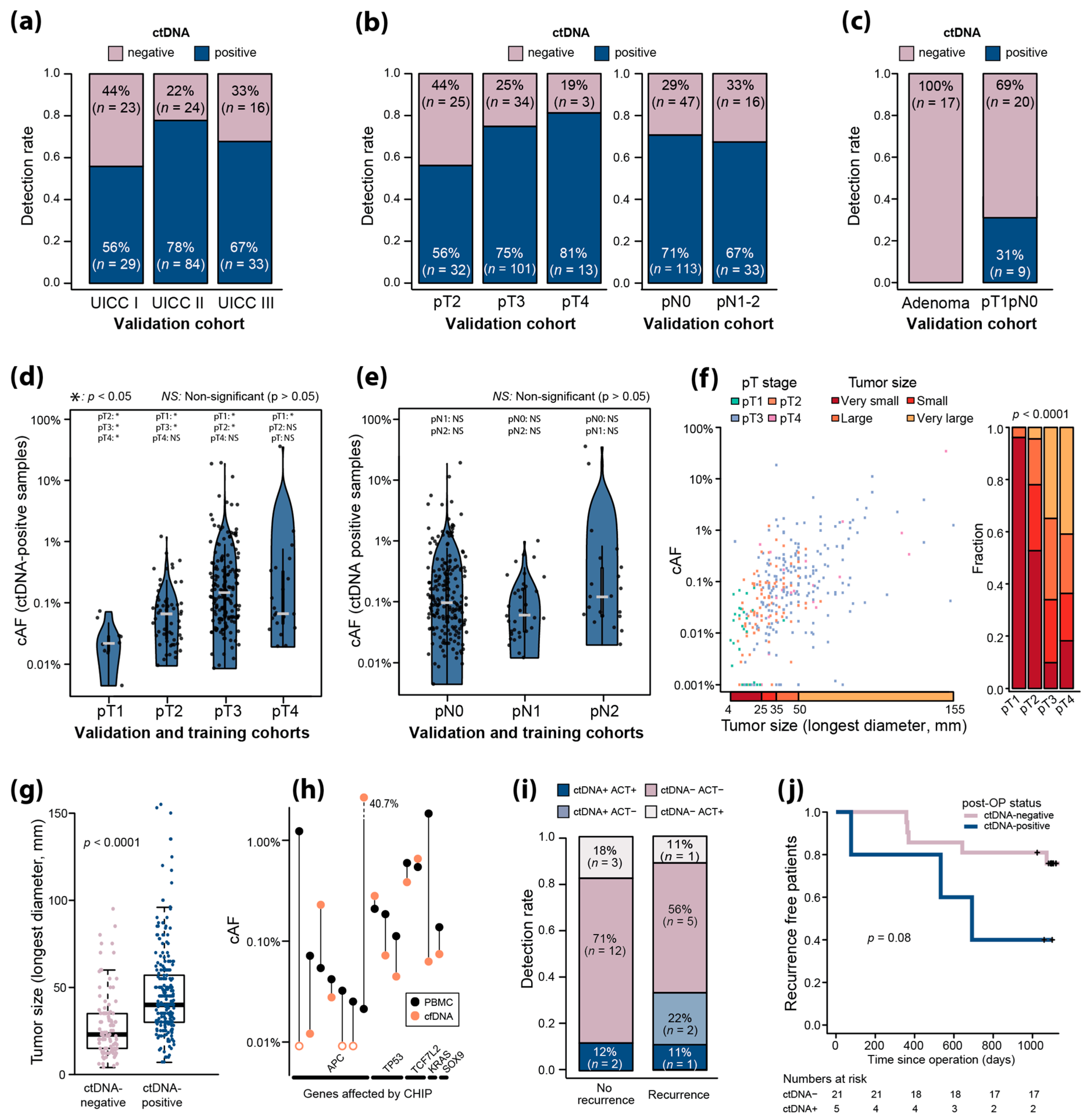
2.7. Validation of UMIseq
2.8. Association between pT, Tumor Size, and ctDNA
2.9. Assessment of Clonal Hematopoiesis of Indeterminate Potential
2.10. UMIseq Applied for Minimal Residual Disease Detection
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Sample Collection and Processing
4.3. Plasma Variant Calling and Estimation of Circulating Allele Frequency
4.4. Blacklisting of SNVs and INDELs
4.5. Flagging of Mutations Associated with Clonal Hematopoiesis
4.6. Limit of Detection Calculation
4.7. In Silico Estimation of the ctDNA Detection Probabilities
4.8. Recurrent COSMIC Mutations
4.9. Analytical Sensitivity Analysis Using a Synthetic Mixture
4.10. Tumor-Informed Model Training of the UMIseq Algorithm
4.11. Assessment of UMIseq Robustness
4.12. Validation of Specificity
4.13. Statistical Considerations and Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Tumor and Normal DNA Sequencing Library Preparation
Appendix A.2. Tumor and Normal DNA Sequencing and Variant Calling
Appendix A.3. Sample Collection for UMIseq
Appendix A.4. UMIseq Library Preparation
Appendix A.5. Targeted Capture of UMIseq Libraries
Appendix A.6. Sequencing and Processing of UMIseq Libraries
Appendix A.7. Conversion Efficiency of cfDNA Libraries
Appendix A.8. Sample Concordance Check
Appendix A.9. Blacklisting of SNVs and INDELs
Appendix A.10. Flagging of Mutations Associated with Clonal Hematopoiesis
Appendix A.11. Limit of Detection Calculation of Individual Mutations
Appendix A.12. In Silico Estimation of the ctDNA Detection Probabilities in Mutation Catalogs Using SNVs or Both SNVs and Non-SNVs
Appendix A.13. Recurrent COSMIC Mutations
Appendix A.14. Analytical Sensitivity Analysis Using a Synthetic Mixture
Appendix A.15. Limit of Detection Calculation for UMIseq
Appendix A.16. Assessment of UMIseq Robustness
- Orthologous method test
- Repeatability test
References
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Hasenleithner, S.O.; Speicher, M.R. A clinician’s handbook for using ctDNA throughout the patient journey. Mol. Cancer 2022, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, T.V.; Tarazona, N.; Frydendahl, A.; Reinert, T.; Gimeno-Valiente, F.; Carbonell-Asins, J.A.; Sharma, S.; Renner, D.; Hafez, D.; Roda, D.; et al. Circulating Tumor DNA in Stage III Colorectal Cancer, beyond Minimal Residual Disease Detection, toward Assessment of Adjuvant Therapy Efficacy and Clinical Behavior of Recurrences. Clin. Cancer Res. 2022, 28, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.; Heider, K.; Ruiz-Valdepenas, A.; Hackinger, S.; Perry, M.; Marsico, G.; Rundell, V.; Wulff, J.; Sharma, G.; Knock, H.; et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann. Oncol. 