
Phenotypic Expression and Outcomes in Patients with the p.Arg301Gln GLA Variant in Anderson–Fabry Disease


 , , , , , , ,
, , , , , , ,  , , , and
, , , and
Abstract
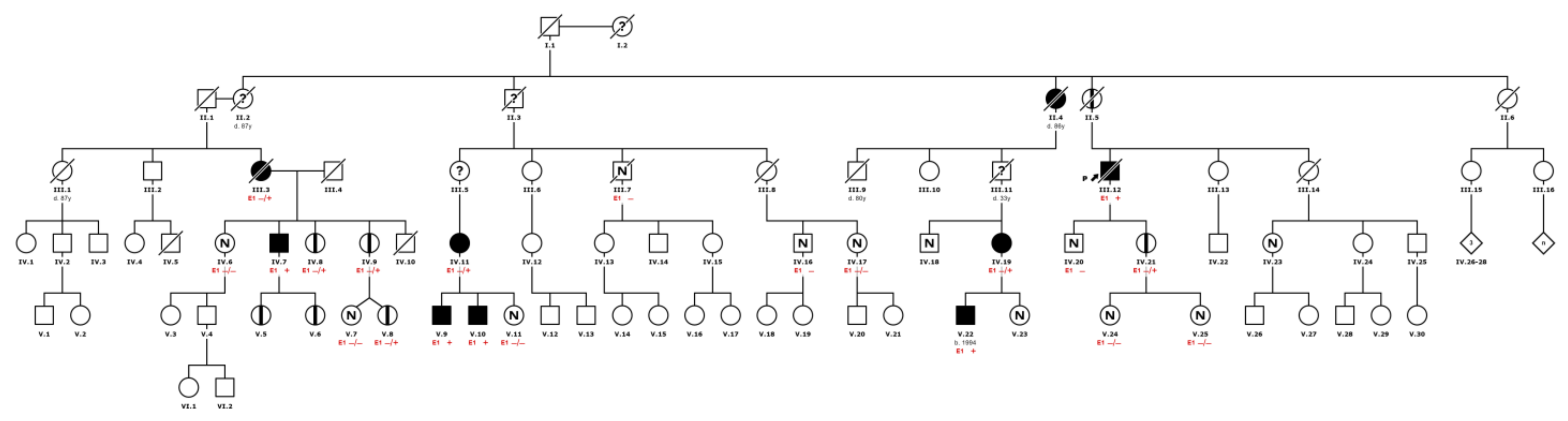
1. Introduction
2. Results
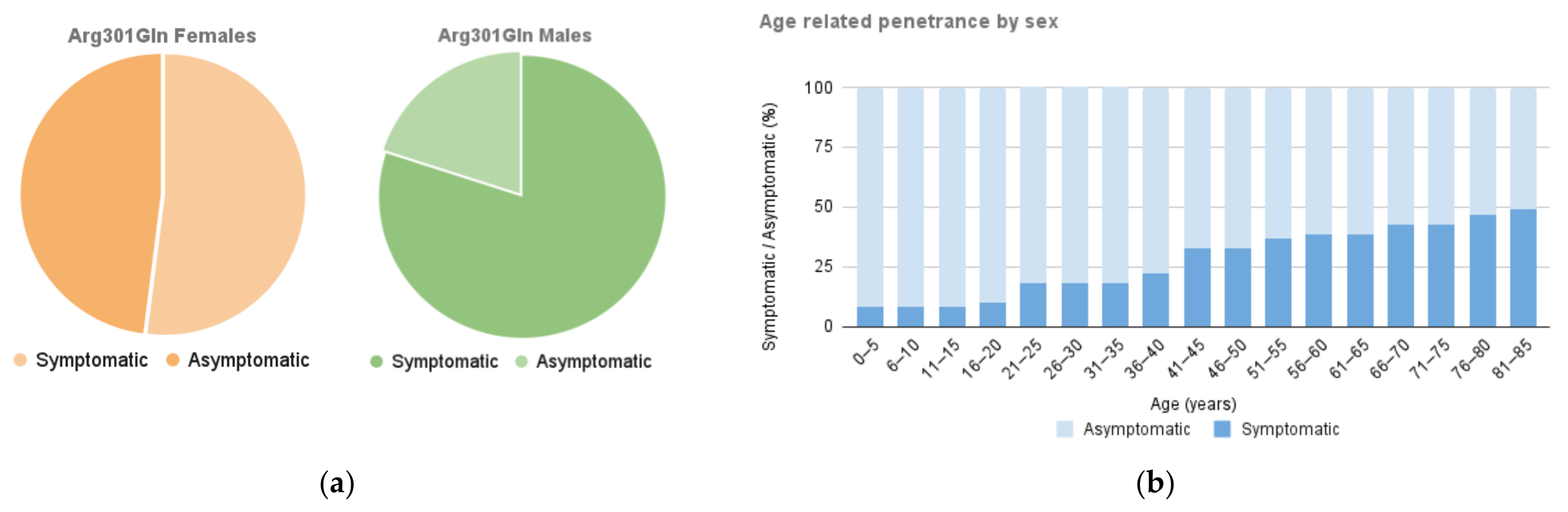
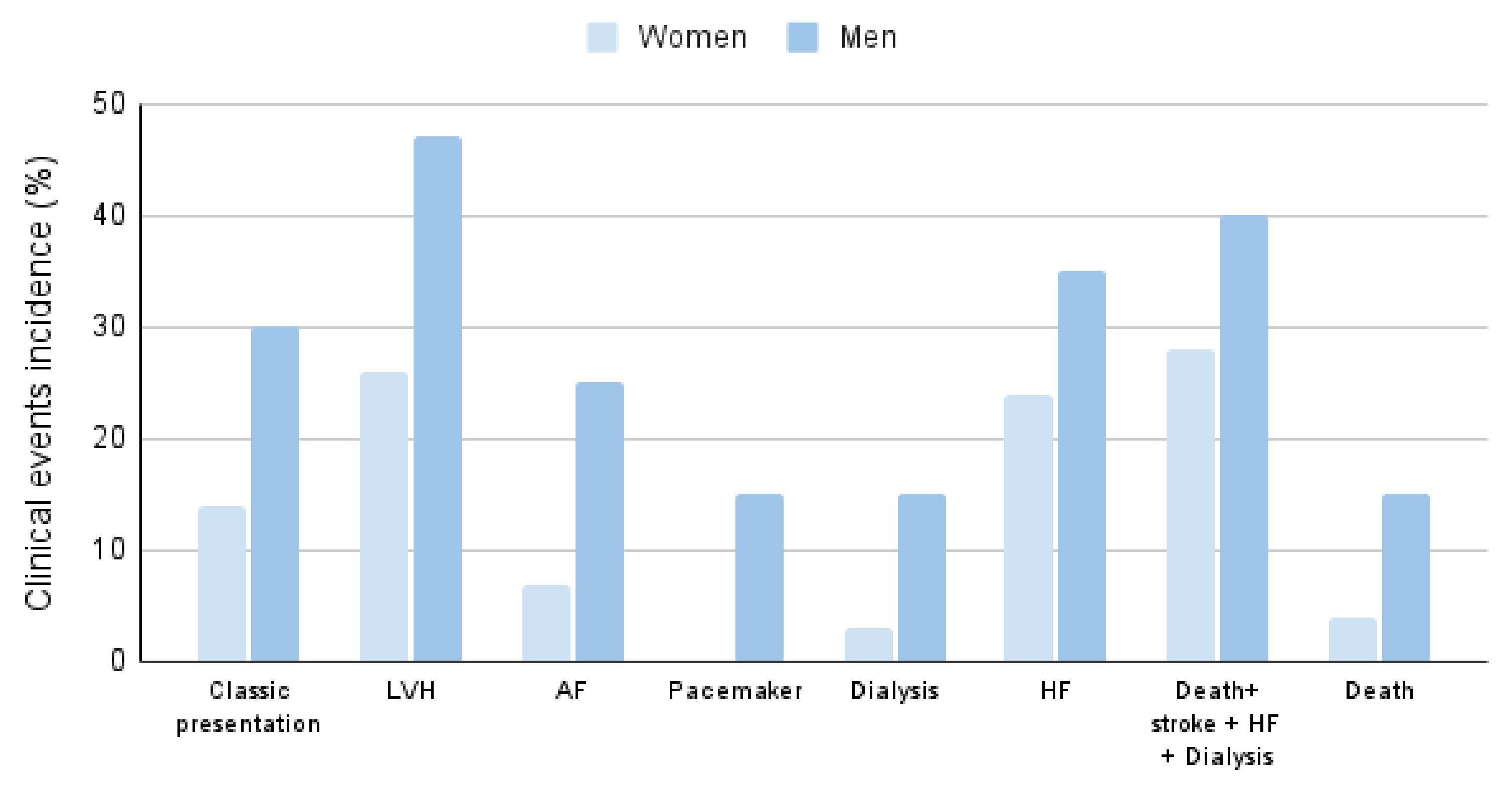
2.1. Organ Involvement and Differences according to Gender
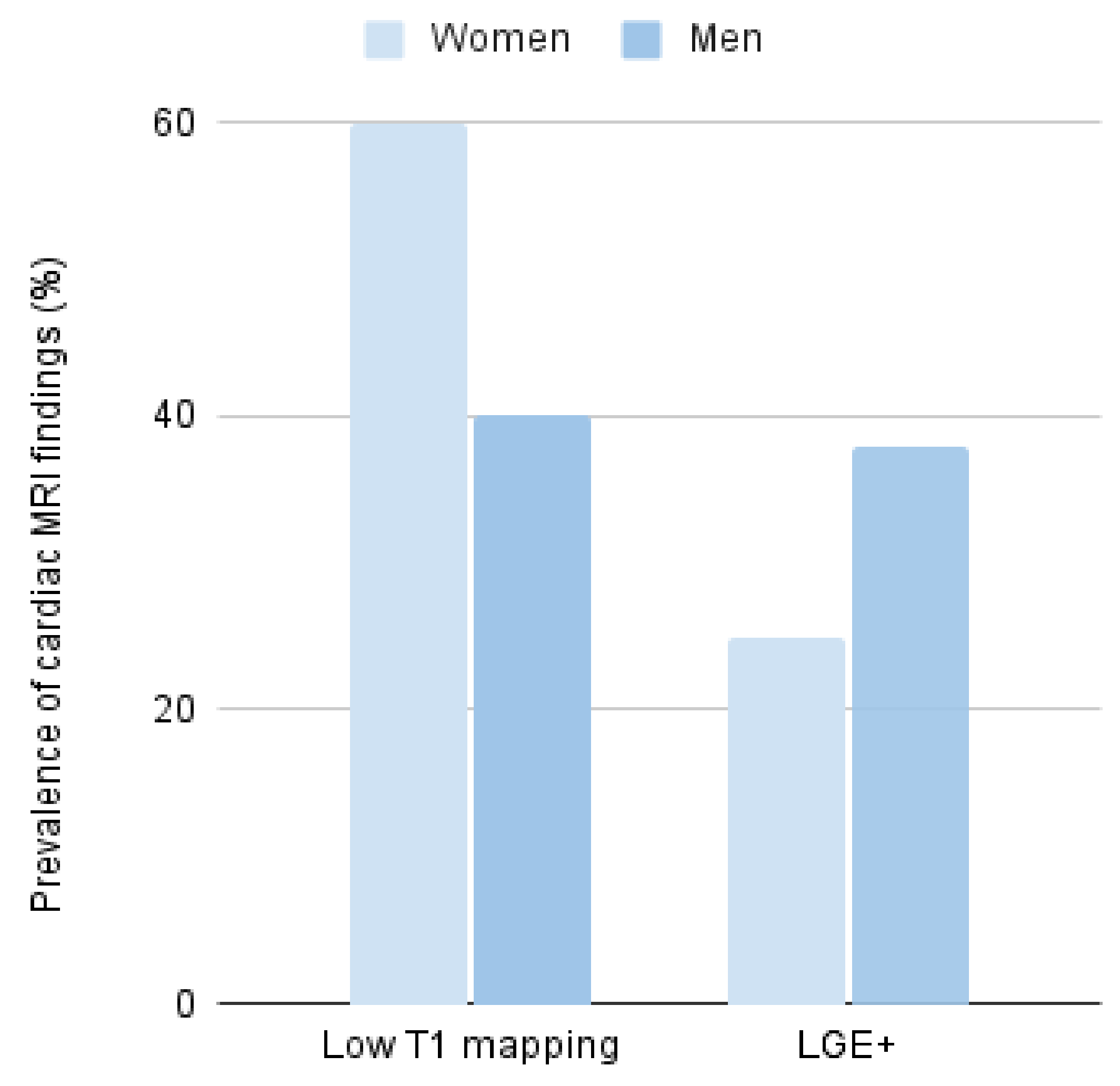
2.2. Biochemical Characteristics According to Gender
2.3. Specific Treatment and Gender Differences
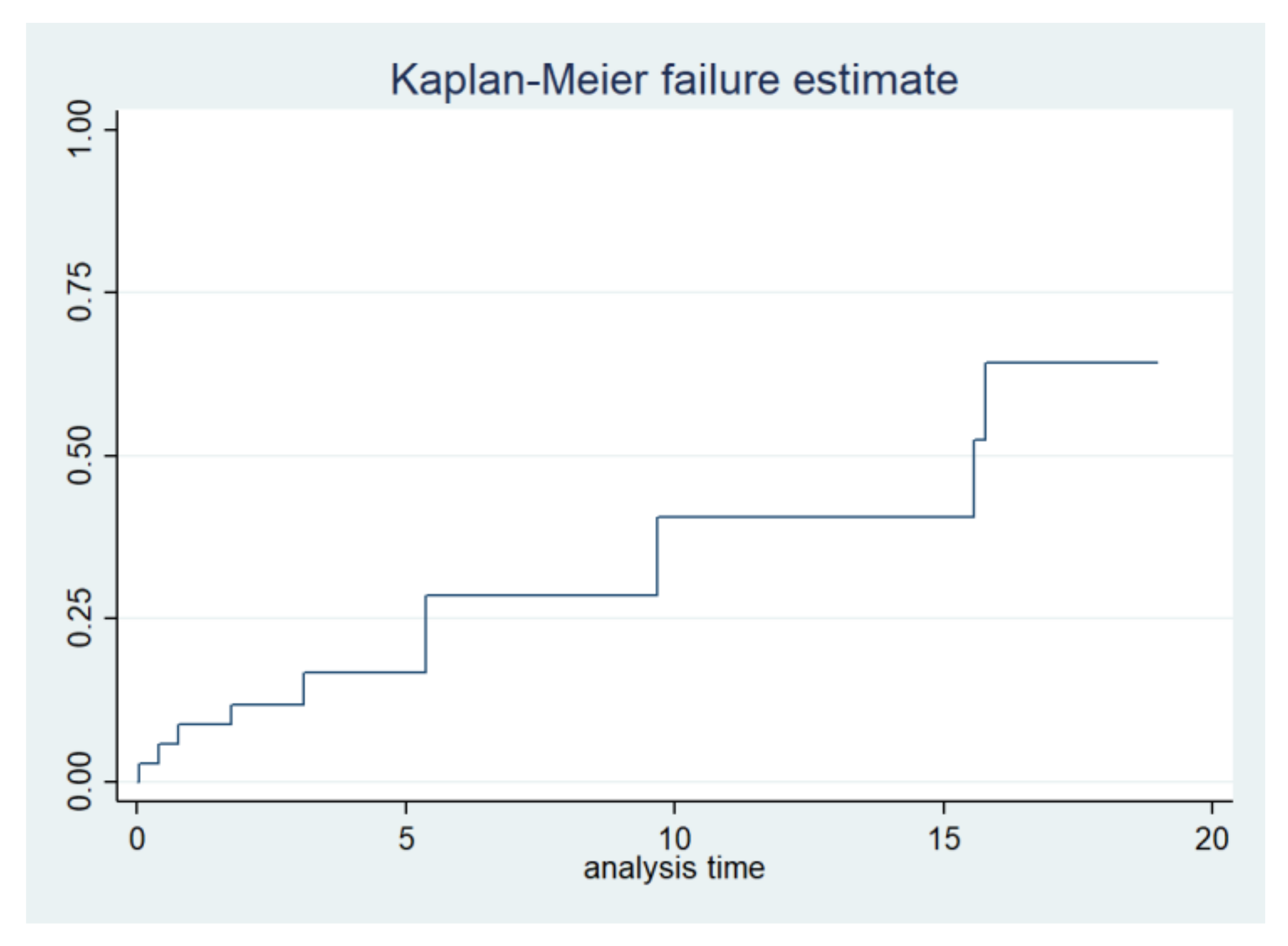
2.4. Life Expectancy and Event-Free Survival
3. Discussion
Limitations
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garman, S.C. Structure-function relationships in alpha-galactosidase A. Acta Paediatr. 2007, 96, 6–16. [Google Scholar] [CrossRef]
- Germain, D.P.; Levade, T.; Hachulla, E.; Knebelmann, B.; Lacombe, D.; Seguin, V.L.; Nguyen, K.; Noël, E.; Rabès, J.P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2022, 101, 390–402. [Google Scholar] [CrossRef]
- Gal, A.; Schäfer, E.; Rohard, I. The genetic basis of Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 33. [Google Scholar]
- Lien, Y.-H.H.; Lai, L.-W.; Lui, C.Y. Unexpected diagnosis of Fabry disease in an 80-year-old man with syncope. Cardiology 2001, 96, 115–116. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nagasawa, T.; Sugimura, K.; Kanno, A.; Tatebe, S.; Aoki, T.; Sato, H.; Kozu, K.; Konno, R.; Nochioka, K.; et al. Clinical Diversity in Patients with Anderson-Fabry Disease with the R301Q Mutation. Intern. Med. 2019, 58, 603–607. [Google Scholar] [CrossRef]
