Abstract
DNA repair pathways play a critical role in genome stability, but in eukaryotic cells, they must operate to repair DNA lesions in the compact and tangled environment of chromatin. Previous studies have shown that the packaging of DNA into nucleosomes, which form the basic building block of chromatin, has a profound impact on DNA repair. In this review, we discuss the principles and mechanisms governing DNA repair in chromatin. We focus on the role of histone post-translational modifications (PTMs) in repair, as well as the molecular mechanisms by which histone mutants affect cellular sensitivity to DNA damage agents and repair activity in chromatin. Importantly, these mechanisms are thought to significantly impact somatic mutation rates in human cancers and potentially contribute to carcinogenesis and other human diseases. For example, a number of the histone mutants studied primarily in yeast have been identified as candidate oncohistone mutations in different cancers. This review highlights these connections and discusses the potential importance of DNA repair in chromatin to human health.
1. Introduction
Cellular DNA is continually disfigured by endogenous and exogenous DNA damaging agents, including reactive oxygen species (ROS), alkylating agents, ultraviolet (UV) radiation, and a myriad of other causes of DNA lesions [1]. Such damage must be efficiently recognized and repaired by cellular repair enzymes in order to prevent the accumulation of mutagenic and cytotoxic DNA lesions. The activities of these repair enzymes are typically coordinated in one or more single- or multi-step repair pathways. These pathways include (1) nucleotide excision repair (NER), which recognizes and excises bulky, helix-distorting DNA lesions [2,3,4,5]; (2) base excision repair (BER), which removes specific classes of small DNA base lesions [6,7,8]; (3) mismatch repair (MMR), which removes DNA mismatches typically arising during replication [9,10]; (4) direct damage reversal, such as the light-dependent repair of UV lesion by photolyase enzymes or repair of O6-methyl-guanine alkylation lesions by the O6-methylguanine DNA methyltransferase suicide enzymes [11,12]; (5) single-strand break repair, which repairs single-stranded DNA gaps [13]; (6) double-strand break (DSB) repair, consisting of homologous recombination (HR), nonhomologous end joining (NHEJ), and other alternative end joining pathways [14,15]; and (7) post replication repair (PRR), which includes translesion DNA synthesis (TLS) by specialized TLS polymerases opposite replication-stalling DNA lesions [16].
In eukaryotic cells, all of these repair pathways must function in the context of a DNA template packaged with histone proteins into nucleosomes [17,18]. Individual nucleosomes are comprised of ~147 bp of DNA wrapped nearly two times around an octamer of histone proteins [19,20,21]. This octamer typically contains two copies each of histones H2A, H2B, H3, and H4. Each of these histone proteins has the same basic core structure, consisting of three alpha helices and two loops, which comprise the histone-fold domain [22]. The individual histones differ substantially in their N-terminal and C-terminal extensions from the core histone-fold domain, which are largely unstructured, but in some cases contain additional secondary structure elements, including an additional C-terminal alpha helix (αC) in histone H2B and an N-terminal helix (αN) in histone H3 [21,22]. The central organizing unit of the histone octamer is a heterotetramer consisting of H3 and H4 proteins. To the central H3/H4 heterotetramer are bound two H2A/H2B heterodimers, each binding to opposite sides or ‘faces’ of the heterotetramer [22].
The nucleosomal DNA is sharply bent as it binds to this histone octamer, with the protein–DNA interactions primarily confined to 14 superhelical locations (SHL) where the minor groove of the DNA is oriented toward the histone octamer (i.e., SHL-6.5 to SHL6.5) [19,20,21]. These interactions primarily occur between the DNA backbone and structural features of the core histone-fold domains (i.e., L1/L2 loops or a1/a1 helices), with the exception of the most distal DNA exit/entry points (SHL ± 6.5), which interact with residues in the H3 αN helix [21]. If the central dyad axis of the nucleosomal DNA is oriented upward, then the H3–H4 heterotetramer generally binds DNA near the top half of the nucleosome, while the H2A/H2B dimers bind DNA near the bottom of the nucleosome (Figure 1A). The N-terminal tails of each histone exit either between the two DNA gyres (i.e., histones H2B and H3) or to the side of one DNA gyre (i.e., histones H2A and H4), extending a considerable length beyond the nucleosome core (Figure 1A). The C-terminal tail of H2A traverses the nucleosome face and exits near the dyad axis at the top of the nucleosome, while the C-terminal tail of H2B forms a unique feature on the nucleosomes surface (see below).
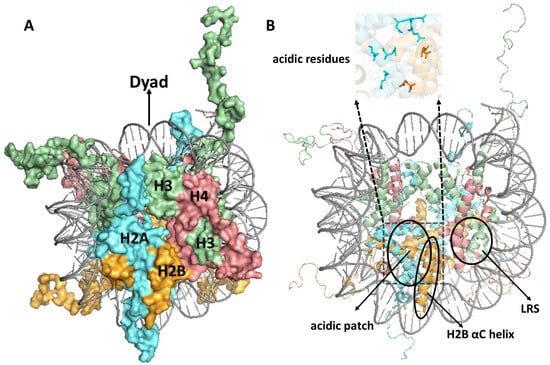
Figure 1.
(A) Structure of the nucleosome. The different histones are indicated as follows: H3, H4, H2A, and H2B are shown in pale green, salmon red, cyan aquamarine, and bright orange, respectively. The central dyad axis is indicated at the top of the nucleosome. (B) Nucleosome structure highlighting unique structural features, including the acidic patch, the histone H2B αC helix, and loss of ribosomal silencing (LRS) surface. Figures were generated using pymol from PDB ID:1KX5.
A nucleosome can be thought of as resembling the wheel of a car, with the DNA being the ‘tire’ that is bound to the lateral surface of the histone octamer, which can be thought of as the ‘rim’ of the wheel (Figure 1A). Using this analogy, the nucleosome ‘face’ or disk surface can be thought of as the side or face of the wheel, containing the spokes and hub. The nucleosome face contains a number of unique features that are often targeted by proteins that interact with nucleosomes. These include (1) the acidic patch, comprised of a number of aspartic acid, glutamic acid, and other H2A/H2B residues which form a deep groove on the nucleosomes surface (Figure 1B) that is targeted by many nucleosome-binding proteins [22,23,24]; (2) the H2B αC helix, which forms a ridge that runs from the center to the bottom of the nucleosome face (Figure 1B) and is ubiquitylated at H2B K123 in yeast or K120 in mammalian cells [22,25]; and (3) the loss of ribosomal silencing (LRS) surface (Figure 1B), consisting of H3/H4 residues that surround the methylated H3 lysine-79 (H3K79) residue [22,26]. Since most DNA in a eukaryotic genome is packaged in nucleosomes, proteins involved in DNA transcription, replication, and repair are thought to exploit these different nucleosome features in order to specifically bind to nucleosomes (as opposed to free histones) and promote these critical DNA metabolic processes in nucleosomal DNA.
2. Impact of Nucleosomes on Genome-Wide DNA Damage, Repair, and Mutagenesis
The wrapping of DNA around the histone octamer to form a nucleosome has profound consequences to DNA damage, repair, and mutagenesis, particularly in tumor genomes. In many cancers, somatic mutations are elevated in nucleosomal DNA relative to flanking linker DNA or nucleosome-free regions [27,28,29]. This pattern can be potentially explained by both biochemical studies and genome-wide repair experiments, indicating that the repair of many DNA lesions is inhibited inside nucleosomes [17,18,27,28,29,30,31,32,33,34,35,36,37,38]. While many cancers showed elevated mutation frequencies in nucleosomes, a few, namely, lung squamous cell carcinoma and lung adenocarcinoma, show the opposite trend with elevated mutations in linker DNA [28]. However, the molecular mechanism responsible for elevated mutation rates in nucleosome-free linker DNA in lung cancers remains an open question.
Recent studies indicate that mutation patterns in cancers are also modulated by the positioning of the minor groove of the DNA helix relative to the histone octamer [27,28,29]. Again, many cancers showed elevated mutation frequencies at positions in the nucleosomal DNA where the minor groove faces the histone octamer (i.e., ‘minor-in’ rotational settings). This can be potentially explained by biochemical and genome-wide studies indicating that BER is particularly inhibited at minor-in positions [17,18,28,32,34,39,40]. In contrast, in certain cancers (e.g., lung cancers and melanoma), somatic mutations are elevated at minor-out rotational settings [28,29], where the minor groove of the nucleosomal DNA faces away from the histone octamer. In the case of skin cancers, such as melanoma, elevated mutation rates at minor-out rotational settings can largely be attributed to elevated levels of UV damage formation at these positions in nucleosomal DNA [28,29,33,41,42]. We and others have found that this nucleosome ‘photofootprint’ is likely caused by DNA bending into the major groove at minor-out positions in nucleosomes [43,44], resulting in a DNA structure more susceptible to forming UV photoproducts such as cyclobutane pyrimidine dimers (CPDs).
