
Regulation of Mertk Surface Expression via ADAM17 and γ-Secretase Proteolytic Processing
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
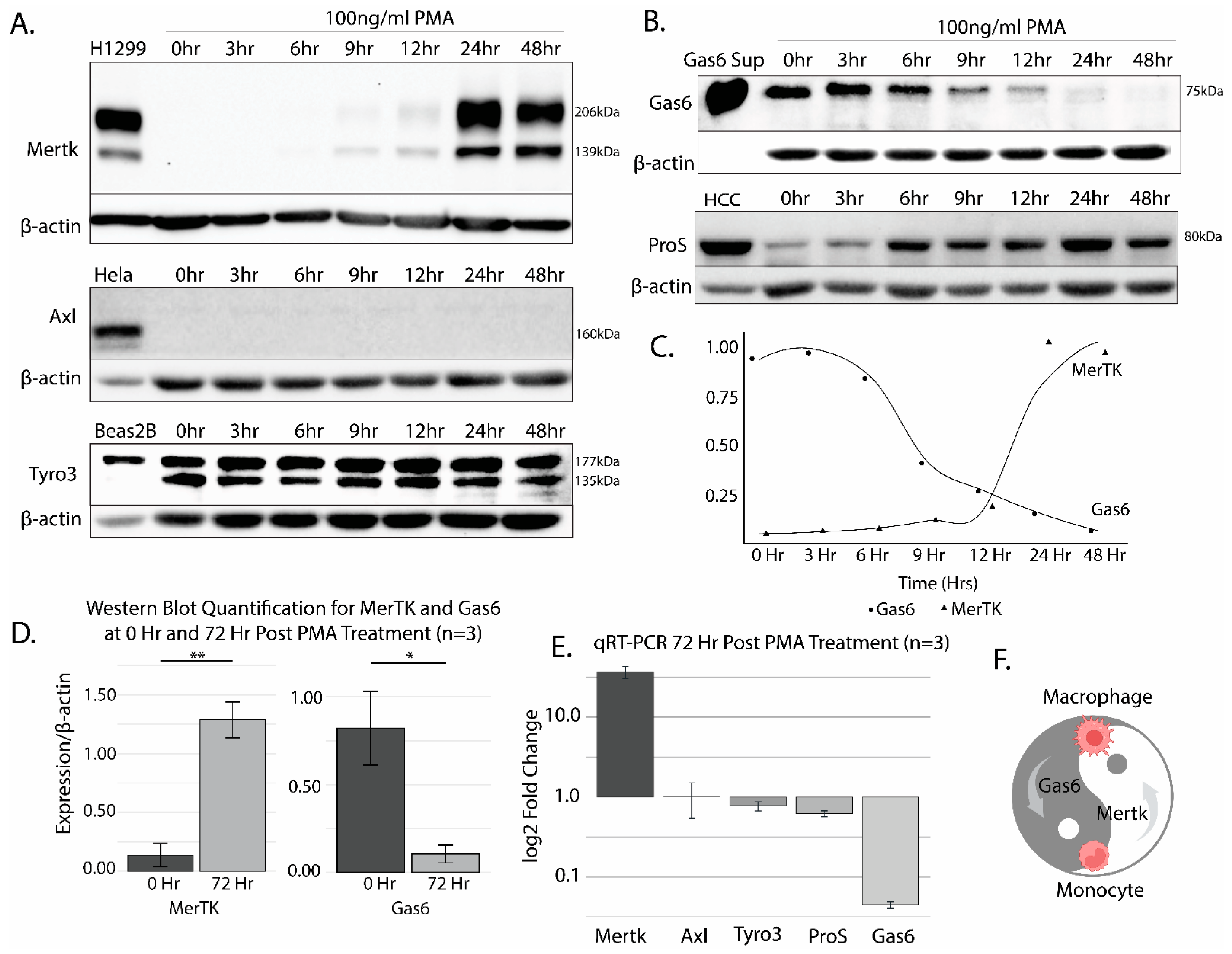
2.1. The Differentiation of THP-1 Cells by PMA Promotes Robust Mertk Up-Regulation at the Transcriptional and Translational Level
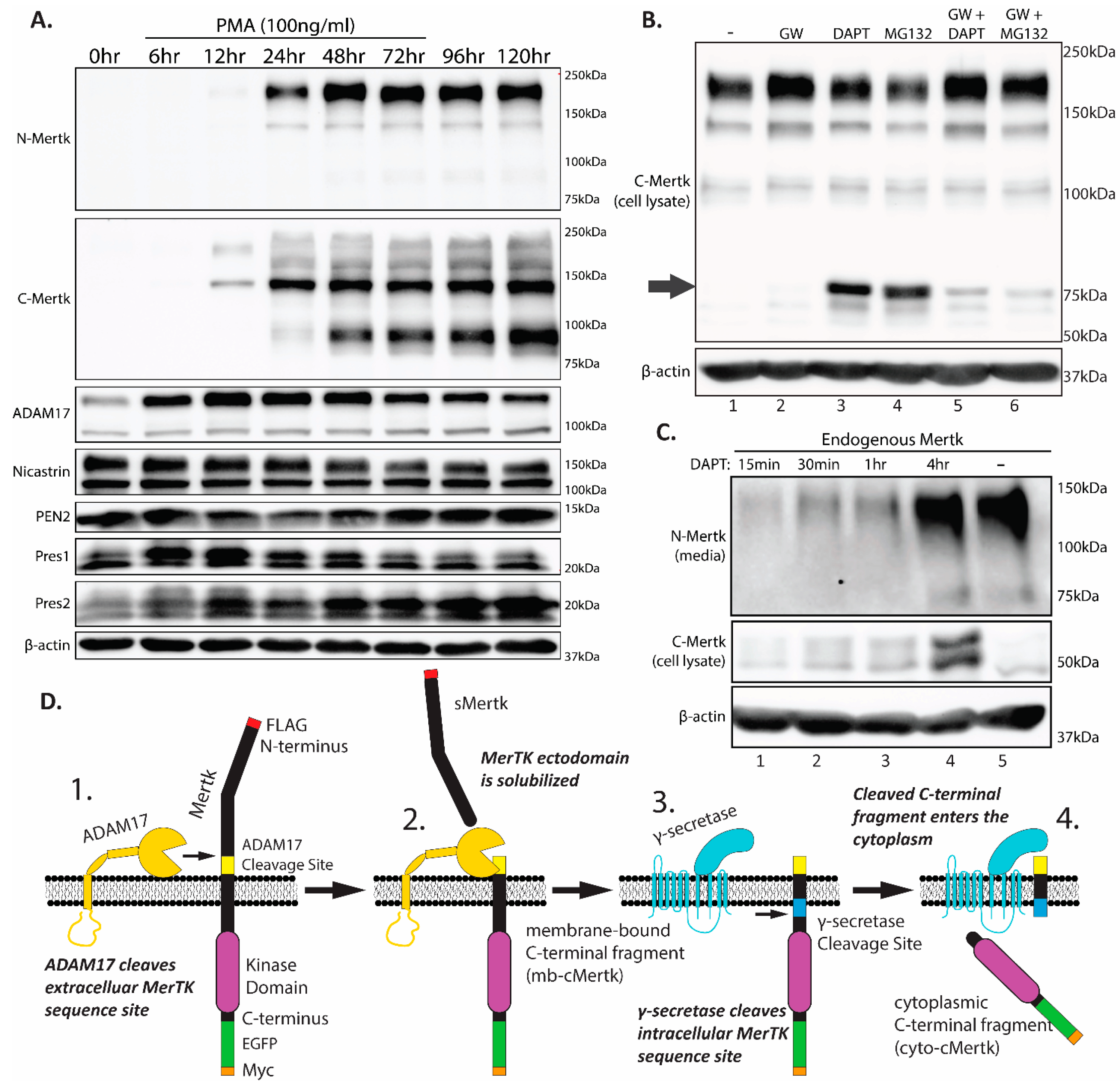
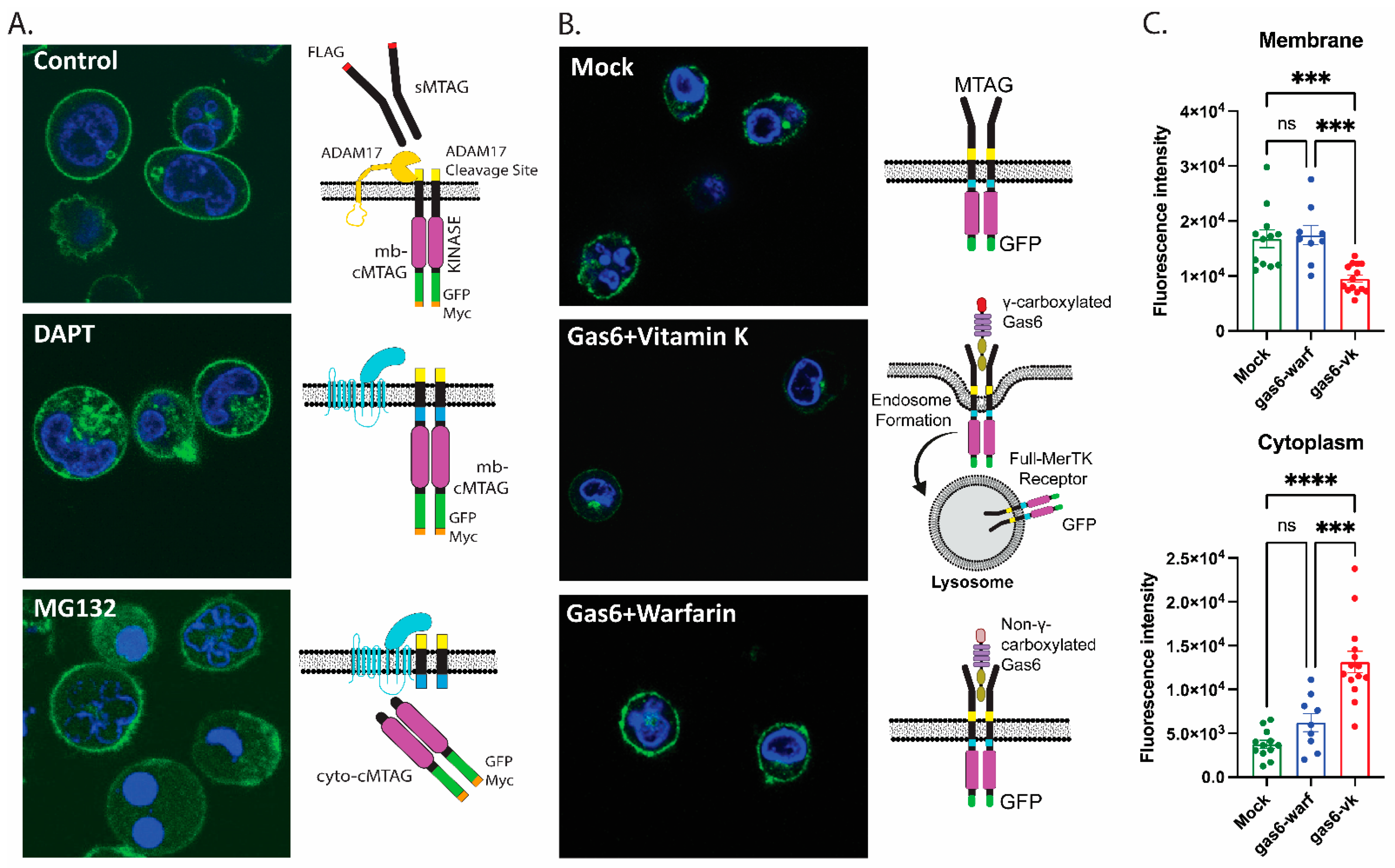
2.2. Homeostatic Regulation of Endogenous Mertk Cleavage by ADAM17 and γ-Secretase
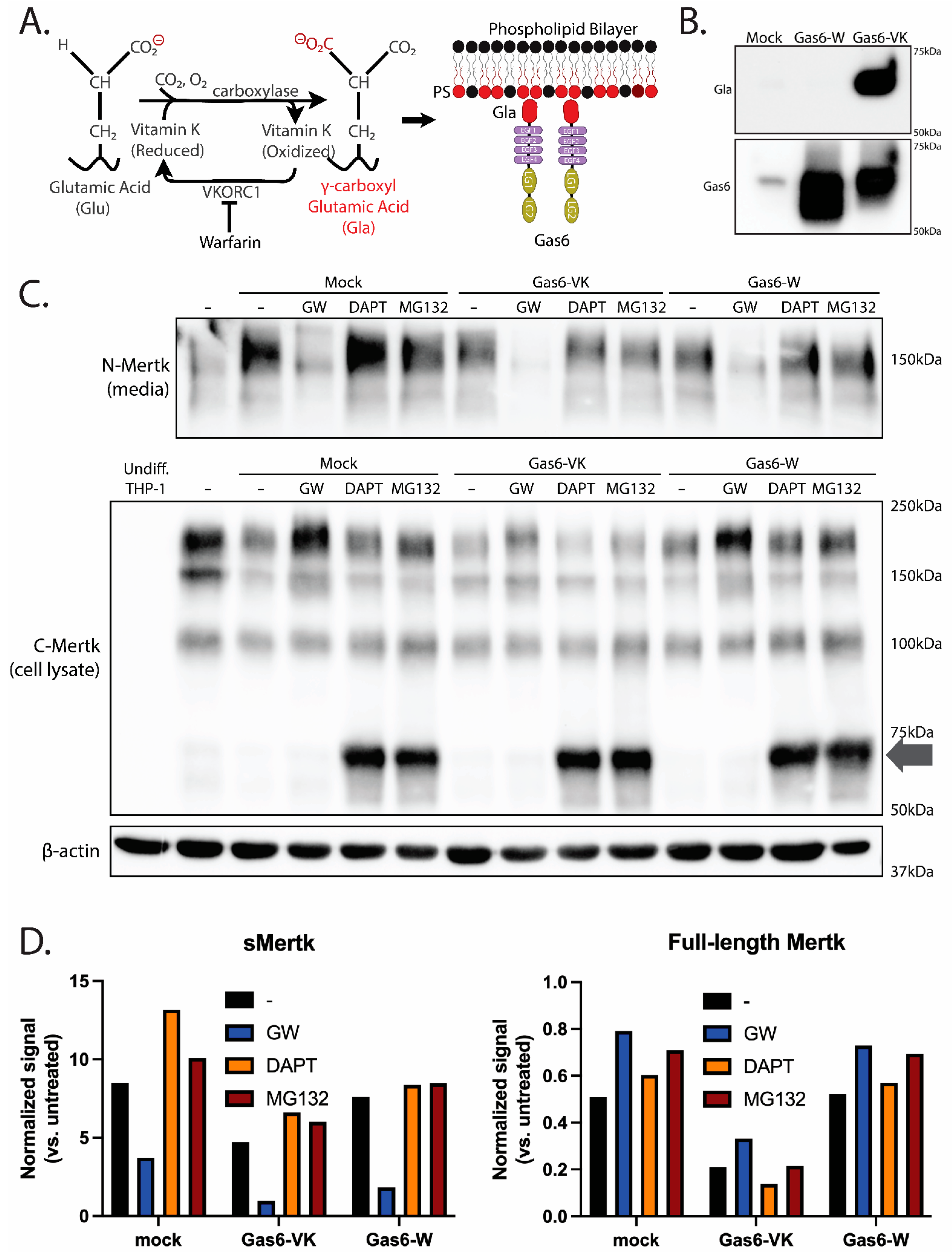
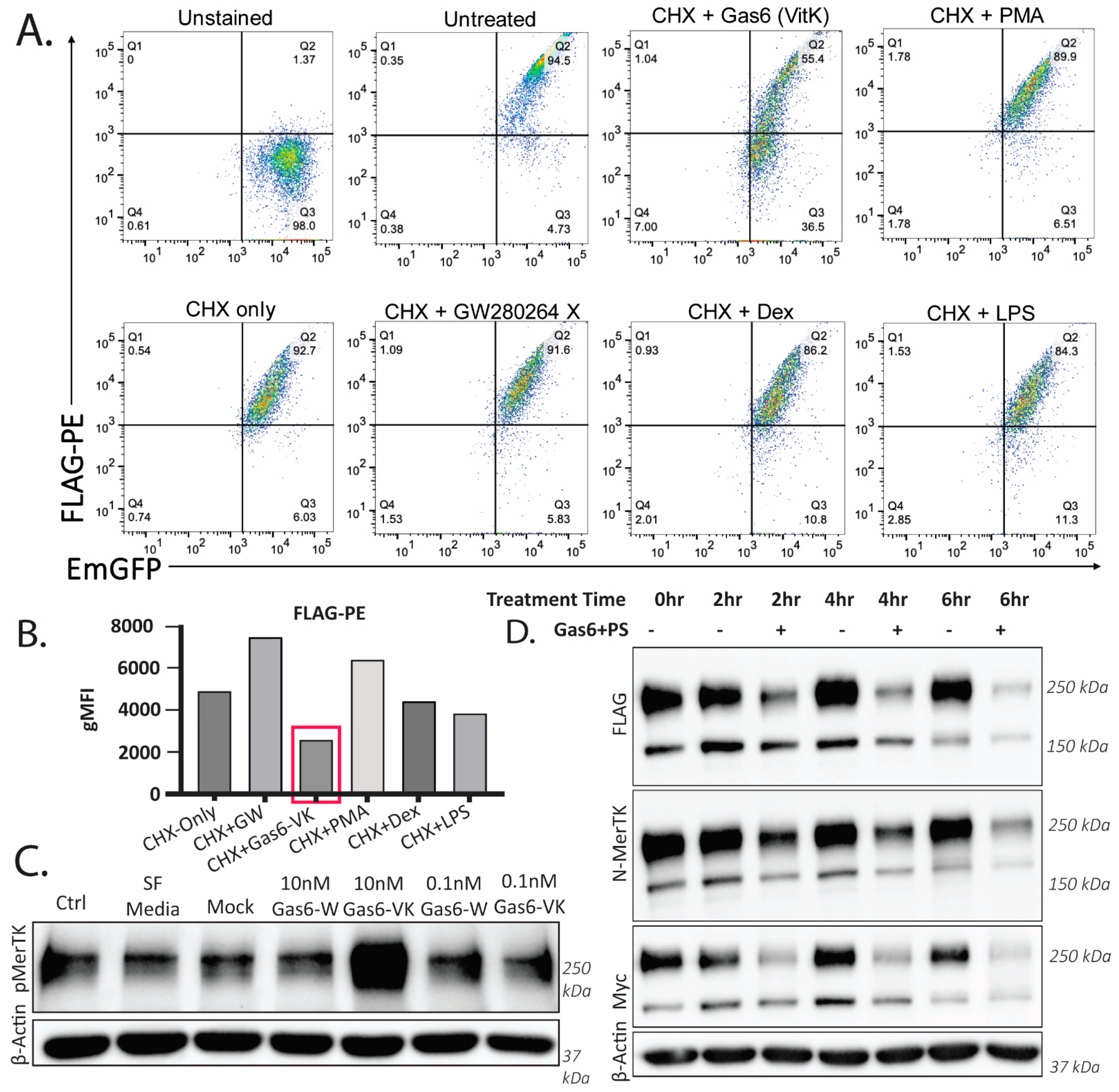
2.3. Ligand-Dependent Activation of Endogenous Mertk and MTag upon Stimulation with Recombinant γ-Carboxylated Gas6
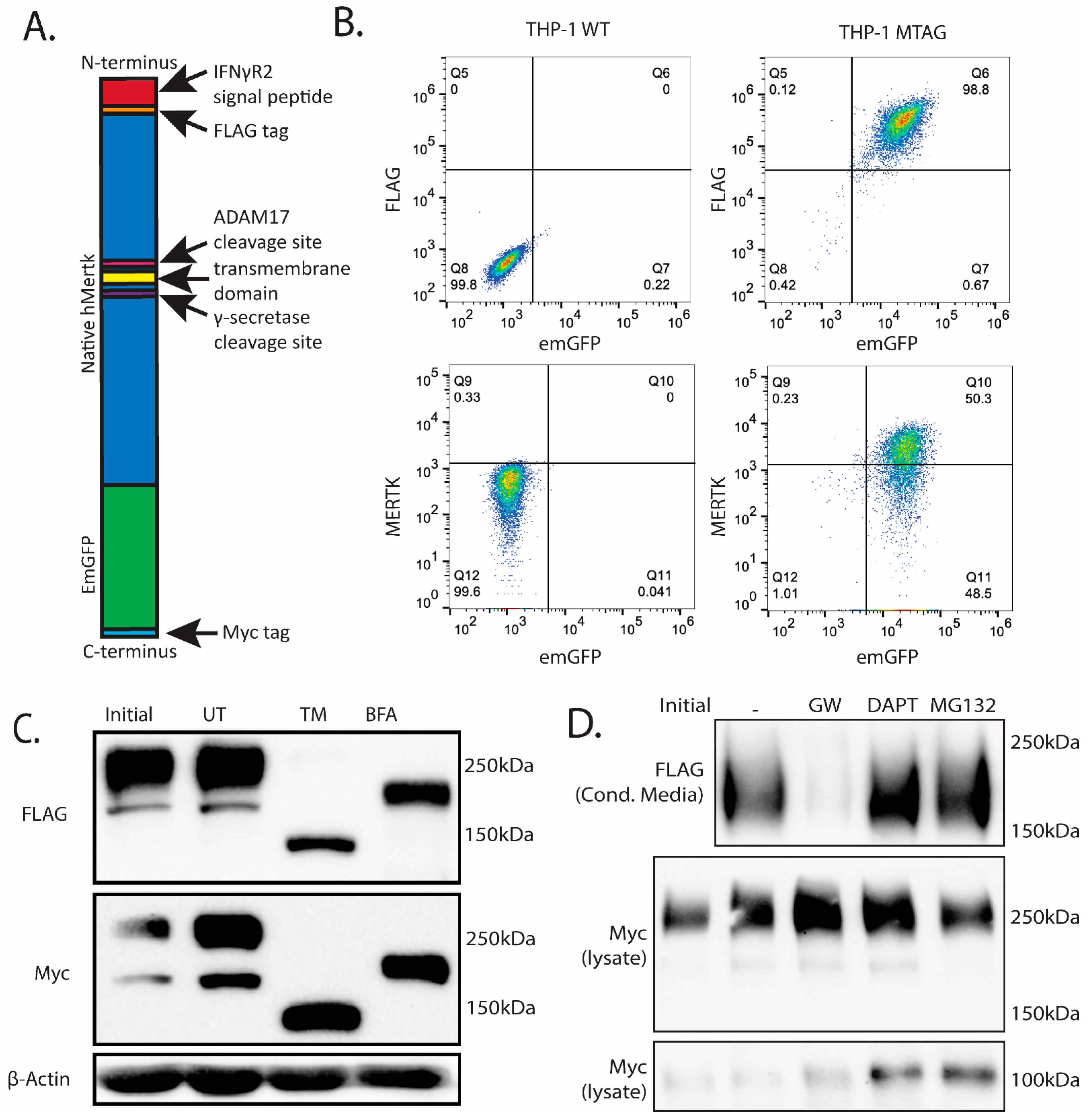
2.4. Stable Ectopic Expression of Flag-Mertk-EGFP-Myc Chimeric Reporter Construct in THP1 Cells
2.5. Gas6-Mediated Activation of Mertk Drives Mertk to a Distinct Proteolytic Itinerary
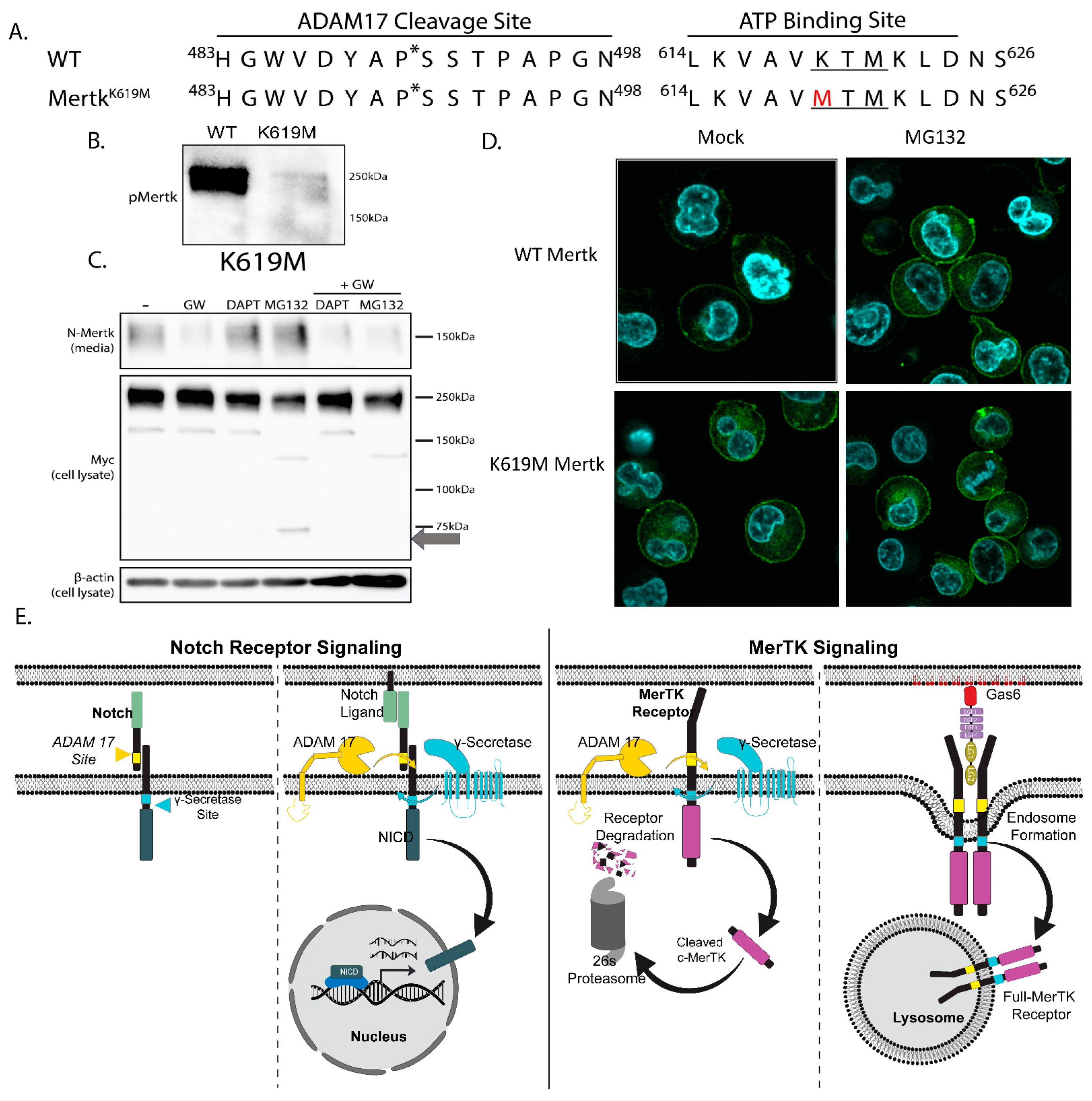
2.6. Mertk K619M Kinase-Dead Mertk Retains Homeostatic Processing by ADAM17 and γ-Secretase
3. Discussion
4. Materials and Methods
4.1. Design of a Flag-hMertk-EGFP-Myc Chimeric Plasmid Construct
4.2. In vitro Cell Culture
4.3. Transduction of THP-1 Cells with the Tagged Mertk Construct
4.4. Monocytic THP-1 cell Differentiation to Macrophages
4.5. C-Terminal Fragment Stabilization
4.6. Western Blotting
Western Blot Antibodies
- -
- ADAM17 (sc-390859, Santa Cruz Biotechnology, Santa Cruz, CA, USA)
- -