- Fabry Mutants List. Available online: http://fabry-database.org/mutants (accessed on 11 March 2024).
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M. Is it Fabry disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Saito, A.; Kimura, T.; Takeuchi, Y.; Matsuda, K.; Fukami, H.; Sato, H.; Iwakura, Y.; Sato, H.; Nagasawa, T. A case of rapid progression of Fabry nephropathy with remarkable glomerulomegaly: A case report and mini literature review of weak response to enzyme replacement therapy (ERT). Ren. Replace. Ther. 2016, 2, 69. [Google Scholar] [CrossRef]
- Schäfer, E.; Baron, K.; Widmer, U.; Deegan, P.; Neumann, H.P.; Sunder-Plassmann, G.; Johansson, J.O.; Whybra, C.; Ries, M.; Pastores, G.M.; et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum. Mutat. 2005, 25, 412. [Google Scholar] [CrossRef]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Fabry Disease Therapy: State-of-the-Art and Current Challenges. Int. J. Mol. Sci. 2020, 22, 206. [Google Scholar] [CrossRef]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicentre, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef]
- Lukas, J.; Cimmaruta, C.; Liguori, L.; Pantoom, S.; Iwanov, K.; Petters, J.; Hund, C.; Bunschkowski, M.; Hermann, A.; Cubellis, M.V.; et al. Assessment of Gene Variant Amenability for Pharmacological Chaperone Therapy with 1-Deoxygalactonojirimycin in Fabry Disease. Int. J. Mol. Sci. 2020, 21, 956. [Google Scholar] [CrossRef]