While many of the studies cited above concern inhibition of DNA excision repair pathways (i.e., NER and BER) by nucleosomes, the packaging of DNA into nucleosomes also impacts other repair pathways. For example, biochemical studies indicate that nucleosomes inhibit repair by the MMR pathway [45], consistent with bioinformatic analysis indicating that inhibition of MMR in nucleosomes may contribute to elevated somatic mutation rates, particularly at minor-in rotational settings, in esophageal cancers [46]. As another example, DNA resection during DSB repair by the HR pathway almost certainly requires nucleosome repositioning and/or eviction [14,47].
Finally, while the packaging of DNA into nucleosomes can inhibit many repair processes, nucleosomes also serve as an essential platform to promote DNA damage signaling (see below). Moreover, many cellular enzymes, including ATP-dependent nucleosome remodeling enzymes and histone modifying enzymes (e.g., histone acetyltransferases, methyltransferases, ubiquitin ligases, etc.) can promote efficient repair of DNA lesions in nucleosomes, as detailed below.
3. Roles of Histone Post-Translational Modifications in Repair
Histone residues are decorated with a variety of post-translational modifications (PTMs) that act in a coordinated and ordered manner to facilitate dynamic changes in chromatin for the tight regulation of cellular processes such as DNA replication, transcription, damage response, and repair [48,49,50]. In recent years, there has been remarkable progress in identification of histone modifications, characterizing their genome-wide distribution and understanding their function in regulating transcription, replication, and repair.
The first key theme that has emerged from these studies Is that many of these histone PTMs have dual functions in distinct cellular processes, such as transcription and repair. For example, acetylation of lysine residues located in the flexible histone N-terminal tails by lysine (K) acetyltransferases (KATs) play a critical role in regulating transcription [48,51,52]. In yeast, Gcn5 and Esa1, which serve as the catalytic subunits of the SAGA [53,54] and NuA4 [55] KAT complexes, respectively, regulate the transcription of many yeast genes by acetylating N-terminal lysine residues in histones H2B and H3 (Gcn5) and H2A and H4 (Esa1) [56]. Histone acetylation by these same KATs also promotes efficient NER of UV damage in yeast [57,58,59,60,61] and, in the case of Gcn5, in human cells [62,63]. Histone acetylation is thought to promote transcription in part by recruiting ATP-dependent chromatin remodeling (ACR) complexes, such as SWI/SNF and RSC in yeast [64,65]. This recruitment is mediated by bromodomains in the subunits of these ACR complexes, as bromodomains specifically bind acetylated lysine residues (e.g., [64,65,66,67]). These same ACR complexes are also required for efficient NER in yeast [68,69,70]; to what extent their recruitment and activity during repair is mediated by histone acetylation remains unclear.
Lysine residues in the structured histone core domain are also acetylated, a primary example being H3 lysine-56 (K56), which is located in the αN helix of histone H3 (Figure 2). H3 K56 is acetylated during the process of histone deposition during replication, and therefore plays overlapping roles in nucleosome assembly, DNA damage response, and checkpoint recovery [71,72,73,74,75,76]. An in vitro study indicates that H3 K56 acetylation may also limit some forms of DNA replication, in this case by DNA polymerase beta, which performs error-prone DNA repair synthesis during BER [77,78]. H3 K56 acetylation has also been reported to promote histone and tRNA gene transcription [79,80,81], as well as NER [82], indicating that this PTM, which directly modulates nucleosomal DNA unwrapping [83,84,85], also multitasks in promoting different cellular functions in chromatin.
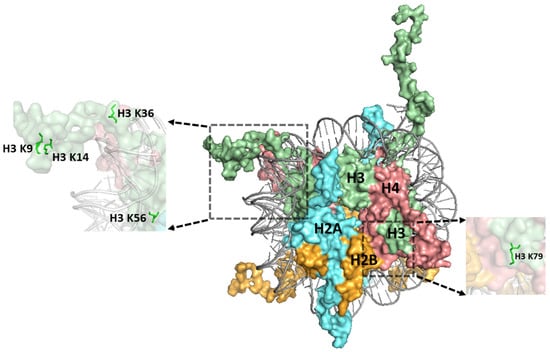
Figure 2.
Structure of the nucleosome highlighting a number of histone residues whose post-translational modifications (PTMs) are implicated in repair. Histone H3 K9, H3 K36, and H3 K79 are methylated and histone residues H3 K9, H3 K14, and H3 K56 are acetylated. Histones are indicated as follows: H3, H4, H2A, and H2B are depicted in pale green, salmon red, cyan aquamarine, and bright orange, respectively. The figure was generated using pymol from PDB ID:1KX5.
Histone lysine residues are also methylated by lysine methyltransferases (KMTs) [48,86], which play a clear role in establishing and maintaining different chromatin states in the genome. A second theme from recent studies is that such pre-existing chromatin states profoundly impact the repair of many different classes of DNA lesions. For example, H3 K9 methylation (Figure 2) is important for the establishment and maintenance of heterochromatic regions [86], and such regions of the genome have elevated somatic mutation rates in many cancers, including skin cancer [87]. Elevated somatic mutation rates can be explained by other studies indicating that such heterochromatic regions are refractory to repair [27,35,88]. H3 K9 methylation also plays critical roles in silencing repeat sequences, including different classes of mobile DNA elements, and regulating tissue-specific gene expression during cellular differentiation [86]. Since genomic regions marked by H3 K9 methylation are typically silenced, and are therefore dispensable for cellular function and homeostasis, repair inhibition in such regions might redirect the limiting cellular repair machinery to instead prioritize fixing damage in more important (and accessible) regions of the genome.
In contrast, trimethylated H3 K36 (H3 K36me3) is enriched at transcribed exon sequences [89,90,91]. Like H3 K9 methylation, H3 K36 methylation (Figure 2) multitasks to perform a number of important cellular functions, including regulating RNA splicing [92,93,94,95] and repressing the initiation of cryptic transcription [92,96,97,98,99]. H3 K36 methylation also plays an important role in regulating DNA repair. For example, a key MMR protein known as MSH6 contains a PWWP domain that specifically binds H3 K36me3, thereby targeting the MMR machinery to preferentially fix replication errors in transcribed exons [100,101,102,103]. Hence, the MMR machinery prioritizes the repair of critical protein-coding exons by recognizing and exploiting pre-existing methylation marks associated with this chromatin state.
Pre-existing histone PTMs have been reported to affect repair even at the level of individual nucleosomes. A genome-wide study of repair of DNA alkylation damage induced by methyl methanesulfonate (MMS) exposure in yeast revealed that pre-existing histone PTMs modulate BER in nucleosomes [32]. This study found that nucleosomes marked by high levels of pre-existing histone acetylation (e.g., H3 K14 acetylation, H3 K23 acetylation, etc.) display more rapid BER of DNA alkylation damage at more distal locations in nucleosomes (i.e., near DNA entry/exit sites in nucleosomes) but paradoxically slower repair of damage near the central nucleosome dyad. Analysis of MMS-induced mutations in yeast revealed a similar pattern, with elevated mutation density in H3 K14 acetylated nucleosomes near the slower-repairing nucleosome dyad [32]. In contrast, nucleosomes marked with H3 K36 methylation show the opposite pattern of BER in nucleosomes, and MMS-induced mutations show a similar trend [32].
A third key theme is that DNA dam”ge a’d its subsequent repair often changes the epigenetic landscape of histone PTMs. Perhaps the best studied example is the phosphorylation of serine-139 residue in the C-terminal tail of the histone variant H2AX adjacent to DNA DSBs, a histone PTM called γH2AX [104,105,106]. Phosphorylation of H2AX by DNA damage signaling kinases such as ATM in human cells, which can extend to nucleosomes as far as a megabase from the DSB, plays an important role in repair [105,107].
Histone ubiquitination, which consists of the covalent attachment of a ubiquitin protein to a target lysine residue by the concerted action of E2 ubiquitin conjugating enzyme and E3 ubiquitin ligase, also plays a critical role in DSB signaling and repair [108,109,110]. At DSBs, the human E3 ubiquitin ligases RNF20/40 mono-ubiquitinate H2B at lysine-120 [111,112,113], while RNF168 E3 ligase catalyzes ubiquitination of H2A (or H2AX) lysine-13 and lysine-15 (K13/K15) [114,115,116,117], in order to promote DNA damage signaling and DSB repair. Similar histone PTM alterations occur at DSBs in yeast, although the canonical yeast histone H2A (which resembles human H2AX) is phosphorylated at serine-129 (S129) and the yeast Bre1 E3 ubiquitin ligase, the homolog of human RNF20/RNF40 dimer complex, ubiquitinates yeast H2B K123 to promote DSB signaling and repair [118,119].
Histone ubiquitination is also altered during NER. The CUL4-DDB-ROC1 E3 ubiquitin ligase complex acts to ubiquitinate lysine residues in histones H3 and H4 in response to UV damage, thereby facilitating the recruitment of a key NER damage sensor known as XPC [120]. On the contrary, H2B K123 is deubiquitinated in yeast in response to RNA polymerase II stalling at UV lesions [121]. Notably, damage-dependent H2B deubiquitination, which promotes efficient NER, is catalyzed in part by the ubiquitin-specific protease Ubp8, a subunit of the Gcn5-containing SAGA complex. Gcn5 has also been reported to stimulate histone H3 acetylation in response to UV damage [60], which promotes efficient NER in chromatin [58,59,122,123]. Hence, two different enzymatic activities in the SAGA complex are thought to alter the histone PTM landscape in order to facilitate repair of UV damage. Thus, chromatin alterations associated with the initial steps of NER (e.g., damage recognition, etc.) can alter the landscape of histone PTMs. Furthermore, the later NER step of re-synthesizing the excised DNA strand can also alter histone PTMs [124,125].
