
Link Protein 1 Is Involved in the Activity-Dependent Modulation of Perineuronal Nets in the Spinal Cord


Abstract
1. Introduction
2. Results
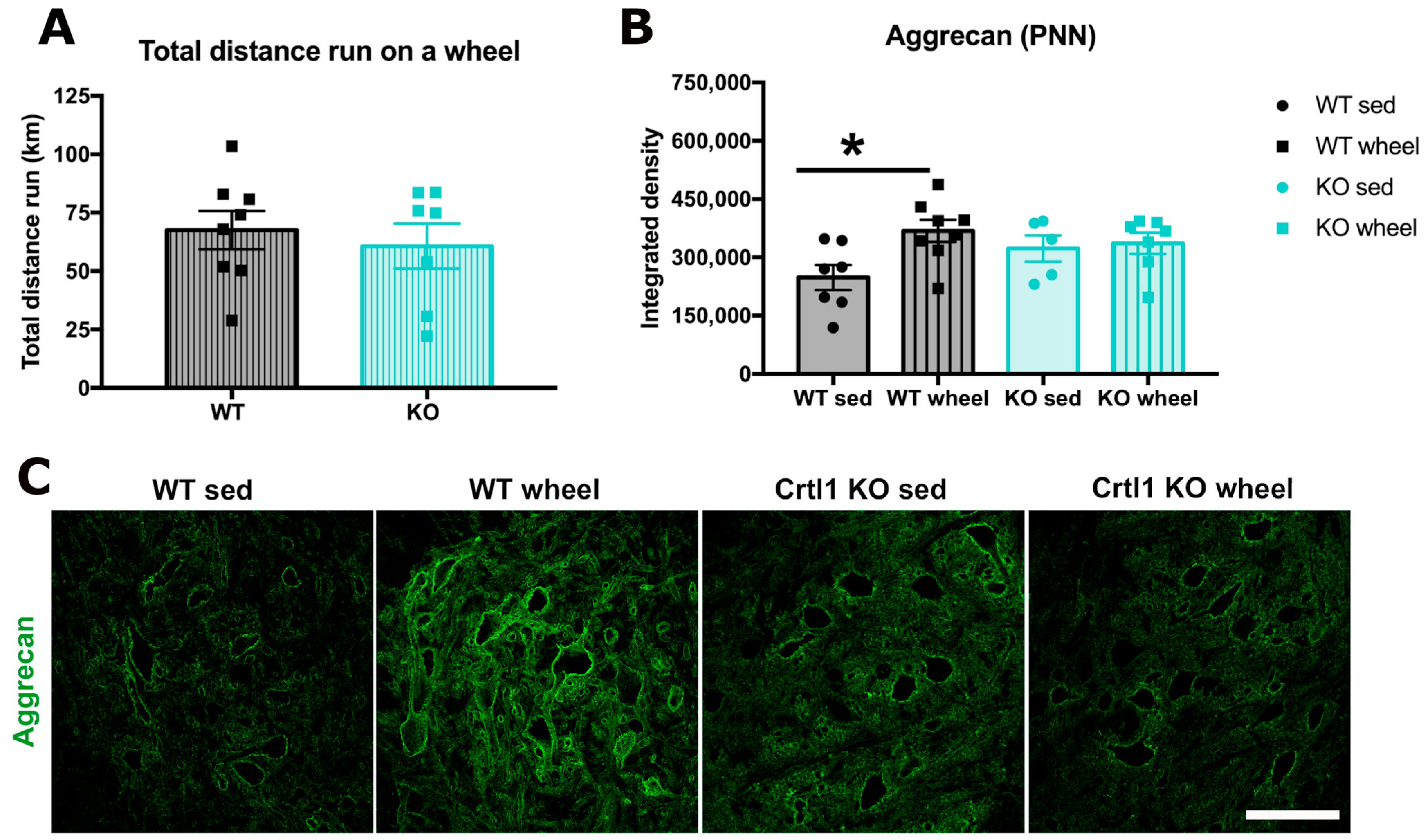
2.1. Link Protein 1 Is Involved in the Activity-Dependent Changes of Perineuronal Nets in the Spinal Cord
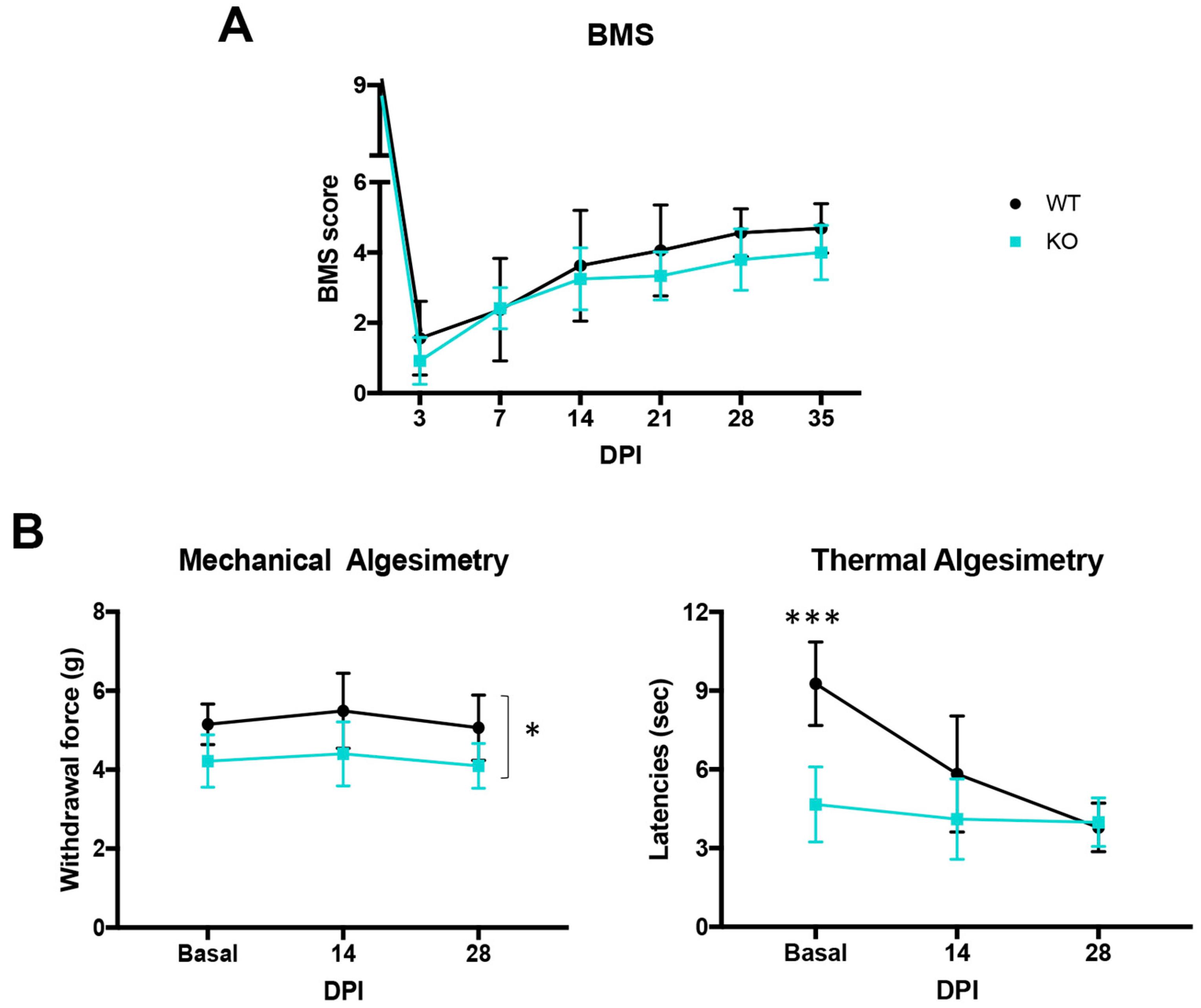
2.2. Uninjured Crtl1 Knock-Out Mice Mimic the Functional Outcomes Observed in the Injured Wild-Type Mice
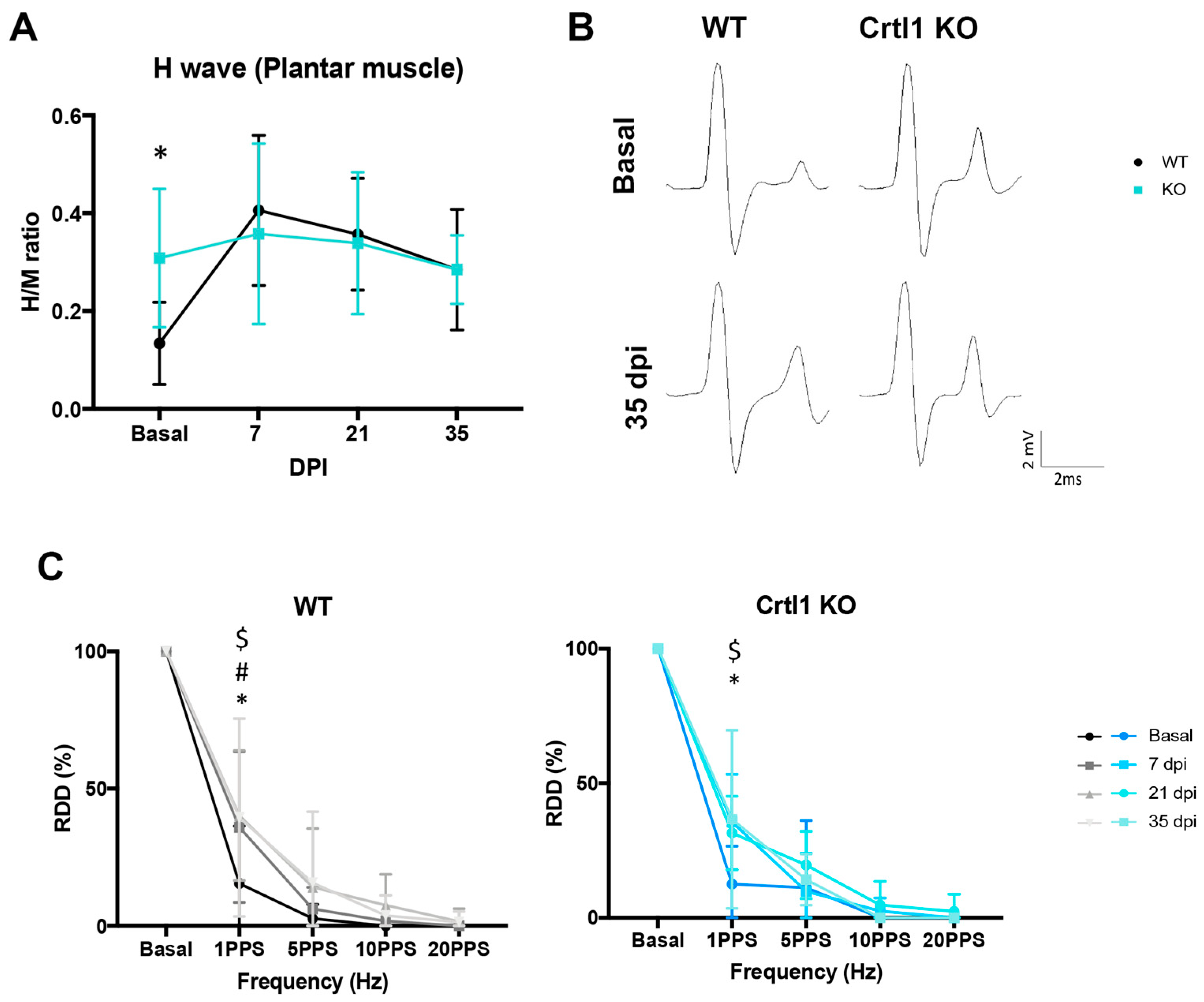
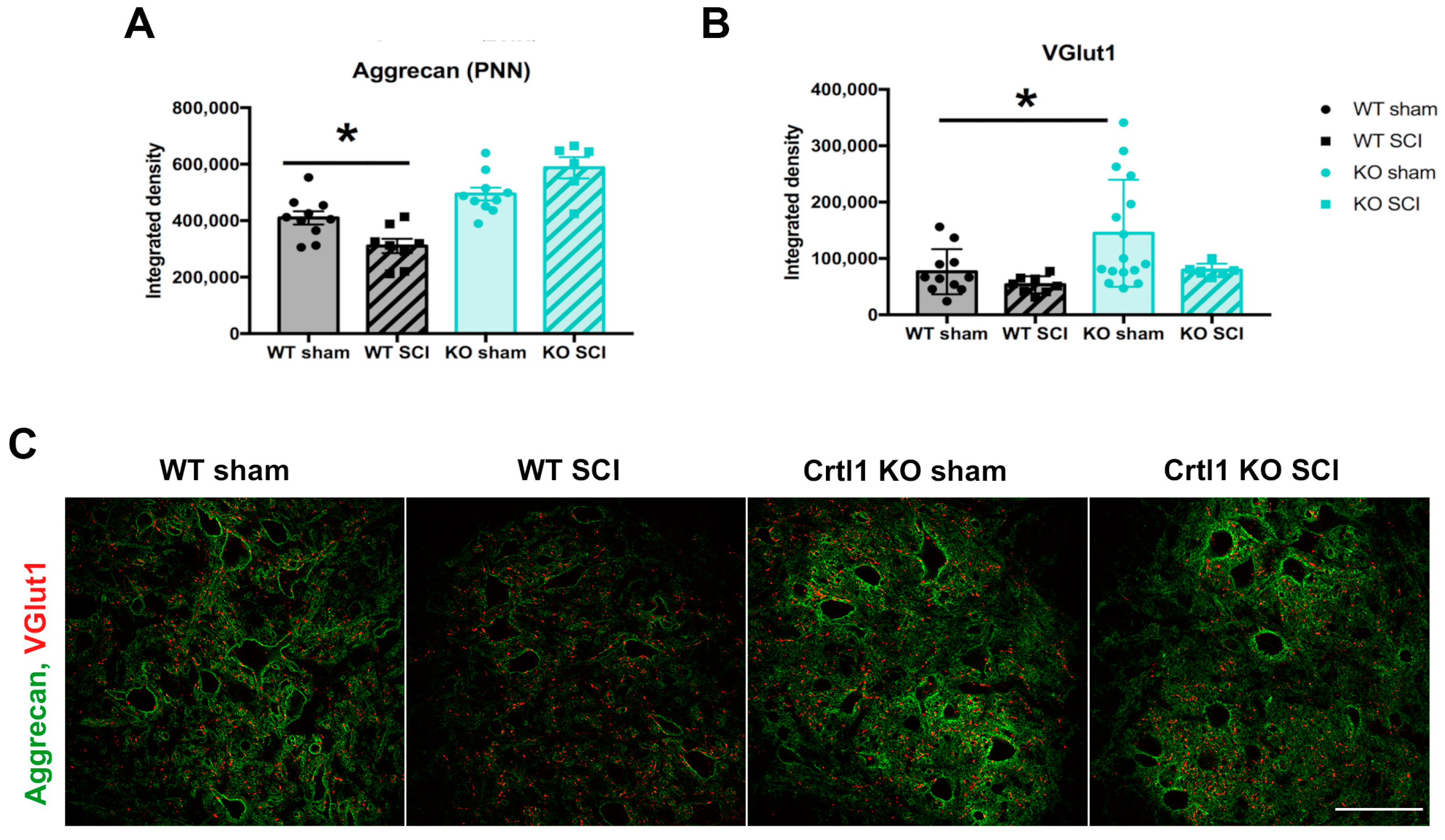
2.3. Lack of Modulation of Perineuronal Nets after Spinal Cord Injury in Crtl1 Knock-Out Mice
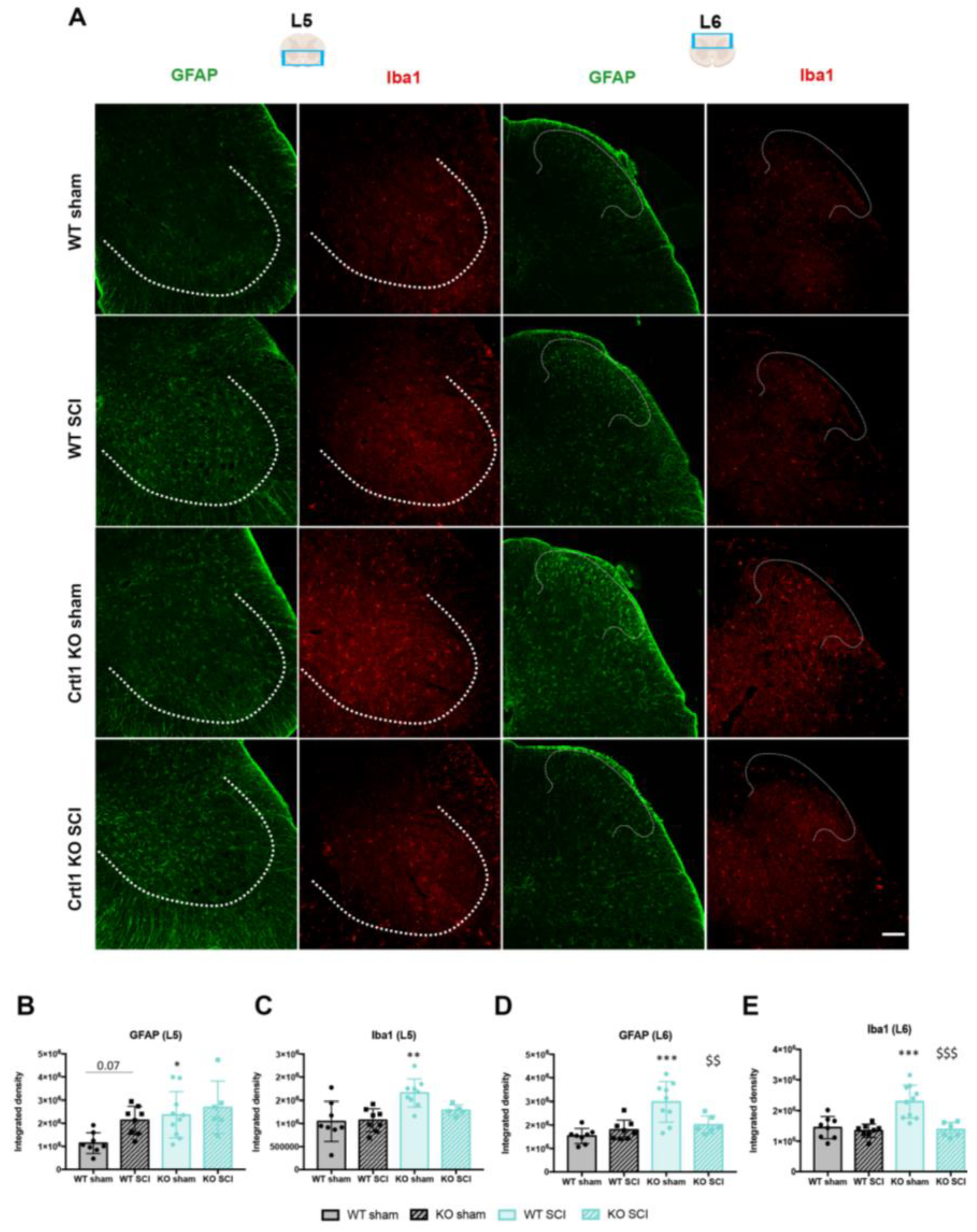
2.4. Lack of Link Protein 1 Modifies Glial Reactivity after Spinal Cord Injury
3. Discussion
3.1. Link Protein 1 Mediates the Activity-Dependent Remodeling of Perineuronal Nets
3.1.1. Remodeling of Spinal Perineuronal Nets after a Locomotor Activity
3.1.2. Remodeling of Spinal Perineuronal Nets after Spinal Cord Injury
3.2. Link Protein 1 Plays a Role in Neural Excitability
4. Materials and Methods
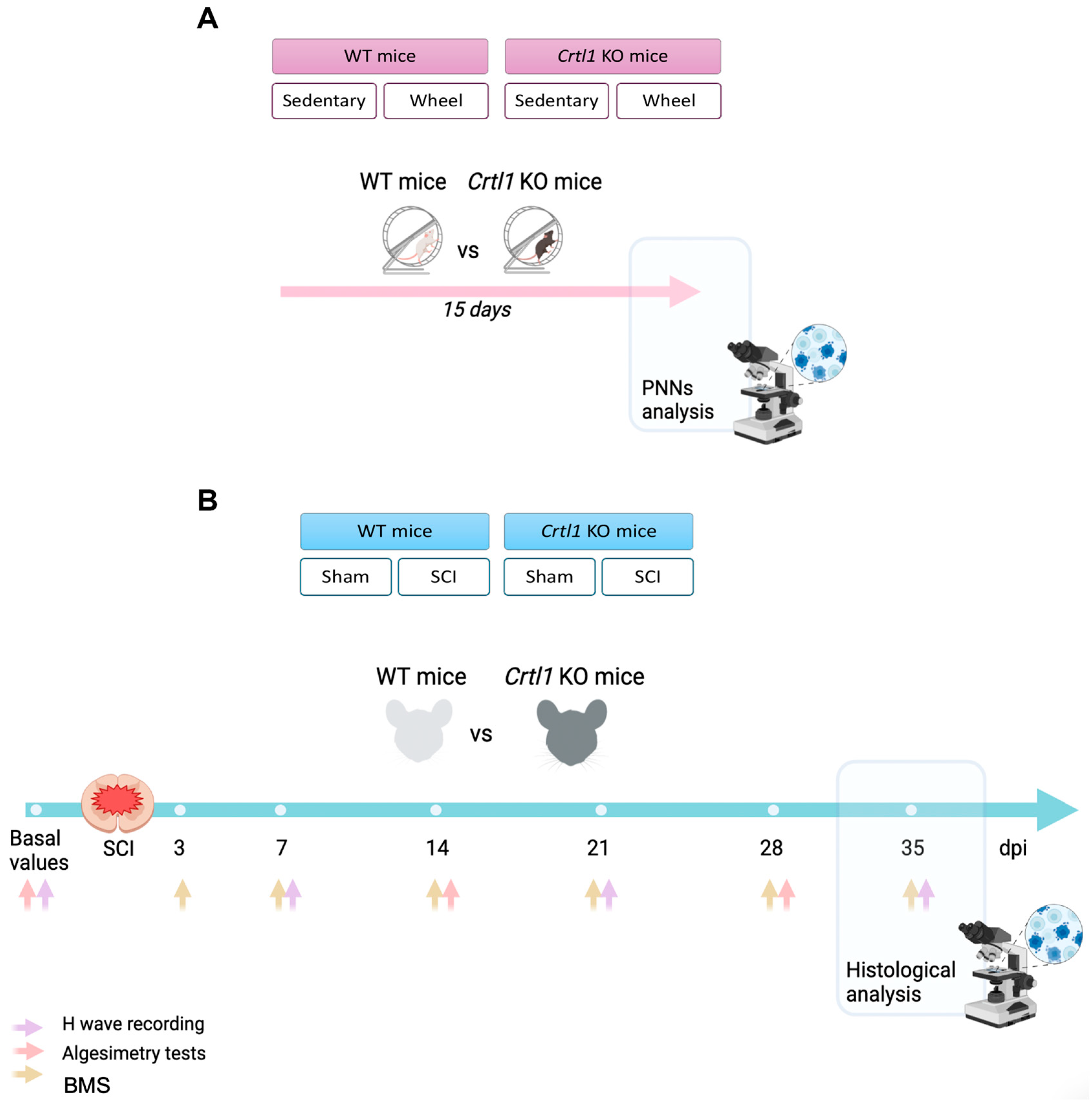
4.1. Experimental Groups
4.2. Spinal Cord Injury
4.3. Activity-Based Therapy Protocol
4.4. Functional Assessment
4.4.1. Locomotion Assessment
4.4.2. Algesimetry Tests
4.4.3. Electrophysiological Tests
4.5. Histological Evaluation
Histological Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tewari, B.P.; Chaunsali, L.; Prim, C.E.; Sontheimer, H. A glial perspective on the extracellular matrix and perineuronal net remodeling in the central nervous system. Front. Cell. Neurosci. 