- γ-secretase antibody sampler kit (#5887, Cell Signaling Technology, Danvers, MA, USA; includes nicastrin, PEN2, Presenilin 1, Presenilin 2)
- -
- N-Mertk (AF891, R&D Systems, Minneapolis, MN, USA)
- -
- C-Mertk (D21F11, Cell Signaling Technology, Danvers, MA, USA)
- -
- B-actin (8H10D10, Cell Signaling Technology, Danvers, MA, USA)
- -
- pMertk (p186-749, Phosphosolutions, Aurora, CO, USA)
- -
- Myc (71D10, Cell Signaling Technology, Danvers, MA, USA)
- -
- Gas6 (SC-376087, Santa Cruz Biotechnology, Santa Cruz, CA, USA)
- -
- FLAG-HRP (637311, Biolegend, San Diego, CA, USA)
4.7. Production of recombinant Gas6
4.8. Gas6/Phosphatidylserine (PS) Treatments
4.9. Flow Cytometry
Flow Antibodies
- -
- Flag PE (637310, Biolegend, CA, USA)
- -
- hMertk (FAB8912A, R&D Systems, MN, USA)
4.10. Confocal Microscopy
4.11. Statistical Significance
4.12. Figures and Illustration
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef]
- McShane, L.; Tabas, I.; Lemke, G.; Kurowska-Stolarska, M.; Maffia, P. TAM receptors in cardiovascular disease. Cardiovasc. Res. 2019, 115, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, L.; Traves, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagorska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef]
- Happonen, K.E.; Burrola, P.G.; Lemke, G. Regulation of brain endothelial cell physiology by the TAM receptor tyrosine kinase Mer. Commun. Biol. 2023, 6, 916. [Google Scholar] [CrossRef]
