
Targeting Molecular Mechanisms of Obesity- and Type 2 Diabetes Mellitus-Induced Skeletal Muscle Atrophy with Nerve Growth Factor

 , ,
, ,  , , and
, , and
Abstract
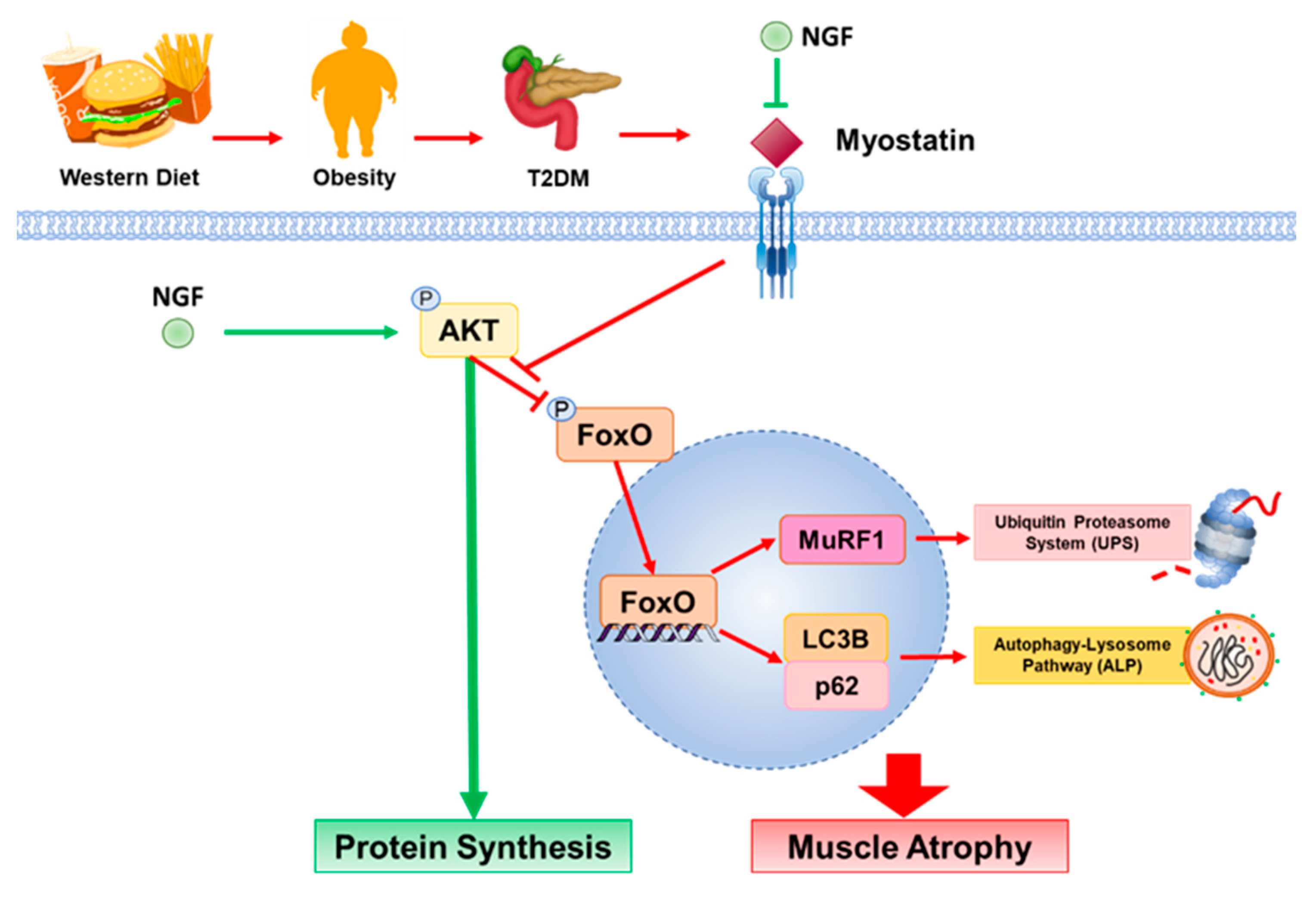
:1. Introduction
2. Results
2.1. Effects of High Fructose and Sucrose (HFS) Diet and