2022, 33, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Kotani, D.; Oki, E.; Nakamura, Y.; Yukami, H.; Mishima, S.; Bando, H.; Shirasu, H.; Yamazaki, K.; Watanabe, J.; Kotaka, M.; et al. Molecular residual disease and efficacy of adjuvant chemotherapy in patients with colorectal cancer. Nat. Med. 2023, 29, 127–134. [Google Scholar] [CrossRef]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuñiga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.B.; Heo, S.; Lim, Y.; Lee, W.; Cho, S.H.; Ahn, J.; Kang, J.K.; Kim, S.Y.; Kim, H.P.; Bang, D.; et al. Personalised circulating tumour DNA assay with large-scale mutation coverage for sensitive minimal residual disease detection in colorectal cancer. Br. J. Cancer 2023, 129, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Heider, K.; Gale, D.; Murphy, S.; Fisher, E.; Mouliere, F.; Ruiz-Valdepenas, A.; Santonja, A.; Morris, J.; Chandrananda, D.; et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med. 2020, 12, eaaz8084. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Drue, S.O.; Rasmussen, M.H.; Frydendahl, A.; Lyskjær, I.; Demuth, C.; Nors, J.; Gotschalck, K.A.; Iversen, L.H.; Andersen, C.L.; et al. DREAMS: Deep read-level error model for sequencing data applied to low-frequency variant calling and circulating tumor DNA detection. Genome Biol. 2023, 24, 99. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Soo, J.; Co Ting Keh, L.; Alig, S.; Chabon, J.J.; Sworder, B.J.; Schultz, A.; Jin, M.C.; Scherer, F.; Garofalo, A.; et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat. Biotechnol. 2021, 39, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Liu, R.; Roberts, E.; Nguyen, E.; Tabrizi, S.; Rhoades, J.; Blewett, T.; Xiong, K.; Gydush, G.; Shea, D.; et al. Single duplex DNA sequencing with CODEC detects mutations with high sensitivity. Nat. Genet. 2023, 55, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Drue, S.O.; Frydendahl, A.; Demuth, C.; Rasmussen, M.H.; Reinert, T.; Pedersen, J.S.; Andersen, C.L. Error Characterization and Statistical Modeling Improves Circulating Tumor DNA Detection by Droplet Digital PCR. Clin. Chem. 2022, 68, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Wang, J.; Zhou, S.; Wang, C.L.; Ye, M.Z.; Wang, X.Y.; Song, Y.; Wang, Y.Q.; Zhang, L.T.; et al. Biological background of the genomic variations of cf-DNA in healthy individuals. Ann. Oncol. 2019, 30, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Venn, O.; Aravanis, A.; Hubbell, E.; Maddala, T.; Beausang, J.F.; Filippova, D.; Gross, S.; Jamshidi, A.; Shen, L.; et al. Prevalence of clonal hematopoiesis of indeterminate potential (CHIP) measured by an ultra-sensitive sequencing assay: Exploratory analysis of the Circulating Cancer Genome Atlas (CCGA) study. J. Clin. Oncol. 2018, 36, 12003. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [PubMed]