- Burstyn-Cohen, T.; Lew, E.D.; Traves, P.G.; Burrola, P.G.; Hash, J.C.; Lemke, G. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron 2012, 76, 1123–1132. [Google Scholar] [CrossRef]
- Prasad, D.; Rothlin, C.V.; Burrola, P.; Burstyn-Cohen, T.; Lu, Q.; Garcia de Frutos, P.; Lemke, G. TAM receptor function in the retinal pigment epithelium. Mol. Cell Neurosci. 2006, 33, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Kasikara, C.; Doran, A.C.; Ramakrishnan, R.; Birge, R.B.; Tabas, I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci. Signal 2018, 11, eaar3721. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Nishi, C.; Toda, S.; Segawa, K.; Nagata, S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol. Cell Biol. 2014, 34, 1512–1520. [Google Scholar] [CrossRef]
- Zizzo, G.; Guerrieri, J.; Dittman, L.M.; Merrill, J.T.; Cohen, P.L. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res. Ther. 2013, 15, R212. [Google Scholar] [CrossRef] [PubMed]
- Dransfield, I.; Zagorska, A.; Lew, E.D.; Michail, K.; Lemke, G. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 2015, 6, e1646. [Google Scholar] [CrossRef] [PubMed]
- Akalu, Y.T.; Mercau, M.E.; Ansems, M.; Hughes, L.D.; Nevin, J.; Alberto, E.J.; Liu, X.N.; He, L.Z.; Alvarado, D.; Keler, T.; et al. Tissue-specific modifier alleles determine Mertk loss-of-function traits. Elife 2022, 11, e80530. [Google Scholar] [CrossRef] [PubMed]
- Hanayama, R.; Tanaka, M.; Miwa, K.; Shinohara, A.; Iwamatsu, A.; Nagata, S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002, 417, 182–187. [Google Scholar] [CrossRef]