Streptozotocin (STZ) Injections on Body Mass and Fasting Blood Glucose Level
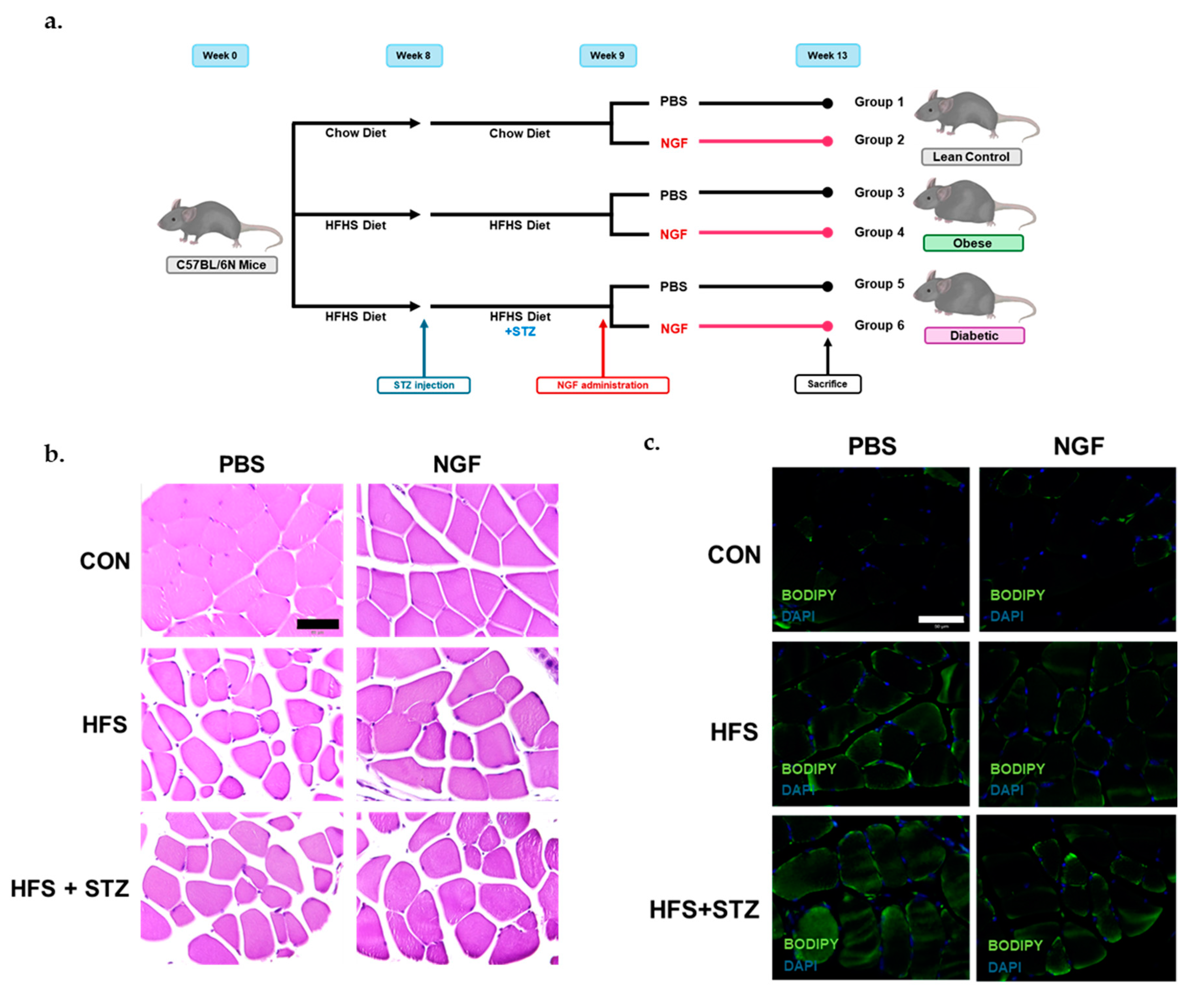
2.2. Skeletal Muscle Morphological Characteristics in Obese and T2DM Mice
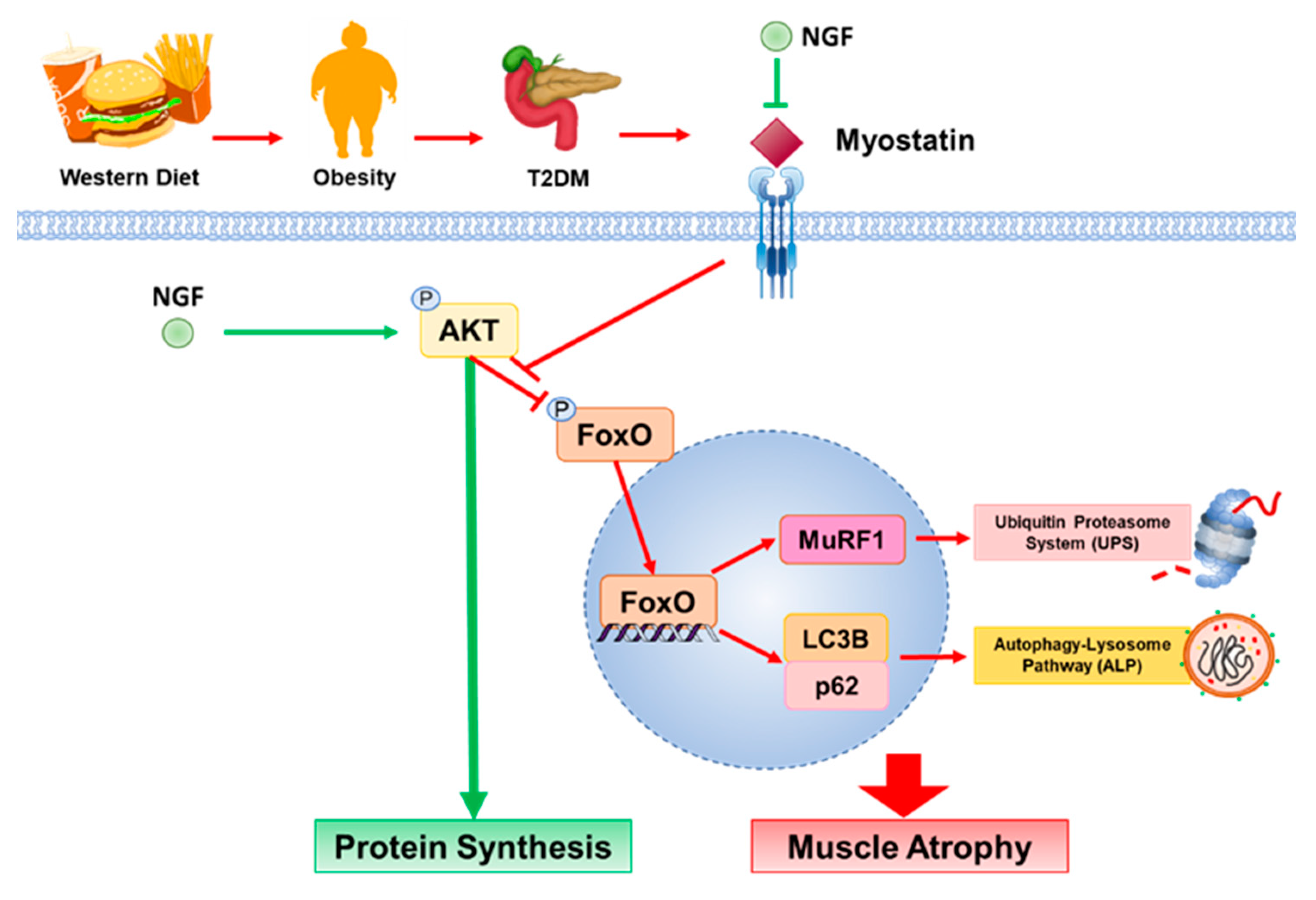
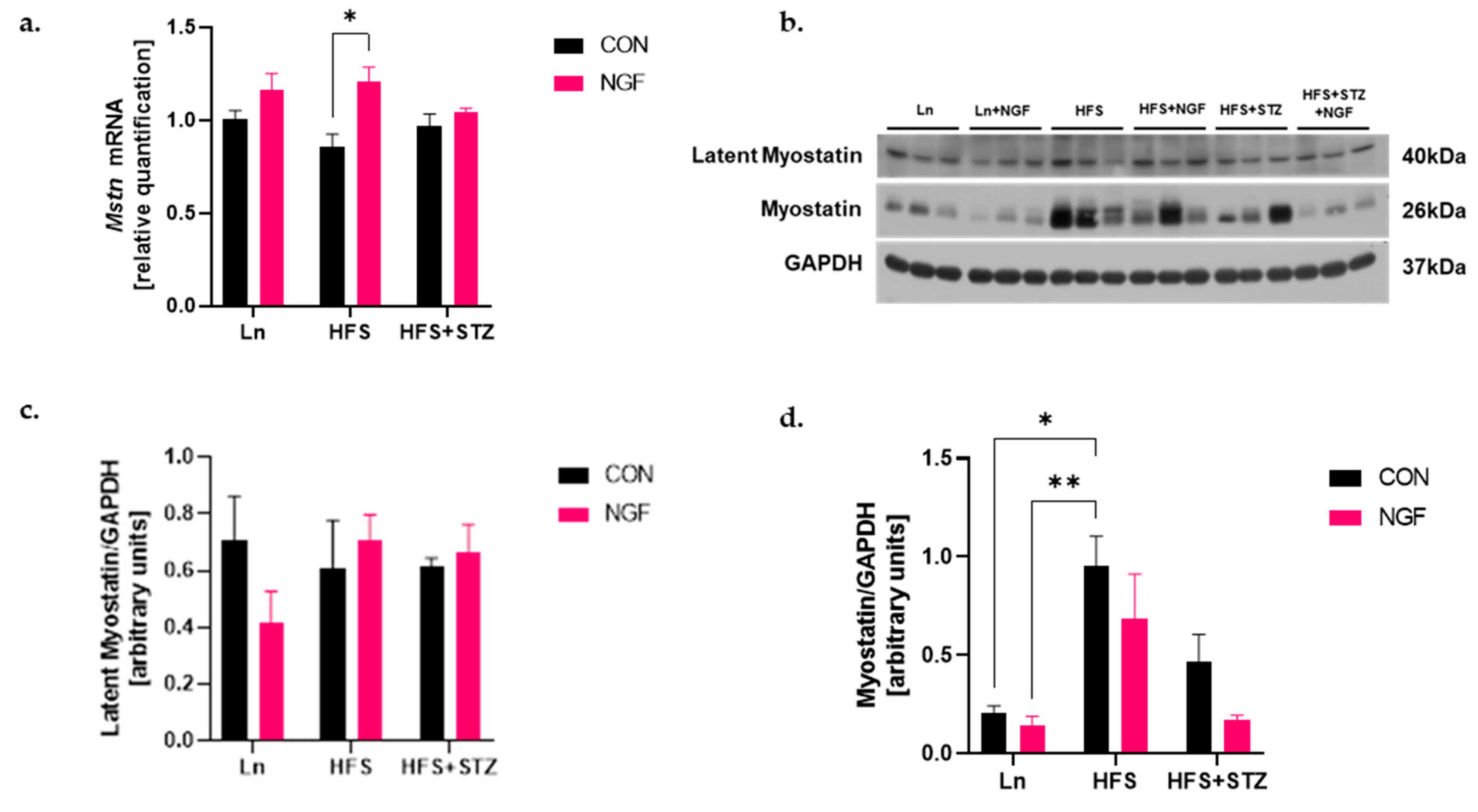