2022, 16, 1022754. [Google Scholar] [CrossRef] [PubMed]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of Ocular Dominance Plasticity in the Adult Visual Cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Oohashi, T.; Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat. Rev. Neurosci. 2019, 20, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.C.; Dick, G.; Wang, D.; Fawcett, J.W. Extracellular matrix and perineuronal nets in CNS repair. Dev. Neurobiol. 2011, 71, 1073–1089. [Google Scholar] [CrossRef] [PubMed]
- Carulli, D.; Pizzorusso, T.; Kwok, J.C.F.; Putignano, E.; Poli, A.; Forostyak, S.; Andrews, M.R.; Deepa, S.S.; Glant, T.T.; Fawcett, J.W. Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain 2010, 133, 2331–2347. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.C.F.; Carulli, D.; Fawcett, J.W. In vitro modeling of perineuronal nets: Hyaluronan synthase and link protein are necessary for their formation and integrity. J. Neurochem. 2010, 114, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Foscarin, S.; Ponchione, D.; Pajaj, E.; Leto, K.; Gawlak, M.; Wilczynski, G.M.; Rossi, F.; Carulli, D. Experience-dependent plasticity and modulation of growth regulatory molecules at central synapses. PLoS ONE 2011, 6, e16666. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ventura, J.; Canal, C.; Hidalgo, J.; Penas, C.; Navarro, X.; Torres-Espin, A.; Fouad, K.; Udina, E. Aberrant perineuronal nets alter spinal circuits, impair motor function, and increase plasticity. Exp. Neurol. 2022, 358, 114220. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Brückner, G.; Dityateva, G.; Grosche, J.; Kleene, R.; Schachner, M. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev. Neurobiol. 2007, 67, 570–588. [Google Scholar] [CrossRef]