Finally, single-stranded DNA breaks and other DNA lesions trigger the recruitment and activation of poly(ADP-ribose) polymerase 1 (PARP1), which catalyzes extensive poly(ADP)ribosylation (PARylation) of PARP1 itself or other target proteins, including histones [48,126]. PARylation promotes the recruitment of XRCC1 and its associated repair proteins, such as LIG3 (DNA Ligase 3), to facilitate single-strand break repair [127,128,129,130,131]. While PARP1 plays a critical role in single-strand break repair and BER, PARP1 also facilitates chromatin remodeling during NER, via the ACR ALC1, through its interactions with the NER damage sensor XPC [132]. Notably, PARP1 activity can significantly deplete cellular stores of key metabolic cofactor NAD+, leading to significant perturbations in cellular metabolism, as well as pathology in individuals with genetic defects in these repair pathways [133,134].
4. Histone Mutants That Affect DNA Repair
Mutants in histone proteins in yeast have been extensively utilized to characterize the role of specific histone domains, residues, and PTMs in different cellular functions, including transcription, replication, chromatin assembly, and DNA repair [49,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152]. These efforts culminated in a trio of reports generating and characterizing libraries of histone mutants in yeast [153,154,155], in which essentially every histone residue was individually mutated (typically to alanine) and the resulting mutant phenotype was characterized. These studies yielded a number of surprising findings. First, relatively few of the histone alanine mutants were lethal in yeast grown in rich media. For example, in histones H2A and H2B, only four residues were essential for yeast viability in rich media, and all of these (yeast H2A Y58, E62, D91, and H2B L109) were located in the acidic patch [153,155] (Figure 1B). Second, many histone mutants showed sensitivity to DNA damaging agents, including the alkylating agent MMS and UV light [153,154,156,157]. Elucidating how these histone mutants perturb DNA damage tolerance, repair, and/or signaling remains an ongoing challenge, although common mechanisms have emerged from recent studies.
First, some histone mutants cause DNA damage sensitivity and repair defects by down-regulating the expression of one or more key DNA repair genes. For example, histone H4 K16R and K16Q mutations rendered yeast cells sensitive to UV damage and cause defects in NER [158]. These mutants were further shown to compromise UV-induced expression of a number of key NER genes (i.e., RAD23, RAD4, RAD16, RAD1, RAD2, and RAD14), which, along with changes in chromatin accessibility, can potentially explain the observed NER defects [158].
Similarly, simultaneous deletion of both the histone H2A and H3 N-terminal tails caused MMS sensitivity and a defect in BER of MMS-induced lesions in yeast [135]. This BER-defect turned out to be caused by reduced expression of the MAG1 gene, which encodes a DNA glycosylase that plays a critical role in initiating BER of DNA alkylation damage. Overexpression of Mag1 in yeast cells lacking the H2A and H3 N-terminal tails rescued the BER defect, but did not restore MMS resistance [135]. Subsequent experiments indicated that the MMS sensitivity of the H2A and H3 tail mutants was epistatic with a mutation in the RAD18 gene, which encodes an E3 ubiquitin ligase that functions in PRR by ubiquitinating proliferating cell nuclear antigen (PCNA). PCNA ubiquitination was compromised in the H2A and H3 tail mutants, suggesting a mechanism by which the histone H2A and H3 N-terminal tails function in PRR of MMS-induced DNA lesions [135].
A second mechanism is that some histone mutants cause DNA damage sensitivity because they are sites of PTMs critical for DNA repair, DNA damage signaling, etc. For instance, the histone H3 K79E mutation, which mutates a lysine in the LRS domain of the nucleosome face (Figure 2 and Figure 3) that is methylated by the Dot1 KMT [159], renders yeast cells sensitive to UV light [160]. Deletion of the yeast DOT1 gene also causes a similar level of UV sensitivity, and epistasis analysis indicates that Dot1 and H3 K79 function in NER and post-replication repair [160]. Subsequent studies [139,161] indicate that Dot1 and H3 K79 function to promote repair by an NER sub-pathway known as global genomic-NER (GG-NER), which directly senses and repairs helix-distorting DNA lesions throughout the genome [4]. However, the mechanism by which H3 K79 methylation by Dot1 promotes GG-NER in yeast remains unclear. DOT1L catalyzed H3 K79 methylation in mammalian cells has been shown to recruit the key GG-NER sensor XPC to UV damage sites to promote efficient NER [162]. Consequently, defects in H3 K79 methylation are associated with increased frequency of UV-induced melanoma development in mice [162], highlighting the potential importance of this pathway in preventing skin carcinogenesis.
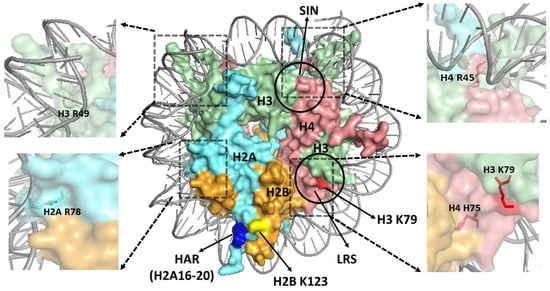
Figure 3.
Structure of a yeast nucleosome, highlighting key regions in the nucleosome surface. The loss of ribosomal silencing (LRS) and Sin− (SIN) domains are indicated with circles. The histone residues H3 K79, H2B K123, and the histone H2A repression (HAR) domain (i.e., H2A residues 16–20) are indicated in red, yellow, and blue color, respectively. The positions of the sprocket arginine residues H2A R78, H3 R49, and H4 R45 are indicated in zoomed images. The location of H3K79 residue (red) in the central region of LRS domain and the neighboring H4 H75 residue (salmon red) is also depicted in a zoomed image. The figure was generated using pymol from PDB ID:1ID3.
While histone H3 K79 regulates GG-NER, the histone H3 K36A mutant in yeast affects a different NER sub-pathway known as transcription coupled-NER (TC-NER) [163], which specifically repairs DNA lesions on the transcribed strand of genes. TC-NER is triggered by RNA polymerase II stalling at a helix-distorting DNA lesion, which is detected by a key TC-NER factor known as CSB in human cells and Rad26 in yeast [2,5,164,165,166]. H3 K36A mutants in yeast show UV sensitivity that is epistatic with mutations in RAD26, indicating that this histone residue functions in TC-NER [163]. H3 K36 is methylated in yeast by the Set2 KMT [167], and deletion of the SET2 gene in yeast also causes UV sensitivity that is epistatic with a RAD26 mutant, and a genome-wide defect in TC-NER of UV damage [163]. Set2 associates with the elongating RNA polymerase II and methylates H3 K36 during transcription elongation [94,168]. We have postulated that RNA polymerase II stalling at a lesion may result in the accumulation of H3 K36 methylation behind the stalled polymerase, potentially serving as an epigenetic signal for TC-NER [163]. Consistent with this model, a previous study suggested that H3 K36 methylation may promote Rad26 recruitment [169].
While mutations in H3 K36 cause UV sensitivity in a wild-type strain background, the H3 K36A mutant paradoxically rescues the UV sensitivity of a GG-NER deficient (rad16∆) yeast strain [163]. Set2-catalyzed histone H3 K36 methylation functions as a signal to recruit lysine deacetylases (KDAC) enzymes, which remove acetylation marks from nucleosomes in the transcribed regions of yeast genes [97,98,99]. This pathway plays a critical role in suppressing cryptic transcription, which initiates from inside of genes and proceeds in either the sense or antisense direction [97,99,170]. Mutants in SET2 or H3 K36 activate cryptic transcription, resulting in antisense transcription of the non-transcribed strand (NTS) of yeast genes. Normally, the NTS is only repaired by GG-NER, but in set2∆ or H3 K36A mutants, it can also be repaired by TC-NER associated with cryptic anti-sense transcription of the NTS [163]. Hence, the partial rescue of UV sensitivity in GG-NER deficient cells is likely due to increased repair resulting from cryptic TC-NER of the NTS of these genes. As Set2 homologs in human cells (i.e., NSD1, NSD2, NSD3 and SETD2) are frequently mutated in human cancers [94,171], these findings may have important implications in carcinogenesis and chemotherapy resistance.
Histone H3 K36 mutations have also been identified in a subset of human cancers and are thought to contribute to carcinogenesis [172,173,174,175,176,177]. One of the most studied of these ‘oncohistone’ mutations is a K36M mutation that occurs in the histone H3 variant H3.3. H3 K36M mutations occur in nearly 90% of cases of a bone cancer called chondroblastoma [178,179] but have also been identified in pediatric soft tissue sarcoma, head and neck squamous cell carcinoma, melanoma, bladder, and colorectal cancer [174,180,181,182]. The H3 K36M is a dominant mutation that binds to and inhibits the activity of SETD2 and other H3 K36 methylating enzymes in human cells [178].