2.3. Obesity Increases Protein Level of Myostatin
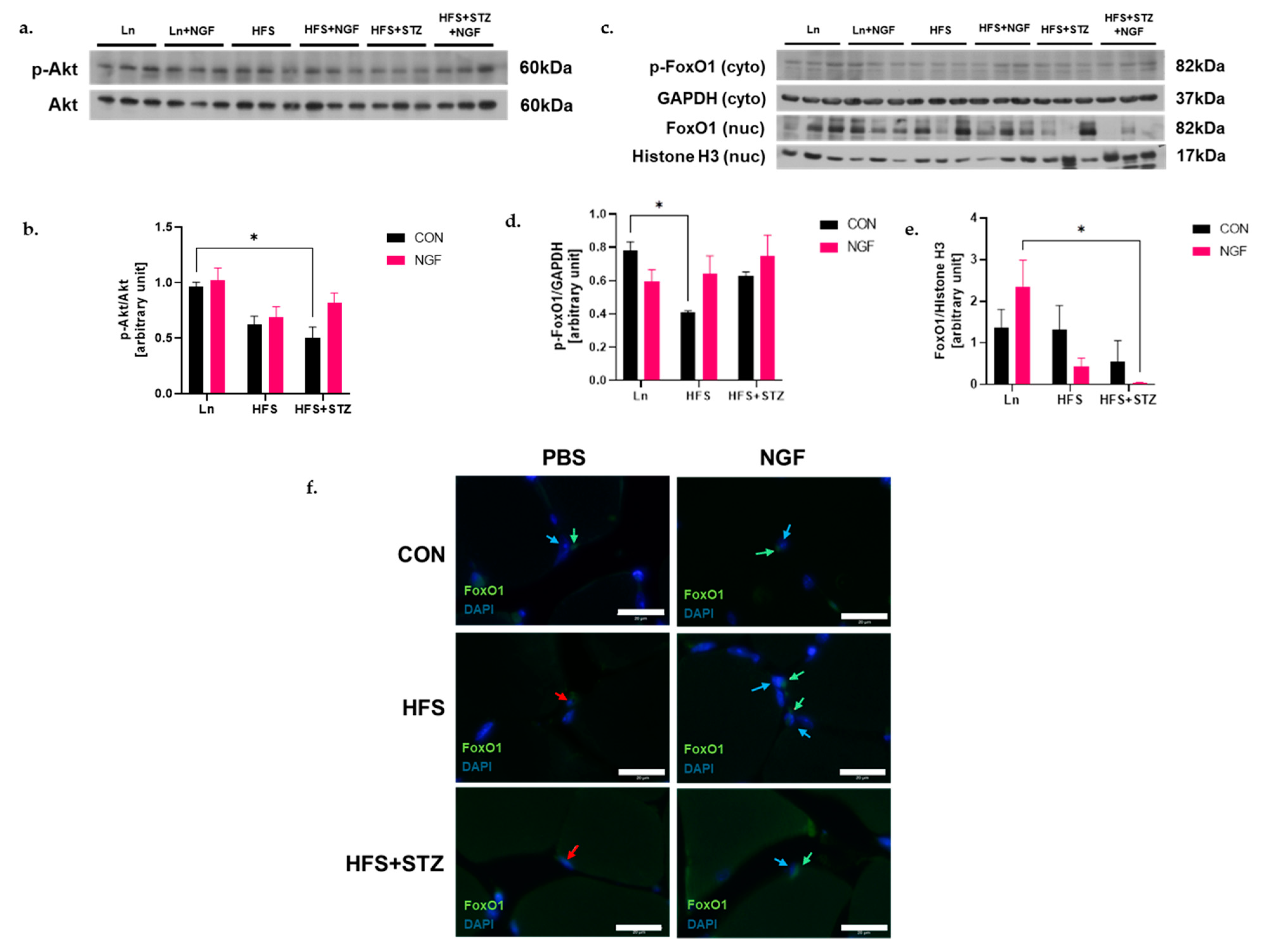
2.4. NGF Prevents Obesity- and Diabetes-Induced Muscle Atrophy through the Akt-Dependent Signaling Pathway
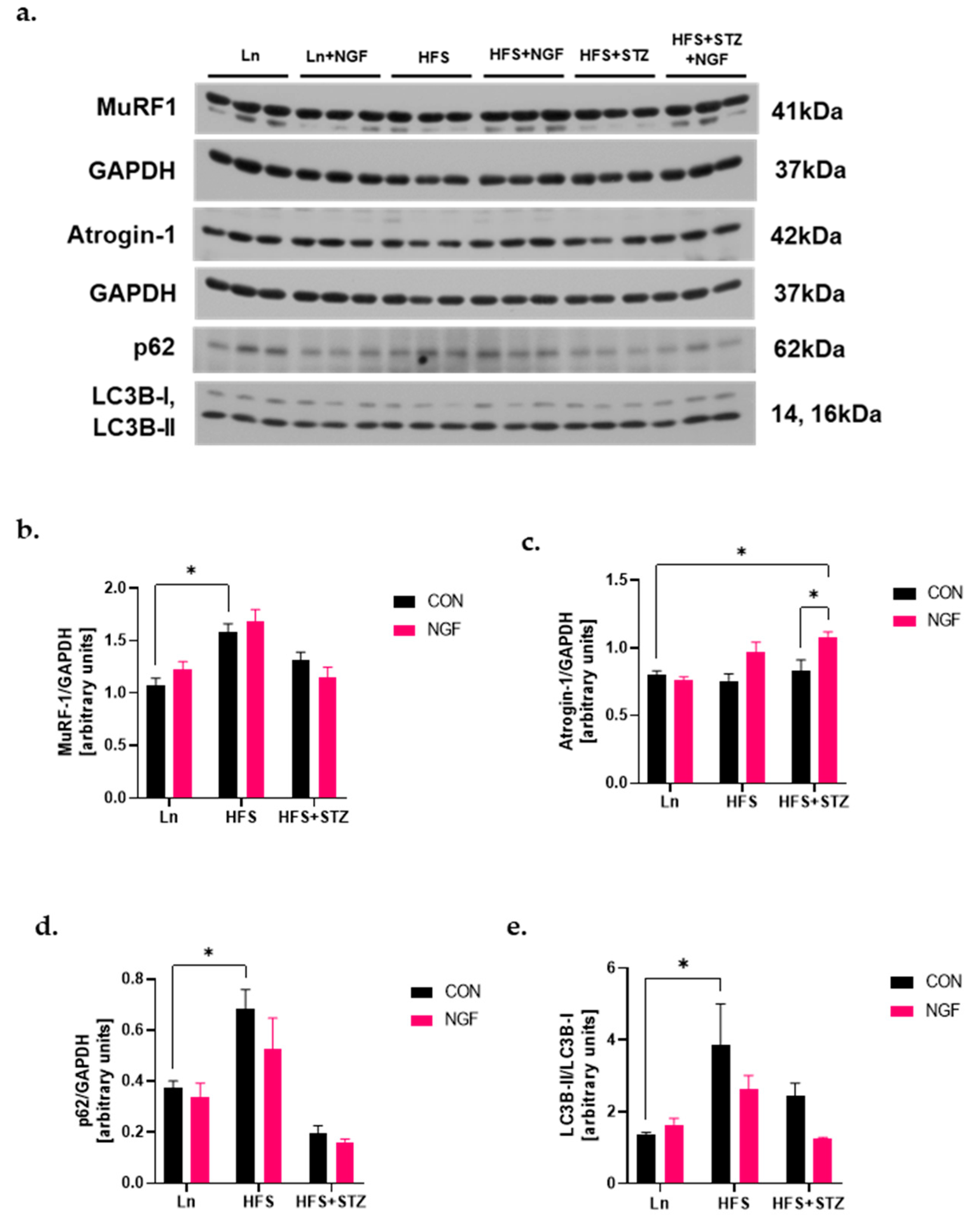
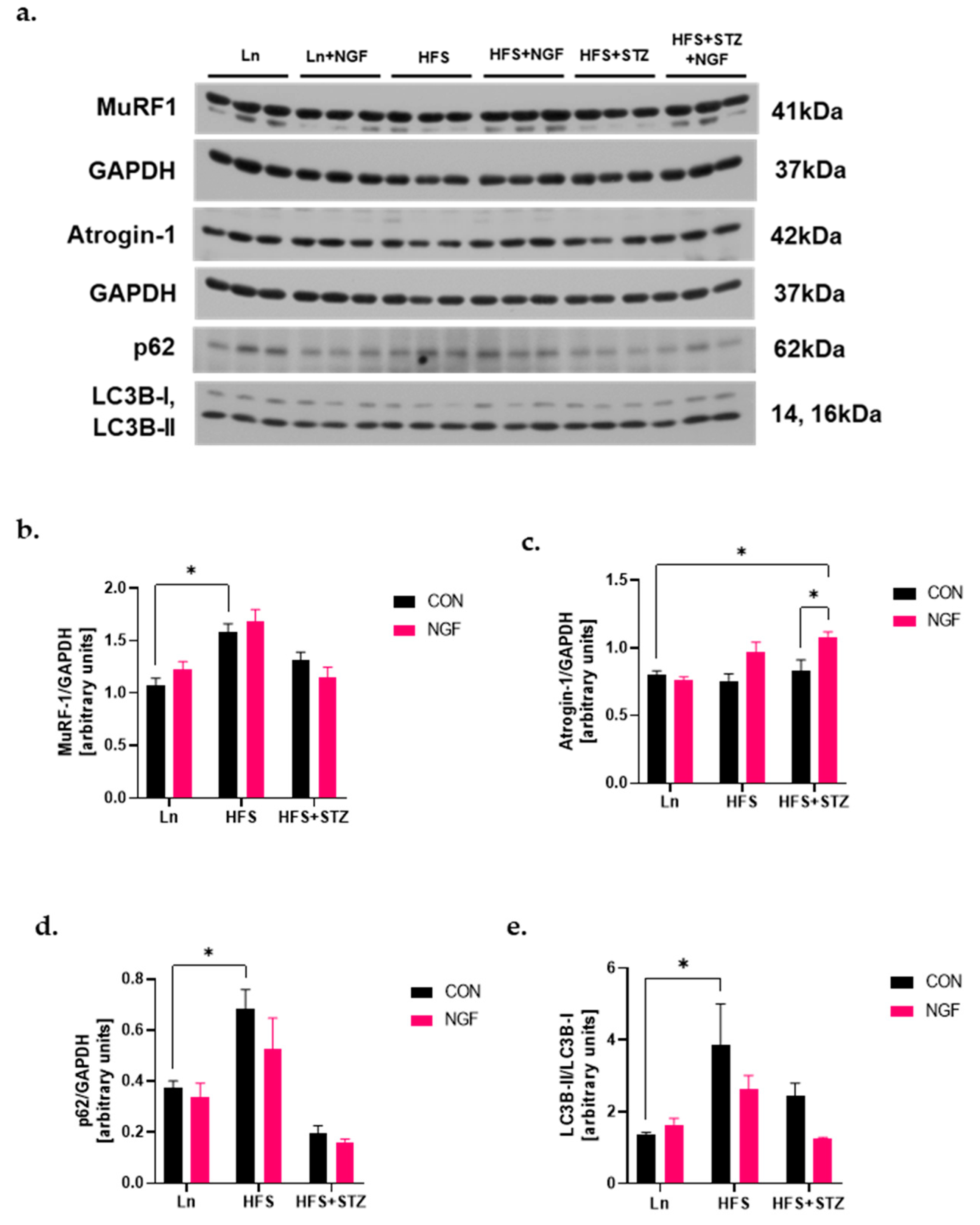
2.5. Levels of Ubiquitin Ligases and Autophagy Markers