- Smith, C.C.; Mauricio, R.; Nobre, L.; Marsh, B.; Wüst, R.C.; Rossiter, H.B.; Ichiyama, R.M. Differential regulation of perineuronal nets in the brain and spinal cord with exercise training. Brain Res. Bull. 2015, 111, 20–26. [Google Scholar] [CrossRef]
- Sánchez-Ventura, J.; Giménez-Llort, L.; Penas, C.; Udina, E. Voluntary wheel running preserves lumbar perineuronal nets, enhances motor functions and prevents hyperreflexia after spinal cord injury. Exp. Neurol. 2021, 336, 113533. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B. Neuropathic pain and spasticity: Intricate consequences of spinal cord injury. Spinal Cord 2017, 55, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Torres-Espín, A.; Beaudry, E.; Fenrich, K.; Fouad, K. Rehabilitative Training in Animal Models of Spinal Cord Injury. J. Neurotrauma 2018, 35, 1970–1985. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.J.; Hughes, D.I.; Polgár, E.; Nagy, G.G.; Mackie, M.; Ottersen, O.P.; Maxwell, D.J. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur. J. Neurosci. 2003, 17, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Hockfield, S. Molecular Evidence for Early Activity-Dependent Development of Hamster Motor Neurons. J. Neurosci. 1988, 8, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.G.; Hockfield, S. Electrical Activity in the Neuromuscular Unit Can Influence the Molecular Development of Motor Neurons. Dev. Biol. 1994, 162, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Carulli, D.; Broersen, R.; de Winter, F.; Muir, E.M.; Mešković, M.; de Waal, M.; de Vries, S.; Boele, H.J.; Canto, C.B.; De Zeeuw, C.I.; et al. Cerebellar plasticity and associative memories are controlled by perineuronal nets. Proc. Natl. Acad. Sci. USA 2020, 117, 6855–6865. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Oohashi, T.; Sata, M.; Bekku, Y.; Hirohata, S.; Nakamura, K.; Yonezawa, T.; Kusachi, S.; Shiratori, Y.; Ninomiya, Y. Lp3/Hapln3, a novel link protein that co-localizes with versican and is coordinately up-regulated by platelet-derived growth factor in arterial smooth muscle cells. Matrix Biol. 2004, 23, 287–298. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oohashi, T.; Edamatsu, M.; Bekku, Y.; Carulli, D. The hyaluronan and proteoglycan link proteins: Organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp. Neurol. 2015, 274, 134–144. [Google Scholar] [CrossRef]
- Bekku, Y.; Su, W.-D.; Hirakawa, S.; Fässler, R.; Ohtsuka, A.; Kang, J.S.; Sanders, J.; Murakami, T.; Ninomiya, Y.; Oohashi, T. Molecular cloning of Bral2, a novel brain-specific link protein, and immunohistochemical colocalization with brevican in perineuronal nets☆. Mol. Cell. Neurosci. 2003, 24, 148–159. [Google Scholar] [CrossRef]
- Bekku, Y.; Saito, M.; Moser, M.; Fuchigami, M.; Maehara, A.; Nakayama, M.; Kusachi, S.; Ninomiya, Y.; Oohashi, T. Bral2 is indispensable for the proper localization of brevican and the structural integrity of the perineuronal net in the brainstem and cerebellum. J. Comp. Neurol. 2012, 520, 1721–1736. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ichiyama, R.M.; Zhao, R.; Andrews, M.R.; Fawcett, J.W. Chondroitinase combined with rehabilitation promotes recovery of forelimb function in rats with chronic spinal cord injury. J. Neurosci. 2011, 31, 9332–9344. [Google Scholar] [CrossRef] [PubMed]