Oncohistone mutations in a neighboring H3 G34 residue have also been identified in human cancers and disrupt H3 K36 methylation. Two of the most common G34 mutants, namely, G34R and G34V, are observed in H3.3 [174] in cerebral cortex tumors [172]. Other variants such as G34W and G34L are found in giant cell tumors of bone [179]. The G34R/V/W variants decrease H3K36me2 and H3K36me3 methylation in cis (i.e., on the same histone H3), but not in trans like the K36M mutation [183,184]. The H3.3 G34R/V/D oncohistone mutants also block interaction with the key mismatch repair protein MSH6 and therefore display a mutator phenotype [184].
While the modification states of the histone residues discussed above (e.g., H3 K36 and H3 K79 methylation) have been extensively studied, proteomics studies have also detected PTMs in other histone residues, which, although obscure, appear to contribute to DNA damage repair or signaling. For example, histone H2B lysine-46 (K46) in mammalian cells was previously reported to be methylated and acetylated [136,185]. Mutation of the homologous H2B residue in yeast to alanine (i.e., H2B K49A) resulted in a UV sensitivity phenotype [136]. Similarly, H2B lysine-108 (K108) in mammalian cells has been reported to be methylated [185], and a mutation in the homologous lysine residue in yeast H2B (H2B K111, see Figure 4), which has also been reported to be methylated [186], results in MMS sensitivity [136]. Notably, subsequent proteomics studies have indicated that H2B K108 methylation is down-regulated in postmortem brain samples from Alzheimer’s disease patients [187]. However, not all histone PTM sites function in the DNA damage response and affect genome stability. For example, H2B lysine-37 (K37) was discovered to be dimethylated in yeast and potentially higher eukaryotes, but mutating this residue did not render yeast cells sensitive to DNA damaging agents [188].
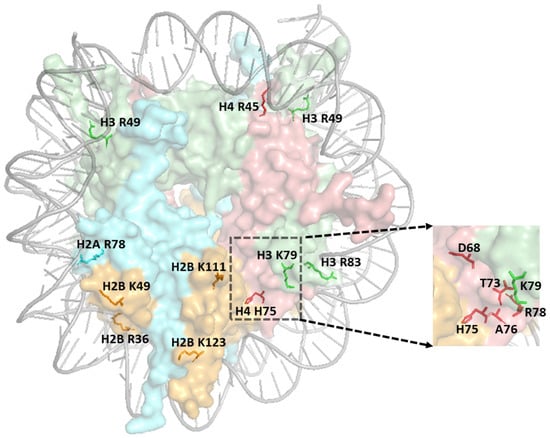
Figure 4.
Structure of nucleosome highlighting histone residues (labeled with histone and residue number) implicated in DNA repair. The zoomed image indicates the residues within the LRS domain. The figure was generated using pymol from PDB ID:1ID3.
A third mechanism is that some histone mutants cause DNA damage sensitivity by altering the modification state of other histone residues. For instance, a previous study indicated that a small domain (residues 16–20) comprising a ‘knuckle’ helix in the histone H2A N-terminal domain represses the expression of many yeast genes, and it was therefore labeled the histone H2A repression (HAR) domain [141,189]. Deletion of the HAR domain in yeast rendered cells UV-sensitive, although the mechanism involved was initially unclear [141]. A subsequent study indicated that the HAR domain is required for histone H2B K123 ubiquitination and H3 K4 methylation, likely because it is adjacent to H2B K123 in the nucleosome structure (Figure 3) and functions as a docking site for H2B ubiquitinating enzymes [190]. Since H2B K123 ubiquitination plays an important role in DNA repair [121,161], and H2B K123 mutants are sensitive to UV irradiation [191], the role of the HAR domain in promoting histone H2B ubiquitination can potentially explain the UV sensitivity of the HAR mutant. Surprisingly, mutations in HAR domain residues, including in residues R17 and S19 of human H2A, are observed in human cancers [174], although their potential role in carcinogenesis is unclear.
A fourth (and final) mechanism by which histone mutants can cause damage sensitivity is by affecting the structure and dynamics of the nucleosome or its binding interfaces with other proteins. One of the key structural features of the nucleosome is that a histone arginine residue inserts into the minor groove of the DNA at each minor-in rotational setting of the nucleosomal DNA, which can be compared to the teeth of a bicycle sprocket (i.e., arginine residues) inserted into the chain (DNA). Many of these ‘sprocket’ arginines have important roles in the structure and dynamics of the nucleosome and also affect DNA damage and repair [192]. For example, H4 R45 is a sprocket arginine that inserts into the DNA minor groove at SHL ± 0.5 (Figure 3), which flank the central dyad (SHL0) [21]. H4 R45C and R45H mutations were originally identified in a yeast genetic screen because they alleviate the requirement for the SWI/SNF ACR complex in transcriptional activation, likely because these mutants increase intrinsic nucleosomal DNA disassociation, mobility, and sliding [193,194], and are therefore called SWI/SNF-independent (Sin−) mutations [26,195]. A subsequent study indicated that histone H4 R45H or R45C mutations promote UV resistance in yeast and facilitate repair of UV damage by NER, likely by enhancing the accessibility of DNA lesions to repair proteins [196,197]. H4 R45C or R45Q mutants are also observed in human cancers and may function as oncohistone mutations [174]. Notably, other Sin− mutations identified in yeast are also prevalent in human cancers, including histone H3 E105K/Q, and have been hypothesized to potentially promote carcinogenesis [174].
Other sprocket arginines also play roles in DNA damage sensitivity and repair, including H2A R78 (SHL ± 5.5) and H3 R49 (SHL ± 6.5, see Figure 3 and Figure 4). Unlike H4 R45C, which promotes UV resistance and repair, alanine mutations in either of these residues result in elevated UV sensitivity in yeast and a defect in NER of UV-induced CPD lesions [192]. Both the H2A R78A and H3 R49A mutants are also MMS sensitive, as are mutants in other histone sprocket arginine residues (i.e., H3 R83A and H2A R43A) [153,154,156,192,198]. However, none of these mutants affect BER of MMS-induced DNA alkylation damage [192], indicating that they likely affect a different repair or damage tolerance pathway.
The mechanism by which the H2A R78 and H3 R49 residues promote NER in chromatin remains unclear. The H2A R78A mutant does not affect expression of any known NER gene, indicating that it may directly affect NER activity [192]. Structural studies and biochemical experiments suggest that H2A R78 may play an important role in interacting with the H2A/H2B chaperones FACT and Nap1 [199,200,201]. It has also been reported that the H2A R77A mutation in human H2A (homologous to H2A R78 in yeast) affects the nucleosome sliding activity of an ACR in vitro [202]. These findings suggest possible mechanisms by which H2A R78 regulates NER in yeast, since both FACT and ACR complexes have been linked to promoting NER activity in chromatin [68,69,132,203,204,205,206,207,208].
One of the sprocket arginine residues that lacked any observable phenotype when mutated was H2B R36 (SHL ± 4.5, see Figure 4) [192]. This residue is located in a highly basic region of histone H2B called the histone H2B repression (HBR) domain, because it plays an important role in transcriptional repression in yeast [209]. While the H2B R36A mutant lacks any observable phenotype, deletion of the entire HBR domain (H2B ∆30–37) causes UV sensitivity in yeast, and previous studies indicate that the HBR mutant affects UV damage formation, as well as repair at specific loci [137,209]. The HBR domain is located between the two DNA gyres as the H2B tail exits the nucleosome core [25]. Biophysical studies indicate that the HBR domain plays an important role in nucleosomal DNA unwrapping and sliding and modulates the activity of BER enzymes in vitro [210]. The HBR domain also plays an important role in FACT-mediated nucleosome disassembly and assembly [211,212], which may potentially explain its role in NER.
While the discussion above focuses on sprocket arginine residues, many other histone residues also affect nucleosome stability, dynamics, and binding interfaces and potentially play a role in DNA damage sensitivity and repair. For example, a recent study used saturation mutagenesis targeted to a small region of histone H4 associated with the LRS domain to identify histone mutants that affected UV sensitivity in yeast. This study identified 24 mutations that either enhanced or reduced UV sensitivity, one of which (histone H4 H75E) significantly decreased GG-NER activity [213]. Although the H4 H75E mutant did not affect the methylation of neighboring H3 K79 (Figure 3), it impaired chromatin binding of the key DNA damage sensor Rad4. Other H4 mutants identified in the screen (e.g., H4 T73D, T73F, T73Y, R78I, D68I, A76T, R78S, and T80L, see Figure 4) affected PRR by TLS DNA polymerases, resulting in reduced UV-induced mutagenesis [214]. This study not only identified new histone mutants that affect repair and the DNA damage response, but also suggests that current histone mutant libraries, which primarily consist of substitutions to alanine, may be missing non-alanine substitutions that have important biological phenotypes. Consistent with this idea, most of the candidate oncohistone mutations identified in human cancers are not alanine substitutions [174,202]
Surprisingly, histone H4 H75 was recently discovered to be mutated (H4 H75R) in a neurological and developmental syndrome, suggesting that it plays important functions in human development [215]. The same study also found that other patients with germline H4 R45C Sin− mutations displayed similar neurological and developmental symptoms [215], although the underlying molecular mechanism responsible for these phenotypes remains unclear.