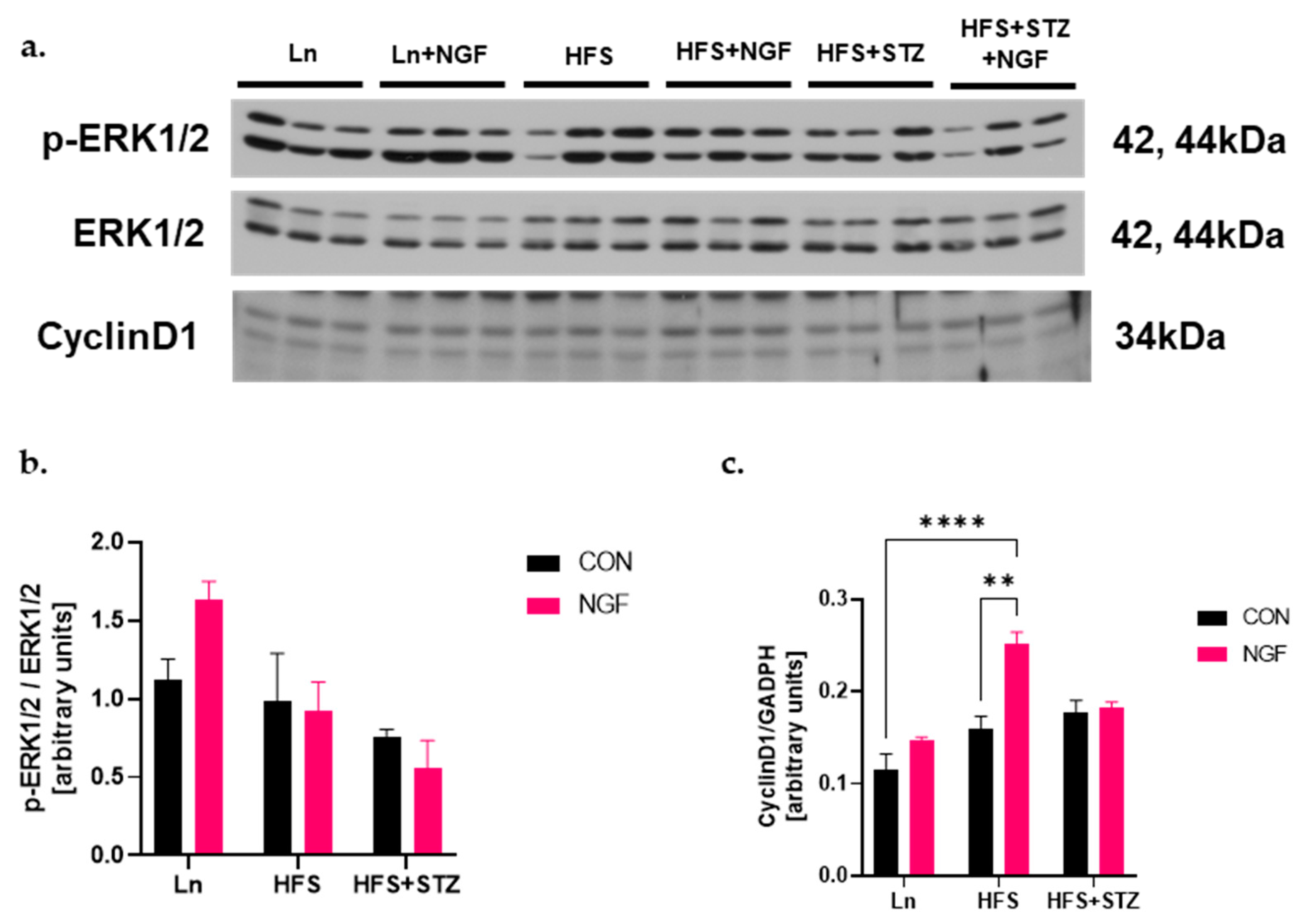
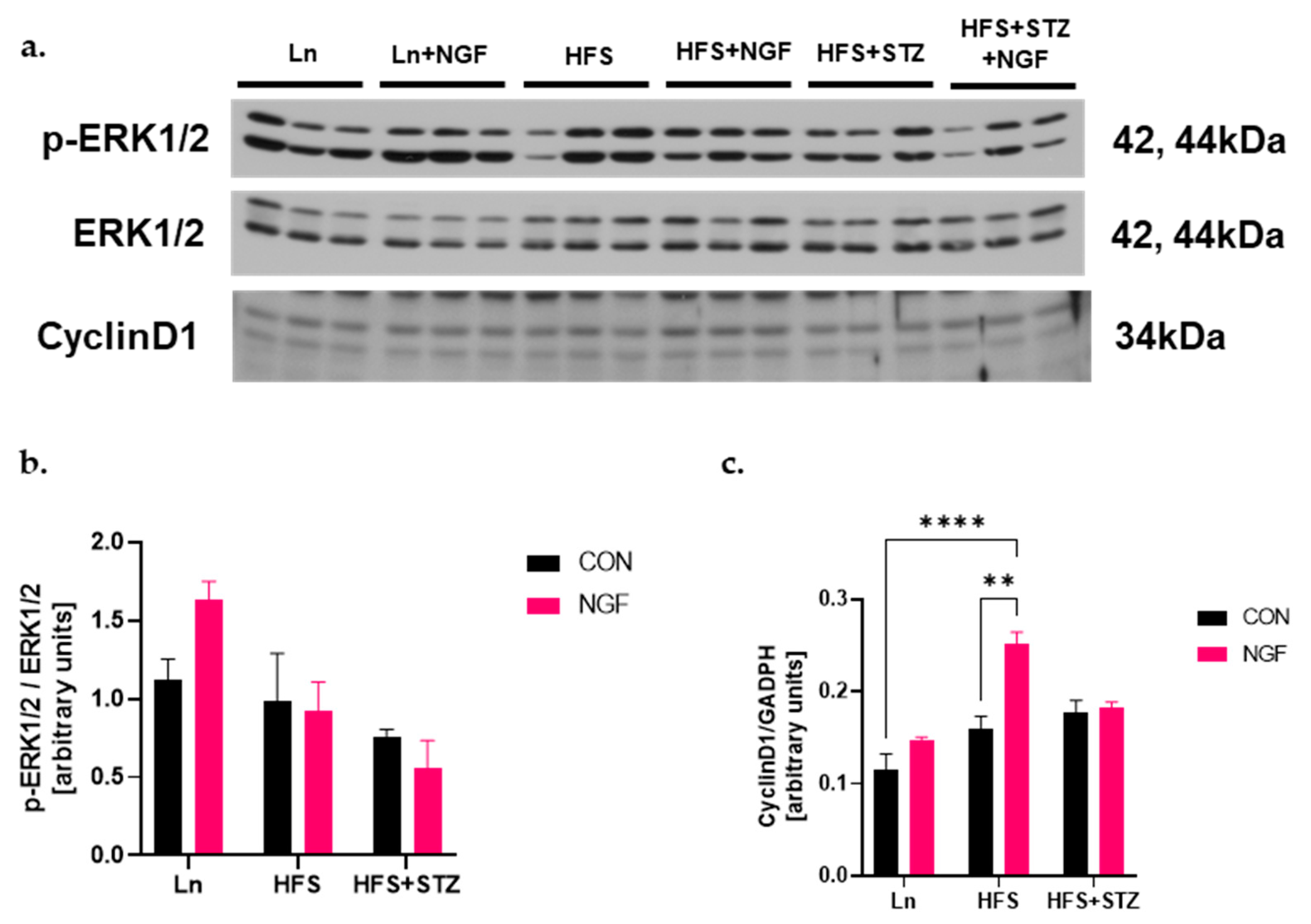
2.6. NGF Promotes Muscle Regeneration Capacity through Cyclin D1
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Diets
4.3. Treatments
4.4. Total Tissue Lysate Preparation
4.5. Tissue Subcellular Fractionation
4.6. Western Blot Analysis
4.7. RT-PCR
4.8. Muscle Histology
4.9. Hematoxylin and Eosin Staining
4.10. BODIPYTM Staining
4.11. Immunofluorescence Staining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Obesity Atlas 2023. World Obesity. Available online: https://s3-eu-west-1.amazonaws.com/wof-files/World_Obesity_Atlas_2023_Report.pdf (accessed on 21 February 2024).
- Centers for Disease Control and Prevention. National Diabetes Statistics Report. Available online: https://www.cdc.gov/diabetes/data/statistics-report/index.html (accessed on 21 February 2024).