- Sakuraba, H.; Tsukimura, T.; Togawa, T.; Tanaka, T.; Ohtsuka, T.; Sato, A.; Shiga, T.; Saito, S.; Ohno, K. Fabry disease in a Japanese population-molecular and biochemical characteristics. Mol. Genet. Metab. Rep. 2018, 17, 73–79. [Google Scholar] [CrossRef]
- Hsu, T.R.; Hung, S.C.; Chang, F.P.; Yu, W.C.; Sung, S.H.; Hsu, C.L.; Dzhagalov, I.; Yang, C.F.; Chu, T.H.; Lee, H.J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence: Experience with a Highly Prevalent Mutation. J. Am. Coll. Cardiol. 2016, 68, 2554–2563. [Google Scholar] [CrossRef]
- Aquaro, G.D.; De Gori, C.; Faggioni, L.; Parisella, M.L.; Aringhieri, G.; Cioni, D.; Lencioni, R.; Neri, E. Cardiac Magnetic Resonance in Fabry Disease: Morphological, Functional, and Tissue Features. Diagnostics 2022, 12, 2652. [Google Scholar] [CrossRef]
- Oliveira, J.P.; Nowak, A.; Barbey, F.; Torres, M.; Nunes, J.P.; Teixeira-E-Costa, F.; Carvalho, F.; Sampaio, S.; Tavares, I.; Pereira, O.; et al. Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: Natural history in males. Eur. J. Med. Genet. 2020, 63, 103703. [Google Scholar] [CrossRef]
- Pica, S.; Sado, D.M.; Maestrini, V.; Fontana, M.; White, S.K.; Treibel, T.; Captur, G.; Anderson, S.; Piechnik, S.K.; Robson, M.D.; et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 99. [Google Scholar] [CrossRef]
- Kolodny, E.; Fellgiebel, A.; Hilz, M.J.; Sims, K.; Caruso, P.; Phan, T.G.; Politei, J.; Manara, R.; Burlina, A. Cerebrovascular involvement in Fabry disease: Current status of knowledge. Stroke 2015, 46, 302–313. [Google Scholar] [CrossRef]