5. Conclusions
In summary, it is clear that the packaging of DNA into nucleosomes profoundly affects both DNA damage formation and repair, and consequently contributes to altered somatic mutation rates in various cancers. Nucleosomes not only act to restrict access to the repair machinery, but can also serve as a key signaling platform in which both pre-existing and repair-associated histone PTMs can facilitate and prioritize repair of DNA lesions. The availability of libraries of histone mutants in yeast has revealed a number of key residues that regulate DNA repair pathways. It is remarkable that a number of these histone mutants characterized by genetic studies in yeast have recently been identified in human cancers and neurological disorders. It will be important in future studies to elucidate the mechanisms by which these and other histone mutants and PTMs potentially contribute to carcinogenesis and other human diseases.
Author Contributions
K.S., J.J.W. and M.A.P. wrote the paper. K.S. generated the figures. All authors have read and agreed to the published version of the manuscript.
Funding
Research in the Wyrick lab is supported by NIEHS grant number R01ES028698, R01ES032814, and R21ES035139. APC was funded by R01ES028698.
Acknowledgments
We are grateful to Peng Mao, Benjamin Morledge-Hampton, and Allysa Sewell for their helpful comments and suggestions. We apologize to authors whose work we did not cite due to the extensive literature in this subject area.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006; p. xxvii. 1118p. [Google Scholar]
- Geijer, M.E.; Marteijn, J.A. What happens at the lesion does not stay at the lesion: Transcription-coupled nucleotide excision repair and the effects of DNA damage on transcription in cis and trans. DNA Repair 2018, 71, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Lindsey-Boltz, L.A.; Li, W.T.; Sancar, A. Molecular Mechanisms of Transcription-Coupled Repair. Annu. Rev. Biochem. 2023, 92, 115–144. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.R. DNA Fragility and Repair: Some Personal Recollections. Annu. Rev. Biochem. 2023, 92, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. Structure and Function of Photolyase and Enzymology: 50th Anniversary. J. Biol. Chem. 2008, 283, 32153–32157. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q. The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer. Cancers 2024, 16, 331. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. DNA single-strand break repair and human genetic disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E. Genome Stability: DNA Repair and Recombination; GS/Garland Science; Taylor & Francis Group: New York, NY, USA, 2014; p. xvi. 399p. [Google Scholar]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Mutter-Rottmayer, E.; Zlatanou, A.; Vaziri, C.; Yang, Y. Mechanisms of Post-Replication DNA Repair. Genes 2017, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Smerdon, M.J.; Wyrick, J.J.; Delaney, S. A half century of exploring DNA excision repair in chromatin. J. Biol. Chem. 2023, 299, 105118. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Hinz, J.M.; Smerdon, M.J. Accessing DNA damage in chromatin: Preparing the chromatin landscape for base excision repair. DNA Repair 2015, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef]
- Makde, R.D.; England, J.R.; Yennawar, H.P.; Tan, S. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 2010, 467, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 2006, 311, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Wyrick, J.J.; Kyriss, M.N.; Davis, W.B. Ascending the nucleosome face: Recognition and function of structured domains in the histone H2A-H2B dimer. Biochim. Biophys. Acta 2012, 1819, 892–901. [Google Scholar] [CrossRef]
- Fry, C.J.; Norris, A.; Cosgrove, M.; Boeke, J.D.; Peterson, C.L. The LRS and SIN domains: Two structurally equivalent but functionally distinct nucleosomal surfaces required for transcriptional silencing. Mol. Cell. Biol. 2006, 26, 9045–9059. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Sabarinathan, R.; Lopez-Bigas, N. Local Determinants of the Mutational Landscape of the Human Genome. Cell 2019, 177, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Muinos, F.; Sabarinathan, R.; Reyes-Salazar, I.; Gonzalez-Perez, A.; Lopez-Bigas, N. Somatic and Germline Mutation Periodicity Follow the Orientation of the DNA Minor Groove around Nucleosomes. Cell 2018, 175, 1074–1087.e18. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Mao, P.; Smerdon, M.J.; Wyrick, J.J.; Roberts, S.A. Nucleosome positions establish an extended mutation signature in melanoma. PLoS Genet. 2018, 14, e1007823. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Sivapragasam, S.; Antony, J.S.; Ulibarri, J.; Hinz, J.M.; Poon, G.M.K.; Wyrick, J.J.; Mao, P. High-resolution mapping demonstrates inhibition of DNA excision repair by transcription factors. eLife 2022, 11, e73943. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. Asymmetric repair of UV damage in nucleosomes imposes a DNA strand polarity on somatic mutations in skin cancer. Genome Res. 2020, 30, 12–21. [Google Scholar] [CrossRef]
- Mao, P.; Brown, A.J.; Malc, E.P.; Mieczkowski, P.A.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome Res. 2017, 27, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 9057–9062. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Smerdon, M.J. The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem. 2013, 288, 13863–13875. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Selby, C.P.; Adar, S.; Adebali, O.; Sancar, A. Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J. Biol. Chem. 2017, 292, 15588–15597. [Google Scholar] [CrossRef]
- Hara, R.; Mo, J.; Sancar, A. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 2000, 20, 9173–9181. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, P.J.; Delaney, S. Chromatin and other obstacles to base excision repair: Potential roles in carcinogenesis. Mutagenesis 2020, 35, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Balliano, A.J.; Hayes, J.J. Base excision repair in chromatin: Insights from reconstituted systems. DNA Repair 2015, 36, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.E.; Caffrey, P.J.; Delaney, S. Initiating base excision repair in chromatin. DNA Repair 2018, 71, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hinz, J.M.; Rodriguez, Y.; Smerdon, M.J. Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. USA 2010, 107, 4646–4651. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.M.; Nissen, K.A.; Smerdon, M.J. UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Proc. Natl. Acad. Sci. USA 1987, 84, 6644–6648. [Google Scholar] [CrossRef]
- Bohm, K.A.; Morledge-Hampton, B.; Stevison, S.; Mao, P.; Roberts, S.A.; Wyrick, J.J. Genome-wide maps of rare and atypical UV photoproducts reveal distinct patterns of damage formation and mutagenesis in yeast chromatin. Proc. Natl. Acad. Sci. USA 2023, 120, e2216907120. [Google Scholar] [CrossRef] [PubMed]
- Stark, B.; Poon, G.M.K.; Wyrick, J.J. Molecular mechanism of UV damage modulation in nucleosomes. Comput. Struct. Biotechnol. J. 2022, 20, 5393–5400. [Google Scholar] [CrossRef] [PubMed]
- Nayis, A.; Liebl, K.; Zacharias, M. Coupling of conformation and CPD damage in nucleosomal DNA. Biophys. Chem. 2023, 300, 107050. [Google Scholar] [CrossRef]
- Li, F.; Tian, L.; Gu, L.Y.; Li, G.M. Evidence That Nucleosomes Inhibit Mismatch Repair in Eukaryotic Cells. J. Biol. Chem. 2009, 284, 33056–33061. [Google Scholar] [CrossRef] [PubMed]
- Morledge-Hampton, B.; Wyrick, J.J. Mutperiod: Analysis of periodic mutation rates in nucleosomes. Comput. Struct. Biotechnol. J. 2021, 19, 4177–4183. [Google Scholar] [CrossRef]
- Gnugge, R.; Reginato, G.; Cejka, P.; Symington, L.S. Sequence and chromatin features guide DNA double-strand break resection initiation. Mol. Cell 2023, 83, 1237–1250.e15. [Google Scholar] [CrossRef] [PubMed]
- Workman, J.L.; Abmayr, S.M. Fundamentals of Chromatin; Springer: New York, NY, USA, 2014; p. xiii. 587p. [Google Scholar]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T.; Reinberg, D. Epigenetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2007; p. 10. 502p. [Google Scholar]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Koutelou, E.; Dent, S.Y.R. Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell 2022, 82, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.A.; Schieltz, D.; Pray-Grant, M.G.; Steger, D.J.; Reese, J.C.; Yates, J.R., 3rd; Workman, J.L. A subset of TAF(II)s are integral components of the SAGA complex required for nucleosome acetylation and transcriptional stimulation. Cell 1998, 94, 45–53. [Google Scholar] [CrossRef]