- Kabel, J.; Henriksen, T.V.; Demuth, C.; Frydendahl, A.; Rasmussen, M.H.; Nors, J.; Birkbak, N.J.; Madsen, A.H.; Løve, U.S.; Andersen, P.V.; et al. Impact of Whole Genome Doubling on Detection of Circulating Tumor DNA in Colorectal Cancer. Cancers 2023, 15, 1136. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, Y.; Lam, V.K.; Shi, Y.; Guan, Y.; Zhang, Y.; Ji, L.; Chen, Y.; Zhao, Y.; Qian, F.; et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis. 2020, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Bredno, J.; Lipson, J.; Venn, O.; Aravanis, A.M.; Jamshidi, A. Clinical correlates of circulating cell-free DNA tumor fraction. PLoS ONE 2021, 16, e0256436. [Google Scholar] [CrossRef]
- Bando, H.; Nakamura, Y.; Taniguchi, H.; Shiozawa, M.; Yasui, H.; Esaki, T.; Kagawa, Y.; Denda, T.; Satoh, T.; Yamazaki, K.; et al. Effects of Metastatic Sites on Circulating Tumor DNA in Patients with Metastatic Colorectal Cancer. JCO Precis. Oncol. 2022, 6, e2100535. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, T.V.; Reinert, T.; Christensen, E.; Sethi, H.; Birkenkamp-Demtröder, K.; Gögenur, M.; Gögenur, I.; Zimmermann, B.G.; Dyrskjøt, L.; Andersen, C.L. The effect of surgical trauma on circulating free DNA levels in cancer patients-implications for studies of circulating tumor DNA. Mol. Oncol. 2020, 14, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Papaemmanuil, E.; Campbell, P.J. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 2014, 30, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Beisel, C.; Rechsteiner, M.; Wild, P.; Schraml, P.; Moch, H.; Beerenwinkel, N. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat. Commun. 2012, 3, 811. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. MBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute, GATK Team. “Markduplicates (Picard)”. Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-MarkDuplicates-Picard- (accessed on 13 June 2023).
- Broad Institute, GATK Team. “HaplotypeCaller”. Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360037225632-HaplotypeCaller (accessed on 22 May 2023).
- Broad Institute, GATK Team. “CNNScoreVariants”. Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360037226672-CNNScoreVariants (accessed on 23 June 2023).
- Broad Institute, GATK Team. “FilterVariantTranches”. Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360040098912-FilterVariantTranches (accessed on 23 June 2023).
- Team, GATK “Mutect2”. Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 (accessed on 1 June 2023).
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Pallisgaard, N.; Spindler, K.L.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Controls to validate plasma samples for cell free DNA quantification. Clin. Chim. Acta 2015, 446, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Mose, L.E.; Perou, C.M.; Parker, J.S. Improved indel detection in DNA and RNA via realignment with ABRA2. Bioinformatics 2019, 35, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Fgbio, Fulcrum Genomics. “Fgbio”. Available online: http://fulcrumgenomics.github.io/fgbio/ (accessed on 23 June 2023).