- Wu, Y.; Singh, S.; Georgescu, M.M.; Birge, R.B. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef]
- Albert, M.L.; Kim, J.I.; Birge, R.B. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat. Cell Biol. 2000, 2, 899–905. [Google Scholar] [CrossRef]
- Toda, S.; Hanayama, R.; Nagata, S. Two-step engulfment of apoptotic cells. Mol. Cell Biol. 2012, 32, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Somersan, S.; Bhardwaj, N. Tethering and tickling: A new role for the phosphatidylserine receptor. J. Cell Biol. 2001, 155, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol. 2001, 11, R795–R805. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Carey, K.; Godowski, P.J. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene 1997, 14, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, H.; Yonemasu, T.; Nakano, T.; Matumoto, K.; Nakamura, T. Identification of Gas6, a putative ligand for Sky and Axl receptor tyrosine kinases, as a novel neurotrophic factor for hippocampal neurons. J. Neurosci. Res. 2002, 68, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol. Cancer Ther. 2015, 14, 2014–2022. [Google Scholar] [CrossRef]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.J.; Wichroski, M.; et al. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef]
- Huelse, J.M.; Fridlyand, D.M.; Earp, S.; DeRyckere, D.; Graham, D.K. MERTK in cancer therapy: Targeting the receptor tyrosine kinase in tumor cells and the immune system. Pharmacol. Ther. 2020, 213, 107577. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer. Cancers 2021, 13, 5639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e359. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.C.; Young, C.; Hicks, D.; Owens, P.; Williams, A.; Vaught, D.B.; Morrison, M.M.; Lim, J.; Williams, M.; Brantley-Sieders, D.M.; et al. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Investig. 2014, 124, 4737–4752. [Google Scholar] [CrossRef] [PubMed]
- Weinger, J.G.; Omari, K.M.; Marsden, K.; Raine, C.S.; Shafit-Zagardo, B. Up-regulation of soluble Axl and Mer receptor tyrosine kinases negatively correlates with Gas6 in established multiple sclerosis lesions. Am. J. Pathol. 2009, 175, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Vaisar, T.; Subramanian, M.; Mautner, L.; Blobel, C.; Tabas, I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 2011, 286, 33335–33344. [Google Scholar] [CrossRef]
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the AXL receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2017, 31, 1382–1397. [Google Scholar] [CrossRef]
- Migdall-Wilson, J.; Bates, C.; Schlegel, J.; Brandao, L.; Linger, R.M.; DeRyckere, D.; Graham, D.K. Prolonged exposure to a Mer ligand in leukemia: Gas6 favors expression of a partial Mer glycoform and reveals a novel role for Mer in the nucleus. PLoS ONE 2012, 7, e31635. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Fluhrer, R.; Haass, C. Intramembrane proteolysis by gamma-secretase. J. Biol. Chem. 2008, 283, 29627–29631. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Inhibition of MMPs and ADAM/ADAMTS. Biochem. Pharmacol. 2019, 165, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P. Therapeutic potential of gamma-secretase inhibitors and modulators. Curr. Top. Med. Chem. 2008, 8, 54–61. [Google Scholar] [CrossRef]
- Kisselev, A.F. Site-Specific Proteasome Inhibitors. Biomolecules 2021, 12, 54. [Google Scholar] [CrossRef]
- Geng, K.; Kumar, S.; Kimani, S.G.; Kholodovych, V.; Kasikara, C.; Mizuno, K.; Sandiford, O.; Rameshwar, P.; Kotenko, S.V.; Birge, R.B. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front. Immunol. 2017, 8, 1521. [Google Scholar] [CrossRef]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef]
- Elbein, A.D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit. Rev. Biochem. 1984, 16, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Verbert, A.; Cacan, R. Trafficking of oligomannosides released during N-glycosylation: A clearing mechanism of the rough endoplasmic reticulum. Biochim. Biophys. Acta 1999, 1473, 137–146. [Google Scholar] [CrossRef]
- Hilliard, B.A.; Zizzo, G.; Ulas, M.; Linan, M.K.; Schreiter, J.; Cohen, P.L. Increased expression of Mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: Correlations with plasma interferon activity and steroid therapy. Arthritis Res. Ther. 2014, 16, R76. [Google Scholar] [CrossRef]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Sansbury, B.E.; Daemen, M.J.; Dorweiler, B.; Spite, M.; Fredman, G.; Tabas, I. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Investig. 2017, 127, 564–568. [Google Scholar] [CrossRef]