- Sims, K.; Politei, J.; Banikazemi, M.; Lee, P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: Natural history data from the Fabry Registry. Stroke 2009, 40, 788–794. [Google Scholar] [CrossRef]
- Zevedo, O.; Gal, A.; Faria, R.; Gaspar, P.; Miltenberger-Miltenyi, G.; Gago, M.F.; Dias, F.; Martins, A.; Rodrigues, J.; Reimão, P.; et al. Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype. Mol. Genet. Metab. 2020, 129, 150–160. [Google Scholar] [CrossRef]
- New York Heart Association. Diseases of the Heart and Blood Vessels: Nomenclature and Criteria for Diagnosis, 6th ed.; Little Brown and Co.: Boston, MA, USA, 1964; p. 114. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Cohort | Male | Female | p-Value | |
|---|---|---|---|---|
| n (%) | 49 | 20 (41%) | 29 (59%) | |
| Age diagnostic, median (IQR) | 40.91 (21.11, 55.90) | 40.09 (19.35, 52.29) | 41.56 (21.57, 57.19) | 0.71 |
| Age symptoms, median (IQR) | 41.51 (21.01, 55.84) | 37.29 (20.5, 49.14) | 46.40 (21.52, 67.98) | 0.32 |
| Time diagnosis-treatment (years). Median (IQR) | 0.91 (0.51, 1.74) | 0.62 (0.40, 1.41) | 1.45 (0.66, 4.29) | 0.21 |
| Symptomatic. n (%) | 31 (63%) | 16 (80%) | 15 (52%) | 0.044 |
| Classical clinical manifestations. n (%) | 10 (20%) | 6 (30%) | 4 (14%) | 0.17 |
| α-GAL A enzyme activity | ||||
| 0: normal | 11(28%) | 0 (%) | 11 (52%) | <0.001 |
| 1: reduced | 21 (54%) | 11 (16%) | 10 (48%) | |
| 2: undetectable | 7 (18%) | 7 (39%) | 0 (0%) | |