- Arbat-Plana, A.; Torres-Espín, A.; Navarro, X.; Udina, E. Activity dependent therapies modulate the spinal changes that motoneurons suffer after a peripheral nerve injury. Exp. Neurol. 2015, 263, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Arbat-Plana, A.; Navarro, X.; Udina, E. Effects of forced, passive, and voluntary exercise on spinal motoneurons changes after peripheral nerve injury. Eur. J. Neurosci. 2017, 46, 2885–2892. [Google Scholar] [CrossRef]
- Alilain, W.J.; Horn, K.P.; Hu, H.; Dick, T.E.; Silver, J. Functional regeneration of respiratory pathways after spinal cord injury. Nature 2011, 475, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Shuto, M.; Funakoshi, K. Chondroitin sulfate expression in perineuronal nets after goldfish spinal cord lesion. Front. Cell. Neurosci. 2018, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Al’joboori, Y.D.; Edgerton, V.R.; Ichiyama, R.M. Effects of rehabilitation on perineural nets and synaptic plasticity following spinal cord transection. Brain Sci. 2020, 10, 824. [Google Scholar] [CrossRef] [PubMed]
- Irvine, S.F.; Kwok, J.C.F. Perineuronal nets in spinal motoneurones: Chondroitin sulphate proteoglycan around alpha motoneurones. Int. J. Mol. Sci. 2018, 19, 1172. [Google Scholar] [CrossRef] [PubMed]
- Galtrey, C.M.; Kwok, J.C.F.; Carulli, D.; Rhodes, K.E.; Fawcett, J.W. Distribution and synthesis of extracellular matrix proteoglycans, hyaluronan, link proteins and tenascin-R in the rat spinal cord. Eur. J. Neurosci. 2008, 27, 1373–1390. [Google Scholar] [CrossRef]
- Dityatev, A.; A Rusakov, D. Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 2011, 21, 353–359. [Google Scholar] [CrossRef]