- Van Greevenbroek, M.M.; Schalkwijk, C.G.; Stehouwer, C.D. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: Causes and consequences. Neth. J. Med. 2013, 71, 174–187. [Google Scholar]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Merz, K.; Thurmond, D.C. Role of skeletal muscle in insulin resistance and glucose uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar]
- Wolfe, R.R. The underappreciated role of muscle in health and disease. Am. J. Clin. Nutr. 2006, 84, 475–482. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Bilodeau, P.A.; Coyne, E.S.; Wing, S.S. The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation. Am. J. Physiol. Cell Physiol. 2016, 311, C392–C403. [Google Scholar] [CrossRef]
- Perry, B.D.; Caldow, M.K.; Brennan-Speranza, T.C.; Sbaraglia, M.; Jerums, G.; Garnham, A.; Wong, C.; Levinger, P.; ul Haq, M.A.; Hare, D.L.; et al. Muscle atrophy in patients with Type 2 Diabetes Mellitus: Roles of inflammatory pathways, physical activity and exercise. Exerc. Immunol. Rev. 2016, 22, 94–109. [Google Scholar]
- Allen, D.L.; Cleary, A.S.; Speaker, K.J.; Lindsay, S.F.; Uyenishi, J.; Reed, J.M.; Madden, M.C.; Mehan, R.S. Myostatin, activin receptor IIb, and follistatin-like-3 gene expression are altered in adipose tissue and skeletal muscle of obese mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E918–E927. [Google Scholar] [CrossRef]
- Wang, F.; Liao, Y.; Li, X.; Ren, C.; Cheng, C.; Ren, Y. Increased circulating myostatin in patients with type 2 diabetes mellitus. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 534–539. [Google Scholar] [CrossRef]
- Lee, E.J.; Ahmad, S.S.; Lim, J.H.; Ahmad, K.; Shaikh, S.; Lee, Y.S.; Park, S.J.; Jin, J.O.; Lee, Y.H.; Choi, I. Interaction of fibromodulin and myostatin to regulate skeletal diabetes, and intracellular lipid accumulation. Cells 2021, 10, 2083. [Google Scholar] [CrossRef]
- Elkina, Y.; Von Haehling, S.; Anker, S.D.; Springer, J. The role of myostatin in muscle wasting: An overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143–151. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hopkinson, N.S.; Kemp, P.R. Myostatin induces autophagy in skeletal muscle in vitro. Biochem. Biophys. Res. Commun. 2011, 415, 632–636. [Google Scholar] [CrossRef]
- Suh, J.; Lee, Y.-S. Myostatin inhibitors: Panacea or predicament for musculoskeletal disorders? J. Bone Metab. 2020, 27, 151–165. [Google Scholar] [CrossRef]
- Klesse, L.J.; Meyers, K.A.; Marshall, C.J.; Parada, L.F. Nerve growth factor induces survival and differentiation through two dis-tinct signaling cascades in PC12 cells. Oncogene 1999, 18, 2055–2068. [Google Scholar] [CrossRef]
- Toti, P.; Villanova, M.; Vatti, R.; Schuerfeld, K.; Stumpo, M.; Barbagli, L.; Malandrini, A.; Costantini, M. Nerve growth factor expression in human dystrophic muscles. Muscle Nerve 2003, 27, 370–373. [Google Scholar] [CrossRef]
- Pallottini, V.; Colardo, M.; Tonini, C.; Martella, N.; Strimpakos, G.; Colella, B.; Tirassa, P.; Bartolomeo, S.D.; Segatto, M. ProNGF/p75NTR axis drives fiber type specification by inducing the fast-glycolytic phenotype in mouse skeletal muscle cells. Cells 2020, 9, 2232. [Google Scholar] [CrossRef]
- Rende, M.; Brizi, E.; Conner, J.; Treves, S.; Censier, K.; Provenzano, C.; Taglialatela, G.; Sanna, P.P.; Donato, R. Nerve growth factor (NGF) influences differentiation and proliferation of myogenic cells in vitro via TrKA. Int. J. Dev. Neurosci. 2000, 18, 869–885. [Google Scholar] [CrossRef]
- Ruberti, F.; Capsoni, S.; Comparini, A.; Di Daniel, E.; Franzot, J.; Gonfloni, S.; Rossi, G.; Berardi, N.; Cattaneo, A. Phenotype knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J. Neurosci. 2000, 20, 2601–2789. [Google Scholar] [CrossRef]
- McFarlane, C.; Langley, B.; Thomas, M.; Hennebry, A.; Plummer, E.; Nicholas, G.; McMahon, C.; Sharma, M.; Kambadur, R. Proteolytic processing of myostatin is auto-regulated during myogenesis. Dev. Biol. 2005, 283, 58–69. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Bhattacharyya, N.; Han, L.-Y.; Hanover, J.A.; Rechler, M.M. Insulin inhibition of transcription stimulated by the forkhead protein Foxo1 is not solely due to nuclear exclusion. Endocrinology 2003, 144, 5615–5622. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Duan, X.; Liao, R.; Little, P.; Gao, G.; Jiang, H.; Lalit, S.; Quirion, R.; Zheng, W. Characterization of intracellular translocation of Forkhead transcription factor O (FoxO) members induced by NGF in PC12 cells. Neurosci. Lett. 2011, 498, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Casimiro, M.C.; Fu, M.; Powell, M.J.; Popov, V.M.; Lindsay, J.; Zani, B.M.; Ciccarelli, C.; Watanabe, G.; Lee, R.J.; et al. Nerve growth factor regulation of cyclin D1 in PC12 cells through a p21RAS extracellular signal-regulated kinase pathway requires cooperative interactions between Sp1 and nuclear factor-κB. Mol. Biol. Cell 2008, 19, 2566–2578. [Google Scholar] [CrossRef] [PubMed]
- Joulia, D.; Bernardi, H.; Garandel, V.; Rabenoelina, F.; Vernus, B.; Cabello, G. Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp. Cell Res. 2003, 286, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248–248.e1. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Phogat, J.; Yadav, A.; Dabur, R. The dependency of autophagy and ubiquitin proteasome system during skeletal muscle atrophy. Biophys. Rev. 2021, 13, 203–219. [Google Scholar] [CrossRef]