- Grant, P.A.; Duggan, L.; Côté, J.; Roberts, S.M.; Brownell, J.E.; Candau, R.; Ohba, R.; Owen-Hughes, T.; Allis, C.D.; Winston, F.; et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 1997, 11, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.; Utley, R.T.; Savard, J.; Clarke, A.; Grant, P.; Brandl, C.J.; Pillus, L.; Workman, J.L.; Cote, J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999, 18, 5108–5119. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Yu, Y.; Ferreiro, J.A.; Waters, R. Histone acetylation, chromatin remodelling, transcription and nucleotide excision repair in S. cerevisiae: Studies with two model genes. DNA Repair 2005, 4, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Yu, Y.; Waters, R. The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers in the MFA2 gene. J. Mol. Biol. 2002, 316, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Evans, K.E.; van Eijk, P.; Bennett, M.; Webster, R.M.; Leadbitter, M.; Teng, Y.; Waters, R.; Jackson, S.P.; Reed, S.H. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome Res. 2016, 26, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Teng, Y.; Liu, H.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.J.; Plummer, D.A.; Wyrick, J.J. NuA4 acetyltransferase is required for efficient nucleotide excision repair in yeast. DNA Repair 2019, 73, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, J.; Mitchell, D.L.; Johnson, D.G. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011, 39, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Moggs, J.G.; Oulad-Abdelghani, M.; Lejeune, F.; Dilworth, F.J.; Stevenin, J.; Almouzni, G.; Tora, L. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J. 2001, 20, 3187–3196. [Google Scholar] [CrossRef]
- Hassan, A.H.; Neely, K.E.; Workman, J.L. Histone acetyltransferase complexes stabilize swi/snf binding to promoter nucleosomes. Cell 2001, 104, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.; Li, B.; Workman, J.L. RSC exploits histone acetylation to abrogate the nucleosomal block to RNA polymerase II elongation. Mol. Cell 2006, 24, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.; Prochasson, P.; Neely, K.E.; Galasinski, S.C.; Chandy, M.; Carrozza, M.J.; Workman, J.L. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 2002, 111, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Szerlong, H.; Erdjument-Bromage, H.; Tempst, P.; Werner, M.; Cairns, B.R. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 2004, 23, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Bohm, K.A.; Hodges, A.J.; Czaja, W.; Selvam, K.; Smerdon, M.J.; Mao, P.; Wyrick, J.J. Distinct roles for RSC and SWI/SNF chromatin remodelers in genomic excision repair. Genome Res. 2021, 31, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Fahy, D.; Smerdon, M.J. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol. 2006, 13, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Srivas, R.; Costelloe, T.; Carvunis, A.R.; Sarkar, S.; Malta, E.; Sun, S.M.; Pool, M.; Licon, K.; van Welsem, T.; van Leeuwen, F.; et al. A UV-induced genetic network links the RSC complex to nucleotide excision repair and shows dose-dependent rewiring. Cell Rep. 2013, 5, 1714–1724. [Google Scholar] [CrossRef][Green Version]
- Yuan, J.; Pu, M.; Zhang, Z.; Lou, Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 2009, 8, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, H.; Wurtele, H.; Davies, B.; Horazdovsky, B.; Verreault, A.; Zhang, Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 2008, 134, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, H.; Hawke, D.; Kobayashi, R.; Verreault, A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 2005, 436, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, R.; Hudson, A.; Jackson, S.P. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 2007, 315, 649–652. [Google Scholar] [CrossRef]
- Han, J.; Zhou, H.; Horazdovsky, B.; Zhang, K.; Xu, R.M.; Zhang, Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007, 315, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008, 134, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Hinz, J.M.; Laughery, M.F.; Wyrick, J.J.; Smerdon, M.J. Site-specific Acetylation of Histone H3 Decreases Polymerase beta Activity on Nucleosome Core Particles In Vitro. J. Biol. Chem. 2016, 291, 11434–11445. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wyrick, J.J. Emerging roles for histone modifications in DNA excision repair. FEMS Yeast Res. 2016, 16, fow090. [Google Scholar] [CrossRef] [PubMed]
- Rufiange, A.; Jacques, P.E.; Bhat, W.; Robert, F.; Nourani, A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol. Cell 2007, 27, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, T.; Liu, C.L.; Erkmann, J.A.; Holik, J.; Grunstein, M.; Kaufman, P.D.; Friedman, N.; Rando, O.J. Cell cycle- and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet. 2008, 4, e1000270. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, K.; Grunstein, M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 2005, 121, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Khan, P.; Chaudhuri, R.N. Acetylation of H3K56 orchestrates UV-responsive chromatin events that generate DNA accessibility during Nucleotide Excision Repair. DNA Repair 2022, 113, 103317. [Google Scholar] [CrossRef] [PubMed]
- Shimko, J.C.; North, J.A.; Bruns, A.N.; Poirier, M.G.; Ottesen, J.J. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 2011, 408, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; North, J.A.; Shimko, J.C.; Forties, R.A.; Ferdinand, M.B.; Manohar, M.; Zhang, M.; Fishel, R.; Ottesen, J.J.; Poirier, M.G. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. USA 2011, 108, 12711–12716. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.; Somers, J.; Webster, R.; Flaus, A.; Owen-Hughes, T. Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol. Cell. Biol. 2007, 27, 4037–4048. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Bockler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef]
- Adar, S.; Hu, J.; Lieb, J.D.; Sancar, A. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E2124–E2133. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Huff, J.T.; Plocik, A.M.; Guthrie, C.; Yamamoto, K.R. Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 2010, 17, 1495–1499. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Fong, N.; Erickson, B.; Bentley, D.L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. USA 2011, 108, 13564–13569. [Google Scholar] [CrossRef]
- DiFiore, J.V.; Ptacek, T.S.; Wang, Y.; Li, B.; Simon, J.M.; Strahl, B.D. Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions. Cell Rep. 2020, 31, 107751. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Douglass, S.M.; Morselli, M.; Obusan, M.B.; Pavlyukov, M.S.; Pellegrini, M.; Johnson, T.L. H3K36 Methylation and the Chromodomain Protein Eaf3 Are Required for Proper Cotranscriptional Spliceosome Assembly. Cell Rep. 2019, 27, 3760–3769.e64. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, S.L.; Strahl, B.D. Shaping the cellular landscape with Set2/SETD2 methylation. Cell. Mol. Life Sci. CMLS 2017, 74, 3317–3334. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, M.R.; Jha, D.K.; Ucles, S.A.; Flood, D.M.; Strahl, B.D.; Stevens, S.W.; Kress, T.L. Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae. RNA Biol. 2016, 13, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Shilatifard, A. A site to remember: H3K36 methylation a mark for histone deacetylation. Mutat. Res. 2007, 618, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Li, B.; Florens, L.; Suganuma, T.; Swanson, S.K.; Lee, K.K.; Shia, W.J.; Anderson, S.; Yates, J.; Washburn, M.P.; et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005, 123, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.C.; Kurdistani, S.K.; Morris, S.A.; Ahn, S.H.; Podolny, V.; Collins, S.R.; Schuldiner, M.; Chin, K.; Punna, T.; Thompson, N.J.; et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 2005, 123, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.A.; Struhl, K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell 2005, 20, 971–978. [Google Scholar] [CrossRef]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Decoding the histone code: Role of H3K36me3 in mismatch repair and implications for cancer susceptibility and therapy. Cancer Res. 2013, 73, 6379–6383. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gu, L.; Li, G.M. H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J. Biol. Chem. 2018, 293, 7811–7823. [Google Scholar] [CrossRef] [PubMed]
- Frigola, J.; Sabarinathan, R.; Mularoni, L.; Muiños, F.; Gonzalez-Perez, A.; López-Bigas, N. Reduced mutation rate in exons due to differential mismatch repair. Nat. Genet. 2017, 49, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. CB 2000, 10, 886–895. [Google Scholar] [CrossRef]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome Instability and gammaH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef] [PubMed]
- Messick, T.E.; Greenberg, R.A. The ubiquitin landscape at DNA double-strand breaks. J. Cell Biol. 2009, 187, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef]
- Song, H.Y.; Shen, R.; Liu, X.W.; Yang, X.G.; Xie, K.; Guo, Z.; Wang, D.G. Histone post-translational modification and the DNA damage response. Genes Dis. 2023, 10, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.N.P.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of Homologous Recombination by RNF20-Dependent H2B Ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef] [PubMed]