- Cawston, T.E.; Young, D.A. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 2010, 339, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Spangenberg, E.E.; Tang, B.; Holmes, T.C.; Green, K.N.; Xu, X. Microglia elimination increases neural circuit connectivity and activity in adult mouse cortex. J. Neurosci. 2021, 41, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- de Castro, R., Jr.; Burns, C.L.; McAdoo, D.J.; Romanic, A.M. Metalloproteinase increases in the injured rat spinal cord. Neuroreport 2000, 11, 3551–3554. [Google Scholar] [CrossRef]
- Zhang, H.; Chang, M.; Hansen, C.N.; Basso, D.M.; Noble-Haeusslein, L.J. Role of Matrix Metalloproteinases and Therapeutic Benefits of Their Inhibition in Spinal Cord Injury. Neurotherapeutics 2011, 8, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.-P.; Miyawaki, A.; Gage, F.H.; Jan, Y.N.; Jan, L.Y. Local generation of glia is a major astrocyte source in postnatal cortex. Nature 2012, 484, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Felix, L.; Stephan, J.; Rose, C.R. Astrocytes of the early postnatal brain. Eur. J. Neurosci. 2021, 54, 5649–5672. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Moon, L.D.F.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, V.; Raman, R.; Capila, I.; Bosques, C.J.; Pojasek, K.; Sasisekharan, R. Biochemical characterization of the chondroitinase ABC I active site. Biochem. J. 2005, 390, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W. The extracellular matrix in plasticity and regeneration after CNS injury and neurodegenerative disease. Prog. Brain Res. 2015, 218, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Tansley, S.; Gu, N.; Guzmán, A.U.; Cai, W.; Wong, C.; Lister, K.C.; Muñoz-Pino, E.; Yousefpour, N.; Roome, R.B.; Heal, J.; et al. Microglia-mediated degradation of perineuronal nets promotes pain. Science 2022, 377, 80–86. [Google Scholar] [CrossRef]