- Henriksen, T.V.; Reinert, T.; Rasmussen, M.H.; Demuth, C.; Løve, U.S.; Madsen, A.H.; Gotschalck, K.A.; Iversen, L.H.; Andersen, C.L. Comparing single-target and multitarget approaches for postoperative circulating tumour DNA detection in stage II–III colorectal cancer patients. Mol. Oncol. 2022, 16, 3654–3665. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Cohort | Validation Cohort | ||||
|---|---|---|---|---|---|
| Characteristic | Stage I–IV CRC | Healthy Controls | Stage I–III CRC | Adenomas and Early CRC | Healthy Controls |
| n | 126 | 37 | 209 | 46 | 24 |
| Gender 1 | |||||
| Female | 48 (38%) | 18 (49%) | 88 (42%) | 20 (43%) | 9 (38%) |
| Male | 78 (62%) | 19 (51%) | 121 (58%) | 26 (57%) | 15 (62%) |
| Age 2 | 71 (64, 78) | 67 (60, 71) | 71 (63, 76) | 65 (59, 73) | 52 (48, 58) |
| UICC stage 1 | |||||
| Adenoma | - | - | - | 17 (37%) | - |
| I | 32 (25%) | - | 52 (25%) | 29 (63%) | - |
| II | 59 (47%) | - | 108 (52%) | - | - |
| III | 28 (22%) | - | 49 (23%) | - | - |
| IV | 7 (6%) | - | - | - | - |
| pT stage 1 | |||||
| Adenoma | - | - | - | 17 (37%) | - |
| pT1 | - | - | 1 (0%) | 29 (63%) | - |
| pT2 | 37 (29%) | - | 57 (27%) | - | - |
| pT3 | 75 (60%) | - | 135 (65%) | - | - |
| pT4 | 8 (6%) | - | 16 (8%) | - | - |
| Unknown | 6 (5%) | - | - | - | - |
| pN stage 1 | |||||
| Adenoma | - | - | - | 17 (37%) | - |
| pN0 | 91 (72%) | - | 160 (77%) | 29 (63%) | - |
| pN1 | 18 (14%) | - | 32 (15%) | - | - |
| pN2 | 11 (9%) | - | 17 (8%) | - | - |
| Unknown | 6 (5%) | - | - | - | - |
| pM stage 1 | |||||
| Adenoma | - | - | - | 17 (37%) | - |
| pM0 | 120 (95%) | - | 209 (100%) | 29 (63%) | - |
| pM1 | 6 (5%) | - | - | - | - |
| Synchronous tumors 1 | - | - | 12 (6%) | ||
| Mutations within capture panel 2 | 3 (2, 4) | - | 3 (2, 4) | 2 (2, 3) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frydendahl, A.; Rasmussen, M.H.; Jensen, S.Ø.; Henriksen, T.V.; Demuth, C.; Diekema, M.; Ditzel, H.J.; Wen, S.W.C.; Pedersen, J.S.; Dyrskjøt, L.; et al. Error-Corrected Deep Targeted Sequencing of Circulating Cell-Free DNA from Colorectal Cancer Patients for Sensitive Detection of Circulating Tumor DNA. Int. J. Mol. Sci. 2024, 25, 4252. https://doi.org/10.3390/ijms25084252
Frydendahl A, Rasmussen MH, Jensen SØ, Henriksen TV, Demuth C, Diekema M, Ditzel HJ, Wen SWC, Pedersen JS, Dyrskjøt L, et al. Error-Corrected Deep Targeted Sequencing of Circulating Cell-Free DNA from Colorectal Cancer Patients for Sensitive Detection of Circulating Tumor DNA. International Journal of Molecular Sciences. 2024; 25(8):4252. https://doi.org/10.3390/ijms25084252
Chicago/Turabian StyleFrydendahl, Amanda, Mads Heilskov Rasmussen, Sarah Østrup Jensen, Tenna Vesterman Henriksen, Christina Demuth, Mathilde Diekema, Henrik Jørn Ditzel, Sara Witting Christensen Wen, Jakob Skou Pedersen, Lars Dyrskjøt, and et al. 2024. "Error-Corrected Deep Targeted Sequencing of Circulating Cell-Free DNA from Colorectal Cancer Patients for Sensitive Detection of Circulating Tumor DNA" International Journal of Molecular Sciences 25, no. 8: 4252. https://doi.org/10.3390/ijms25084252