- Kadowaki, M.; Karim, M.R. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009, 452, 199–213. [Google Scholar] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Wang, D.-T.; Yang, Y.-J.; Huang, R.-H.; Zhang, Z.-H.; Lin, X. Myostatin activates the ubiquitin-proteasome and autophagy-lysosome systems contributing to muscle wasting in chronic kidney disease. Oxidative Med. Cell. Longev. 2015, 2015, 684965. [Google Scholar] [CrossRef]
- Bassi, D.; Bueno, P.d.G.; Nonaka, K.O.; Selistre-Araujo, H.S.; Leal, A.M.d.O. Exercise alters myostatin protein expression in sedentary and exercised streptozotocin-diabetic rats. Arq. Bras. Endocrinol. Metabol. 2015, 59, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.P.; Ibebunjo, C.; A Grasser, W.; Paralkar, V.M. Myostatin expression in age and denervation-induced skeletal muscle atrophy. J. Musculoskelet. Neuronal. Interact. 2003, 3, 8–16. [Google Scholar] [PubMed]
- Jeanplong, F.; Bass, J.; Smith, H.; Kirk, S.; Kambadur, R.; Sharma, M.; Oldham, J. Prolonged underfeeding of sheep increases myostatin and myogenic regulatory factor Myf-5 in skeletal muscle while IGF-I and myogenin are repressed. J. Endocrinol. 2003, 176, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Silvis, C.; Nystrom, G.; Frost, R.A. Regulation of myostatin by glucocorticoids after thermal injury. FASEB J. 2001, 15, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, C.-W.; Wang, R.; Tien, P.; Dale, R.; Sun, L.-Q. Myostatin antisense RNA-mediated muscle growth in normal and cancer cachexia mice. Gene Ther. 2007, 15, 155–160. [Google Scholar]
- Smith, I.J.; Aversa, Z.; Alamdari, N.; Petkova, V.; Hasselgren, P. Sepsis downregulates myostatin mRNA levels without altering myostatin protein levels in skeletal muscle. J. Cell. Biochem. 2010, 111, 1059–1073. [Google Scholar] [CrossRef]
- Carlson, C.J.; Booth, F.W.; Gordon, S.E.; Shimkus, K.L.; Shirazi-Fard, Y.; Wiggs, M.P.; Ullah, S.T.; Pohlenz, C.; Gatlin, D.M.; Carroll, C.C.; et al. Skeletal muscle myostatin mRNA expression is fiber-type specific and increases during hindlimb unloading. Am. J. Physiol. 1999, 277, R601–R606. [Google Scholar] [CrossRef]
- Meloche, S.; Pouysségur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Qian, T.; Le, Q.; Sun, X.; Wu, J.; Chen, J.; Yu, X.; Xu, J. NGF promotes cell cycle progression by regulating D-type cyclins via PI3K/Akt and MAPK/Erk activation in human corneal epithelial cells. Mol. Vis. 2012, 18, 758–764. [Google Scholar]
- Thomas, M.; Langley, B.; Berry, C.; Sharma, M.; Kirk, S.; Bass, J.; Kambadur, R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 2000, 275, 40235–40243. [Google Scholar] [CrossRef]
- Oldham, J.M.; Osepchook, C.C.; Jeanplong, F.; Falconer, S.J.; Matthews, K.G.; Conaglen, J.V.; Gerrard, D.F.; Smith, H.K.; Wilkins, R.J.; Bass, J.J.; et al. The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J. Physiol. 2009, 587, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.H.; Wang, N.Q.; Yin, L.; Chen, X.; Hou, B.Y.; Qiang, G.F.; Chan, C.B.; Yang, X.Y.; Du, G.H. Effect of skeletal muscle phenotype and gender on fasting-induced myokine expression in mice. Biochem. Biophys. Res. Commun. 2019, 514, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.N.; Reddy, N.M.; Patil, K.R.; Nakhate, K.T.; Ojha, S.; Patil, C.R.; Agrawal, Y.O. Challenges and issues with streptozotocin-induced diabetes—A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Interact. 2016, 244, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Frey, W.H.; Liu, J.; Chen, X.; Thorne, R.G.; Fawcett, J.R.; Ala, T.A.; Rahman, Y.E. Delivery of 125I-NGF to the brain via the olfactory route. J. Alzheimer’s Dis. 1998, 1, 35–44. [Google Scholar]
- Dimauro, I.; Pearson, T.; Caporossi, D.; Jackson, M.J. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC. Res. Notes. 2012, 20, 513. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Seq (5′–3′) | RefSeq |
|---|---|---|
| Mstn | FW: GCCATGATCTTGCTGTAACCT RV: CAGTCAAGCCCAAAGTCTCT /56-FAM/TCAGCCCAT/ZEN/CTTCTCCTGGTCCT/3IABkFQ/ | NM_010834 |
| Actb | FW: GATTACTGCTCTGGCTCCTAG RV: GACTCATCGTACTCCTGCTTG /5SUN/CTGGCCTCA/ZEN/CTGTCCACCTTCC/3IABkFQ/ | NM_007393 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jun, L.; Ding, X.-W.; Robinson, M.; Jafari, H.; Knight, E.; Geetha, T.; Greene, M.W.; Babu, J.R. Targeting Molecular Mechanisms of Obesity- and Type 2 Diabetes Mellitus-Induced Skeletal Muscle Atrophy with Nerve Growth Factor. Int. J. Mol. Sci. 2024, 25, 4307. https://doi.org/10.3390/ijms25084307
Jun L, Ding X-W, Robinson M, Jafari H, Knight E, Geetha T, Greene MW, Babu JR. Targeting Molecular Mechanisms of Obesity- and Type 2 Diabetes Mellitus-Induced Skeletal Muscle Atrophy with Nerve Growth Factor. International Journal of Molecular Sciences. 2024; 25(8):4307. https://doi.org/10.3390/ijms25084307
Chicago/Turabian StyleJun, Lauren, Xiao-Wen Ding, Megan Robinson, Hassan Jafari, Emily Knight, Thangiah Geetha, Michael W. Greene, and Jeganathan Ramesh Babu. 2024. "Targeting Molecular Mechanisms of Obesity- and Type 2 Diabetes Mellitus-Induced Skeletal Muscle Atrophy with Nerve Growth Factor" International Journal of Molecular Sciences 25, no. 8: 4307. https://doi.org/10.3390/ijms25084307
APA StyleJun, L., Ding, X.-W., Robinson, M., Jafari, H., Knight, E., Geetha, T., Greene, M. W., & Babu, J. R. (2024). Targeting Molecular Mechanisms of Obesity- and Type 2 Diabetes Mellitus-Induced Skeletal Muscle Atrophy with Nerve Growth Factor. International Journal of Molecular Sciences, 25(8), 4307. https://doi.org/10.3390/ijms25084307