| LysoGb3 increased. n (%) | 30 (81%) | 15 (94%) | 15 (71%) | 0.086 |
| Neuropathic pain. n (%) | 5 (10%) | 3 (15%) | 2 (7%) | 0.36 |
| Acroparesthesia. n (%) | 11 (22%) | 6 (30%) | 5 (17%) | 0.29 |
| Hypohidrosis. n (%) | 2 (4%) | 0 (0%) | 2 (7%) | 0.23 |
| CNS involvement. n (%) | 6 (12%) | 3 (15%) | 3 (10%) | 0.63 |
| Cornea Verticillata. n (%) | 2 (4%) | 1 (5%) | 1 (3%) | 0.79 |
| Hearing impairment. n (%) | 7 (14%) | 4 (20%) | 3 (10%) | 0.34 |
| Angiokeratoma. n (%) | 4 (8%) | 2 (10%) | 2 (7%) | 0.7 |
| Gastrointestinal manifestations. n (%) | 11 (22%) | 6 (30%) | 5 (17%) | 0.29 |
| Proteinuria. n (%) | 18 (38%) | 9 (45%) | 9 (32%) | 0.36 |
| Hypertension. n (%) | 15 (36%) | 9 (45%) | 9 (32%) | 0.36 |
| Diabetes. n (%) | 3 (6%) | 2 (10%) | 1 (3%) | 0.35 |
| PR < 121 ms. n (%) | 9 (18%) | 3 (15%) | 6 (12%) | 0.61 |
| LVH EKG. n (%) | 15 (36%) | 8 (47%) | 7 (28%) | 0.21 |
| LVH on echo/MRI. n (%) | 16 (35%) | 9 (47%) | 7 (26%) | 0.13 |
| T-wave inversion | 25 (58%) | 14 (82%) | 11 (42%) | 0.009 |
| Complete Bundle branch block. n (%) | ||||
| 0: none | 44 (90%) | 15 (75%) | 29 (100%) | |
| 1: left | 3 (6%) | 3 (15%) | 0 (0%) | 0.008 |
| 2: right | 2 (4%) | 2 (10%) | 0 (0%) | |
| AV Block. n (%) | ||||
| 0: none | 45 (92%) | 18 (90%) | 27 (93%) | |
| 1º | 4 (8%) | 2 (10%) | 2 (7%) | 1 |
| 2º | 0 (0%) | 0 (0%) | 0 (0%) | |
| 3º | 0 (0%) | 0 (0%) | 0 (0%) | |
| Atrial fibrillation of flutter. n (%) | 7 (14%) | 5 (35%) | 2 (7%) | 0.075 |
| Pacemaker. n (%) | 3 (6%) | 3 (15%) | 0 (0%) | 0.031 |