- Wu, J.; Ekman, C.; Jonsen, A.; Sturfelt, G.; Bengtsson, A.A.; Gottsater, A.; Lindblad, B.; Lindqvist, E.; Saxne, T.; Dahlback, B. Increased plasma levels of the soluble Mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res. Ther. 2011, 13, R62. [Google Scholar] [CrossRef]
- Morales, A.; Rojo Rello, S.; Cristobal, H.; Fiz-Lopez, A.; Arribas, E.; Mari, M.; Tutusaus, A.; de la Cal-Sabater, P.; Nicolaes, G.A.F.; Ortiz-Perez, J.T.; et al. Growth Arrest-Specific Factor 6 (GAS6) Is Increased in COVID-19 Patients and Predicts Clinical Outcome. Biomedicines 2021, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Azbazdar, Y.; Karabicici, M.; Erdal, E.; Ozhan, G. Regulation of Wnt Signaling Pathways at the Plasma Membrane and Their Misregulation in Cancer. Front. Cell Dev. Biol. 2021, 9, 631623. [Google Scholar] [CrossRef]
- Bland, C.E.; Kimberly, P.; Rand, M.D. Notch-induced proteolysis and nuclear localization of the Delta ligand. J. Biol. Chem. 2003, 278, 13607–13610. [Google Scholar] [CrossRef] [PubMed]
- Delwig, A.; Bland, C.; Beem-Miller, M.; Kimberly, P.; Rand, M.D. Endocytosis-independent mechanisms of Delta ligand proteolysis. Exp. Cell Res. 2006, 312, 1345–1360. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Alvarado, G.; Li, W. Tubby regulates microglial phagocytosis through MerTK. J. Neuroimmunol. 2012, 252, 40–48. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, T.D.; Koller, B.H.; Earp, H.S.; Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 1999, 162, 3498–3503. [Google Scholar] [CrossRef]