- Boulenguez, P.; Liabeuf, S.; Bos, R.; Bras, H.; Jean-Xavier, C.; Brocard, C.; Stil, A.; Darbon, P.; Cattaert, D.; Delpire, E.; et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 2010, 16, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neurosci. 2012, 18, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Raineteau, O.; Schwab, M.E. Plasticity of motor systems after incomplete spinal cord injury. Nat. Rev. Neurosci. 2001, 2, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Cameron, W.E.; Núez-Abades, P.A. Physiological changes accompanying anatomical remodeling of mammalian motoneurons during postnatal development. Brain Res. Bull. 2000, 53, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Czipri, M.; Otto, J.M.; Cs-Szabó, G.; Kamath, R.V.; Vermes, C.; Firneisz, G.; Kolman, K.J.; Watanabe, H.; Li, Y.; Roughley, P.J.; et al. Genetic rescue of chondrodysplasia and the perinatal lethal effect of cartilage link protein deficiency. J. Biol. Chem. 2003, 278, 39214–39223. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.; Hoab, S.M.; Waiteb, P.M.E. Effects of different anesthetics on the paired-pulse depression of the H reflex in adult rat. Exp. Neurol. 2002, 177, 494–502. [Google Scholar] [CrossRef][Green Version]
- Friese, A.; Kaltschmidt, J.A.; Ladle, D.R.; Sigrist, M.; Jessell, T.M.; Arber, S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc. Natl. Acad. Sci. USA 2009, 106, 13588–13593. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Secondary Antibodies | ||||||
|---|---|---|---|---|---|---|---|