- So, C.C.; Ramachandran, S.; Martin, A. E3 Ubiquitin Ligases RNF20 and RNF40 Are Required for Double-Stranded Break (DSB) Repair: Evidence for Monoubiquitination of Histone H2B Lysine 120 as a Novel Axis of DSB Signaling and Repair. Mol. Cell. Biol. 2019, 39, e00488-18. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Pinato, S.; Scandiuzzi, C.; Arnaudo, N.; Citterio, E.; Gaudino, G.; Penengo, L. RNF168, a new RING finger, MIU-containing protein that modifies chromatin by ubiquitination of histones H2A and H2AX. BMC Mol. Biol. 2009, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Horn, V.; Uckelmann, M.; Zhang, H.Y.; Eerland, J.; Aarsman, I.; le Paige, U.B.; Davidovich, C.; Sixma, T.K.; van Ingen, H. Structural basis of specific H2A K13/K15 ubiquitination by RNF168. Nat. Commun. 2019, 10, 1751. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.H.; Li, D.; Lu, Z.; Liu, G.X.; Wang, M.; Xing, P.Y.; Wang, M.; Dong, Y.; Wang, X.J.; Li, J.Y.; et al. Bre1-dependent H2B ubiquitination promotes homologous recombination by stimulating histone eviction at DNA breaks. Nucleic Acids Res. 2018, 46, 11326–11339. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yan, J.; Wang, X.; Chen, J.; Wang, X.; Dong, Y.; Zhang, S.; Gan, X.; Huang, J.; Chen, X. RPA-mediated recruitment of Bre1 couples histone H2B ubiquitination to DNA replication and repair. Proc. Natl. Acad. Sci. USA 2021, 118, e2017497118. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef]
- Mao, P.; Meas, R.; Dorgan, K.M.; Smerdon, M.J. UV damage-induced RNA polymerase II stalling stimulates H2B deubiquitylation. Proc. Natl. Acad. Sci. USA 2014, 111, 12811–12816. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Teng, Y.; Waters, R.; Reed, S.H. How chromatin is remodelled during DNA repair of UV-induced DNA damage in Saccharomyces cerevisiae. PLoS Genet. 2011, 7, e1002124. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Liu, H.; Gill, H.W.; Yu, Y.; Waters, R.; Reed, S.H. Saccharomyces cerevisiae Rad16 mediates ultraviolet-dependent histone H3 acetylation required for efficient global genome nucleotide-excision repair. EMBO Rep. 2008, 9, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Almouzni, G. Chromatin dynamics after DNA damage: The legacy of the access-repair-restore model. DNA Repair 2015, 36, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Roche, D.; Almouzni, G. New histone incorporation marks sites of UV repair in human cells. Cell 2006, 127, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [Google Scholar] [CrossRef] [PubMed]
- Breslin, C.; Hornyak, P.; Ridley, A.; Rulten, S.L.; Hanzlikova, H.; Oliver, A.W.; Caldecott, K.W. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015, 43, 6934–6944. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. XRCC1 protein; Form and function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef] [PubMed]
- Blessing, C.; Apelt, K.; van den Heuvel, D.; Gonzalez-Leal, C.; Rother, M.B.; van der Woude, M.; González-Prieto, R.; Yifrach, A.; Parnas, A.; Shah, R.G.; et al. XPC-PARP complexes engage the chromatin remodeler ALC1 to catalyze global genome DNA damage repair. Nat. Commun. 2022, 13, 4762. [Google Scholar] [CrossRef] [PubMed]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Meas, R.; Smerdon, M.J.; Wyrick, J.J. The amino-terminal tails of histones H2A and H3 coordinate efficient base excision repair, DNA damage signaling and postreplication repair in Saccharomyces cerevisiae. Nucleic Acids Res. 2015, 43, 4990–5001. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kyriss, M.N.; Jin, Y.; Gallegos, I.J.; Sanford, J.A.; Wyrick, J.J. Novel functional residues in the core domain of histone H2B regulate yeast gene expression and silencing and affect the response to DNA damage. Mol. Cell. Biol. 2010, 30, 3503–3518. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nag, R.; Kyriss, M.; Smerdon, J.W.; Wyrick, J.J.; Smerdon, M.J. A cassette of N-terminal amino acids of histone H2B are required for efficient cell survival, DNA repair and Swi/Snf binding in UV irradiated yeast. Nucleic Acids Res. 2010, 38, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Rodriguez, A.M.; Wyrick, J.J. Genetic and genomewide analysis of simultaneous mutations in acetylated and methylated lysine residues in histone H3 in Saccharomyces cerevisiae. Genetics 2009, 181, 461–472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chaudhuri, S.; Wyrick, J.J.; Smerdon, M.J. Histone H3 Lys79 methylation is required for efficient nucleotide excision repair in a silenced locus of Saccharomyces cerevisiae. Nucleic Acids Res. 2009, 37, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Rodriguez, A.M.; Stanton, J.D.; Kitazono, A.A.; Wyrick, J.J. Simultaneous mutation of methylated lysine residues in histone H3 causes enhanced gene silencing, cell cycle defects, and cell lethality in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 6832–6841. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.A.; Wyrick, J.J. Regulation of gene transcription by the histone H2A N-terminal domain. Mol. Cell. Biol. 2007, 27, 7641–7648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, A.M.; Pouchnik, D.J.; Walker, J.L.; Wyrick, J.J. Redundant roles for histone H3 N-terminal lysine residues in subtelomeric gene repression in Saccharomyces cerevisiae. Genetics 2004, 167, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Lenfant, F.; Mann, R.K.; Thomsen, B.; Ling, X.; Grunstein, M. All four core histone N-termini contain sequences required for the repression of basal transcription in yeast. EMBO J. 1996, 15, 3974–3985. [Google Scholar] [CrossRef]
- Ling, X.; Harkness, T.A.; Schultz, M.C.; Fisher-Adams, G.; Grunstein, M. Yeast histone H3 and H4 amino termini are important for nucleosome assembly in vivo and in vitro: Redundant and position-independent functions in assembly but not in gene regulation. Genes Dev. 1996, 10, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.S.; Mann, R.K.; Grunstein, M. Yeast histone H3 and H4 N termini function through different GAL1 regulatory elements to repress and activate transcription. Proc. Natl. Acad. Sci. USA 1995, 92, 5664–5668. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Laroche, T.; Strahl-Bolsinger, S.; Gasser, S.M.; Grunstein, M. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: A molecular model for the formation of heterochromatin in yeast. Cell 1995, 80, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.F.; Altschuler, S.J.; Wu, L.F.; Rando, O.J. Genomic characterization reveals a simple histone H4 acetylation code. Proc. Natl. Acad. Sci. USA 2005, 102, 5501–5506. [Google Scholar] [CrossRef] [PubMed]
- Recht, J.; Dunn, B.; Raff, A.; Osley, M.A. Functional analysis of histones H2A and H2B in transcriptional repression in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 2545–2553. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jensen, K.; Santisteban, M.S.; Urekar, C.; Smith, M.M. Histone H2A.Z acid patch residues required for deposition and function. Mol. Genet. Genom. MGG 2011, 285, 287–296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahn, S.H.; Cheung, W.L.; Hsu, J.Y.; Diaz, R.L.; Smith, M.M.; Allis, C.D. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 2005, 120, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sabet, N.; Tong, F.; Madigan, J.P.; Volo, S.; Smith, M.M.; Morse, R.H. Global and specific transcriptional repression by the histone H3 amino terminus in yeast. Proc. Natl. Acad. Sci. USA 2003, 100, 4084–4089. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M.; Yang, P.; Santisteban, M.S.; Boone, P.W.; Goldstein, A.T.; Megee, P.C. A novel histone H4 mutant defective in nuclear division and mitotic chromosome transmission. Mol. Cell. Biol. 1996, 16, 1017–1026. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsubara, K.; Sano, N.; Umehara, T.; Horikoshi, M. Global analysis of functional surfaces of core histones with comprehensive point mutants. Genes Cells Devoted Mol. Cell. Mech. 2007, 12, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Hyland, E.M.; Yuan, D.S.; Huang, H.; Bader, J.S.; Boeke, J.D. Probing nucleosome function: A highly versatile library of synthetic histone H3 and H4 mutants. Cell 2008, 134, 1066–1078. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, S.; Sanderson, B.W.; Delventhal, K.M.; Bradford, W.D.; Staehling-Hampton, K.; Shilatifard, A. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 2008, 15, 881–888. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sakamoto, M.; Noguchi, S.; Kawashima, S.; Okada, Y.; Enomoto, T.; Seki, M.; Horikoshi, M. Global analysis of mutual interaction surfaces of nucleosomes with comprehensive point mutants. Genes Cells Devoted Mol. Cell. Mech. 2009, 14, 1271–1330. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Maertens, A.M.; Hyland, E.M.; Dai, J.; Norris, A.; Boeke, J.D.; Bader, J.S. HistoneHits: A database for histone mutations and their phenotypes. Genome Res. 2009, 19, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Khan, P.; Nag Chaudhuri, R. Regulated acetylation and deacetylation of H4 K16 is essential for efficient NER in Saccharomyces cerevisiae. DNA Repair 2018, 72, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, F.; Gafken, P.R.; Gottschling, D.E. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 2002, 109, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Bostelman, L.J.; Keller, A.M.; Albrecht, A.M.; Arat, A.; Thompson, J.S. Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair 2007, 6, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Tatum, D.; Li, S. Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J. Biol. Chem. 2011, 286, 17530–17535. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Chen, S.; Wang, H.; Yin, C.; Han, C.; Peng, C.; Liu, Z.; Wan, L.; Zhang, X.; Zhang, J.; et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat. Commun. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Selvam, K.; Plummer, D.A.; Mao, P.; Wyrick, J.J. Set2 histone methyltransferase regulates transcription coupled-nucleotide excision repair in yeast. PLoS Genet. 2022, 18, e1010085. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Hoeijmakers, J.H.J.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Moreno, N.N.; Olthof, A.M.; Svejstrup, J.Q. Transcription-Coupled Nucleotide Excision Repair and the Transcriptional Response to UV-Induced DNA Damage. Annu. Rev. Biochem. 2023, 92, 81–113. [Google Scholar] [CrossRef]