| NSVT. n (%) | 4 (8%) | 4 (20%) | 0 (0%) | 0.013 |
| VT. n (%) | 2 (4%) | 2 (10%) | 0 (0%) | 0.087 |
| Echo: max_wall_thickness (mm. Median (IQR) | 11 (9, 17) | 13 (9, 20) | 9.5 (8, 13) | 0.048 |
| Echo LVEF %. Median (IQR) | 63 (59, 70) | 64.5 (58.5, 69.5) | 62 (60, 71) | 0.79 |
| Diastolic pattern. n (%) 0: normal | 14 (30%) | 4 (21%) | 10 (37%) | |
| 1: impairment | 21 (46%) | 7 (37%) | 14 (52%) | 0.095 |
| 2: pseudonormal | 10 (22%) | 7 (37%) | 3 (11%) | |
| 3: restrictive | 1 (2%) | 1 (5%) | 0 (0%) | |
| E/e. Median (IQR) | 7.9 (6, 13) | 0.5 (5.9, 14.32) | 7 (6, 11) | 0.38 |
| Atrial dilatation (%) | 22 (47%) | 11 (55%) | 11 (41%) | 0.33 |
| Global longitudinal strain %. Median (IQR) | 19 (14.1, 20.62) | 11.95 (8.55, 18.31) | 20 (19, 21.4) | 0.021 |
| Cardiac MRI wall thickness (mm). Median (IQR) | 11 (9, 17) | 16 (10, 18) | 10.5 (8, 15) | 0.11 |
| LGE +. n/MRI (%) | 10/32 (31%) | 6/16 (37.5%) | 4/16 (25%) | 0.45 |
| Low T1 mapping. n/MRI (%) | 10/21 (48%) | 4/10 (40%) | 6/11 (54%) | 0.52 |
| Treatment. n (%) 0: none | 25 (52%) | 4 (21%) | 21 (72%) | 0.003 |
| 1: agalsidase A | 5 (10%) | 4 (21%) | 1 (3%) | |
| 2: agalsidase B | 6 (13%) | 4 (21%) | 2 (7%) | |
| 3: migalastat | 12 (25%) | 7 (37%) | 55 (17%) | |
| Death, Stroke, HF, or Renal Replacement. n (%) | 16 (33%) | 8 (40%) | 8 (28%) | 0.36 |
| Death. n (%) | 4 (9%) | 3 (15%) | 1 (4%) | 0.17 |
| Stroke. n (%) | 4 (8%) | 2 (25%) | 1 (4%) | 0.16 |
| Heart Failure. n (%) | 14 (29%) | 7 (35%) | 7 (24%) | 0.41 |