| Name | Dilution | Host | Reference | Name | Dilution | Host | Reference |
| Aggrecan | 1:250 | Rabbit | AB1031—Millipore (Burlington, MA, USA) | Alexa 488 | 1:200 | Donkey x Rabbit | A21206—Invitrogen (Waltham, MA, USA) |
| VGlut1 | 1:300 | Guinea Pig | AB5905—Millipore | Cys3 | 1:200 | Donkey x Guinea Pig | 706-165-148—Jackson (Scottsdale, AZ, USA) |
| GFAP | 1:1000 | Rabbit | AB5804—Millipore | Alexa 488 | 1:200 | Donkey x Rabbit | A21206—Invitrogen |
| Iba1 | 1:300 | Goat | AB5076—Abcam (Cambridge, UK) | Alexa 594 | 1:200 | Donkey x Goat | A11058—Invitrogen |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Ventura, J.; Lago, N.; Penas, C.; Navarro, X.; Udina, E. Link Protein 1 Is Involved in the Activity-Dependent Modulation of Perineuronal Nets in the Spinal Cord. Int. J. Mol. Sci. 2024, 25, 4267. https://doi.org/10.3390/ijms25084267
Sánchez-Ventura J, Lago N, Penas C, Navarro X, Udina E. Link Protein 1 Is Involved in the Activity-Dependent Modulation of Perineuronal Nets in the Spinal Cord. International Journal of Molecular Sciences. 2024; 25(8):4267. https://doi.org/10.3390/ijms25084267
Chicago/Turabian StyleSánchez-Ventura, Judith, Natalia Lago, Clara Penas, Xavier Navarro, and Esther Udina. 2024. "Link Protein 1 Is Involved in the Activity-Dependent Modulation of Perineuronal Nets in the Spinal Cord" International Journal of Molecular Sciences 25, no. 8: 4267. https://doi.org/10.3390/ijms25084267
APA StyleSánchez-Ventura, J., Lago, N., Penas, C., Navarro, X., & Udina, E. (2024). Link Protein 1 Is Involved in the Activity-Dependent Modulation of Perineuronal Nets in the Spinal Cord. International Journal of Molecular Sciences, 25(8), 4267. https://doi.org/10.3390/ijms25084267