- Li, S. Transcription coupled nucleotide excision repair in the yeast Saccharomyces cerevisiae: The ambiguous role of Rad26. DNA Repair 2015, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Kizer, K.O.; Phatnani, H.P.; Shibata, Y.; Hall, H.; Greenleaf, A.L.; Strahl, B.D. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 2005, 25, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Chaurasia, P.; Lahudkar, S.; Durairaj, G.; Shukla, A.; Bhaumik, S.R. Rad26p, a transcription-coupled repair factor, is recruited to the site of DNA lesion in an elongating RNA polymerase II-dependent manner in vivo. Nucleic Acids Res. 2010, 38, 1461–1477. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Li, H.; Gogol, M.M.; Workman, J.L. Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat. Commun. 2016, 7, 13610. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Feng, L.J.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Tabar, V. Histone Mutations in Cancer. Annu. Rev. Cancer Biol. 2018, 2, 337–351. [Google Scholar] [CrossRef]
- Espinoza Pereira, K.N.; Shan, J.; Licht, J.D.; Bennett, R.L. Histone mutations in cancer. Biochem. Soc. Trans. 2023, 51, 1749–1763. [Google Scholar] [CrossRef] [PubMed]
- Amatori, S.; Tavolaro, S.; Gambardella, S.; Fanelli, M. The dark side of histones: Genomic organization and role of oncohistones in cancer. Clin. Epigenet. 2021, 13, 71. [Google Scholar] [CrossRef]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenet. Chromatin 2014, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Shi, J.; Shi, X.; Li, W.; Wen, H. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J. Mol. Biol. 2018, 430, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.C.; Nielsen, E.C.; Matthiesen, R.; Jensen, L.H.; Sehested, M.; Finn, P.; Grauslund, M.; Hansen, A.M.; Jensen, O.N. Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteom. MCP 2006, 5, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Gafken, P.R.; Tsukiyama, T. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat. Struct. Mol. Biol. 2010, 17, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.W.; Turko, I.V. Histone post-translational modifications in frontal cortex from human donors with Alzheimer’s disease. Clin. Proteom. 2015, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.E.; Zhou, L.; Parra, M.A.; Chen, X.; Strahl, B.D. Identification of lysine 37 of histone H2B as a novel site of methylation. PLoS ONE 2011, 6, e16244. [Google Scholar] [CrossRef] [PubMed]
- Wyrick, J.J.; Parra, M.A. The role of histone H2A and H2B post-translational modifications in transcription: A genomic perspective. Biochim. Biophys. Acta 2009, 1789, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wyrick, J.J.; Reese, J.C. Novel trans-tail regulation of H2B ubiquitylation and H3K4 methylation by the N terminus of histone H2A. Mol. Cell. Biol. 2010, 30, 3635–3645. [Google Scholar] [CrossRef] [PubMed]
- Rossodivita, A.A.; Boudoures, A.L.; Mecoli, J.P.; Steenkiste, E.M.; Karl, A.L.; Vines, E.M.; Cole, A.M.; Ansbro, M.R.; Thompson, J.S. Histone H3 K79 methylation states play distinct roles in UV-induced sister chromatid exchange and cell cycle checkpoint arrest in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 6286–6299. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.J.; Gallegos, I.J.; Laughery, M.F.; Meas, R.; Tran, L.; Wyrick, J.J. Histone Sprocket Arginine Residues Are Important for Gene Expression, DNA Repair, and Cell Viability in Saccharomyces cerevisiae. Genetics 2015, 200, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Muthurajan, U.M.; Bao, Y.; Forsberg, L.J.; Edayathumangalam, R.S.; Dyer, P.N.; White, C.L.; Luger, K. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004, 23, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Flaus, A.; Rencurel, C.; Ferreira, H.; Wiechens, N.; Owen-Hughes, T. Sin mutations alter inherent nucleosome mobility. EMBO J. 2004, 23, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kruger, W.; Peterson, C.L.; Sil, A.; Coburn, C.; Arents, G.; Moudrianakis, E.N.; Herskowitz, I. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995, 9, 2770–2779. [Google Scholar] [CrossRef]
- Nag, R.; Smerdon, M.J. Altering the chromatin landscape for nucleotide excision repair. Mutat. Res. 2009, 682, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Nag, R.; Gong, F.; Fahy, D.; Smerdon, M.J. A single amino acid change in histone H4 enhances UV survival and DNA repair in yeast. Nucleic Acids Res. 2008, 36, 3857–3866. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Hyland, E.M.; Norris, A.; Boeke, J.D. Yin and Yang of histone H2B roles in silencing and longevity: A tale of two arginines. Genetics 2010, 186, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.J.; Gloss, L.M.; Wyrick, J.J. Residues in the Nucleosome Acidic Patch Regulate Histone Occupancy and Are Important for FACT Binding in Saccharomyces cerevisiae. Genetics 2017, 206, 1339–1348. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kemble, D.J.; McCullough, L.L.; Whitby, F.G.; Formosa, T.; Hill, C.P. FACT Disrupts Nucleosome Structure by Binding H2A-H2B with Conserved Peptide Motifs. Mol. Cell 2015, 60, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Hondele, M.; Stuwe, T.; Hassler, M.; Halbach, F.; Bowman, A.; Zhang, E.T.; Nijmeijer, B.; Kotthoff, C.; Rybin, V.; Amlacher, S.; et al. Structural basis of histone H2A-H2B recognition by the essential chaperone FACT. Nature 2013, 499, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Bagert, J.D.; Mitchener, M.M.; Patriotis, A.L.; Dul, B.E.; Wojcik, F.; Nacev, B.A.; Feng, L.; Allis, C.D.; Muir, T.W. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol. 2021, 17, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Wienholz, F.; Zhou, D.; Turkyilmaz, Y.; Schwertman, P.; Tresini, M.; Pines, A.; van Toorn, M.; Bezstarosti, K.; Demmers, J.A.A.; Marteijn, J.A. FACT subunit Spt16 controls UVSSA recruitment to lesion-stalled RNA Pol II and stimulates TC-NER. Nucleic Acids Res. 2019, 47, 4011–4025. [Google Scholar] [CrossRef]
- Mandemaker, I.K.; Vermeulen, W.; Marteijn, J.A. Gearing up chromatin: A role for chromatin remodeling during the transcriptional restart upon DNA damage. Nucleus 2014, 5, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Dinant, C.; Ampatziadis-Michailidis, G.; Lans, H.; Tresini, M.; Lagarou, A.; Grosbart, M.; Theil, A.F.; van Cappellen, W.A.; Kimura, H.; Bartek, J.; et al. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell 2013, 51, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Fahy, D.; Liu, H.; Wang, W.; Smerdon, M.J. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle 2008, 7, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Rüthemann, P.; Balbo Pogliano, C.; Codilupi, T.; Garajovà, Z.; Naegeli, H. Chromatin remodeler CHD1 promotes XPC-to-TFIIH handover of nucleosomal UV lesions in nucleotide excision repair. EMBO J. 2017, 36, 3372–3386. [Google Scholar] [CrossRef] [PubMed]
- Hara, R.; Sancar, A. The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol. Cell. Biol. 2002, 22, 6779–6787. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.A.; Kerr, D.; Fahy, D.; Pouchnik, D.J.; Wyrick, J.J. Deciphering the roles of the histone H2B N-terminal domain in genome-wide transcription. Mol. Cell. Biol. 2006, 26, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Duan, M.; Wyrick, J.J.; Smerdon, M.J. A cassette of basic amino acids in histone H2B regulates nucleosome dynamics and access to DNA damage. J. Biol. Chem. 2018, 293, 7376–7386. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Crickard, J.B.; Srikanth, A.; Reese, J.C. A highly conserved region within H2B is important for FACT to act on nucleosomes. Mol. Cell. Biol. 2014, 34, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Kyriss, M.N.; Hodges, A.J.; Duan, M.; Morris, R.T.; Lavine, M.D.; Topping, T.B.; Gloss, L.M.; Wyrick, J.J. A basic domain in the histone H2B N-terminal tail is important for nucleosome assembly by FACT. Nucleic Acids Res. 2016, 44, 9142–9152. [Google Scholar] [CrossRef] [PubMed]
- Selvam, K.; Rahman, S.A.; Li, S. Histone H4 H75E mutation attenuates global genomic and Rad26-independent transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2019, 47, 7392–7401. [Google Scholar] [CrossRef] [PubMed]
- Selvam, K.; Rahman, S.A.; Forrester, D.; Bao, A.; Lieu, M.; Li, S. Histone H4 LRS mutations can attenuate UV mutagenesis without affecting PCNA ubiquitination or sumoylation. DNA Repair 2020, 95, 102959. [Google Scholar] [CrossRef] [PubMed]
- Tessadori, F.; Duran, K.; Knapp, K.; Fellner, M.; Deciphering Developmental Disorders, S.; Smithson, S.; Beleza Meireles, A.; Elting, M.W.; Waisfisz, Q.; O’Donnell-Luria, A.; et al. Recurrent de novo missense variants across multiple histone H4 genes underlie a neurodevelopmental syndrome. Am. J. Hum. Genet. 2022, 109, 750–758. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).