| Dialysis. n (%) | 4 (8%) | 3 (15%) | 1 (3%) | 0.15 |
| NTproBNP (pg/mL). Median (IQR) | 219 (70.5, 765) | 292 (57, 899) | 129 (74, 592) | 0.44 |
| Patient | Gender | Country | Cornea Verticillate | Stroke | Proteinuria | Dialysis | HF or LVH | Pacemaker | NSVT/VT |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  |  | |||||||
| 2 | | |  | ||||||
| 3 | | |  | ||||||
| 4 | | | | | |||||
| 5 |  | | |||||||
| 6 | | | |||||||
| 7 | | | |||||||
| 8 | | | |||||||
| 9 | | | | |  | ||||
| 10 | | | |||||||
| 11 | | | | ||||||
| 12 | | |  | | | ||||
| 13 | | | |||||||
| 14 | | | |||||||
| 15 | | | | ||||||
| 16 | | | | | | ||||
| 17 | | | | | |||||
| 18 | | | | ||||||
| 19 | | | |||||||
| 20 | | | | ||||||
| 21 | | | | |  | | |||
| 22 | | | |||||||
| 23 | | | |  | | ||||
| 24 | | | | | | ||||
| 25 | | | | | | ||||
| 26 | | | |||||||
| 27 | | | |||||||
| 28 | | | |||||||
| 29 | | | | ||||||
| 30 | | | | | |||||
| 31 | | | | ||||||
| 32 | | | |||||||
| 33 | | |  | | | | | ||
| 34 | | | |||||||
| 35 | | | | ||||||
| 36 | | | | ||||||
| 37 | |  | | | |||||
| 38 | | | |||||||
| 39 | | | | ||||||
| 40 | | | |||||||
| 41 | | | | ||||||
| 42 | | | |||||||
| 43 | |  | | | | ||||
| 44 | | | | ||||||
| 45 | | | | | |||||
| 46 | | | | | | ||||
| 47 | | | | | | | | ||
| 48 | | | | ||||||
| 49 | | | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, R.; Rico-Ramírez, Y.; Hermida-Ameijeiras, Á.; Abdullah, I.M.S.; Lau, K.; Alvarez-Rubio, J.; Fortuny, E.; Martínez-Monzonís, A.; Nowak, A.; Nordbeck, P.; et al. Phenotypic Expression and Outcomes in Patients with the p.Arg301Gln GLA Variant in Anderson–Fabry Disease. Int. J. Mol. Sci. 2024, 25, 4299. https://doi.org/10.3390/ijms25084299
Blanco R, Rico-Ramírez Y, Hermida-Ameijeiras Á, Abdullah IMS, Lau K, Alvarez-Rubio J, Fortuny E, Martínez-Monzonís A, Nowak A, Nordbeck P, et al. Phenotypic Expression and Outcomes in Patients with the p.Arg301Gln GLA Variant in Anderson–Fabry Disease. International Journal of Molecular Sciences. 2024; 25(8):4299. https://doi.org/10.3390/ijms25084299
Chicago/Turabian StyleBlanco, Rocío, Yolanda Rico-Ramírez, Álvaro Hermida-Ameijeiras, Israa Mahmoud Sanad Abdullah, Kolja Lau, Jorge Alvarez-Rubio, Elena Fortuny, Amparo Martínez-Monzonís, Albina Nowak, Peter Nordbeck, and et al. 2024. "Phenotypic Expression and Outcomes in Patients with the p.Arg301Gln GLA Variant in Anderson–Fabry Disease" International Journal of Molecular Sciences 25, no. 8: 4299. https://doi.org/10.3390/ijms25084299
APA StyleBlanco, R., Rico-Ramírez, Y., Hermida-Ameijeiras, Á., Abdullah, I. M. S., Lau, K., Alvarez-Rubio, J., Fortuny, E., Martínez-Monzonís, A., Nowak, A., Nordbeck, P., Veras-Burgos, C., Pons-Llinares, J., Rossi, E., Caimi-Martínez, F., Bosch-Rovira, T., Alamar-Cervera, M., Ruiz-Pizarro, V., Torres-Juan, L., Heine-Suñer, D., & Ripoll-Vera, T. (2024). Phenotypic Expression and Outcomes in Patients with the p.Arg301Gln GLA Variant in Anderson–Fabry Disease. International Journal of Molecular Sciences, 25(8), 4299. https://doi.org/10.3390/ijms25084299