- Gao, L.; He, C.; Yang, A.; Zhou, H.; Lu, Q.; Birge, R.B.; Wu, Y. Receptor tyrosine kinases Tyro3, Axl, and Mertk differentially contribute to antibody-induced arthritis. Cell Commun. Signal 2023, 21, 195. [Google Scholar] [CrossRef]
- Kimani, S.G.; Geng, K.; Kasikara, C.; Kumar, S.; Sriram, G.; Wu, Y.; Birge, R.B. Contribution of Defective PS Recognition and Efferocytosis to Chronic Inflammation and Autoimmunity. Front. Immunol. 2014, 5, 566. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e407. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer 5′ -> 3′ | Reverse Primer 5′ -> 3′ |
|---|---|---|
| hMerTK | CAGGAAGATGGGACCTCTCTG | GGCTGAAGTCTTTCATGCACGC |
| hAxl | GTTTGGAGCTGTGATGGAAGGC | CGCTTCACTCAGGAAATCCTCC |
| hTyro3 | CACTGAGCTGGCTGACTAAGCCCC | AATGCATGCACTTAAGCAGCAGGG |
| hGas6 | GCCTTCTACAGCCTGGACTAC | TCTTGAGTTTCTTCGTGGAGTG |
| hProS | CCATTCCAGACCAGTGTAG | GGTAACTTCCAGGTGTATTATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lahey, K.C.; Varsanyi, C.; Wang, Z.; Aquib, A.; Gadiyar, V.; Rodrigues, A.A.; Pulica, R.; Desind, S.; Davra, V.; Calianese, D.C.; et al. Regulation of Mertk Surface Expression via ADAM17 and γ-Secretase Proteolytic Processing. Int. J. Mol. Sci. 2024, 25, 4404. https://doi.org/10.3390/ijms25084404
Lahey KC, Varsanyi C, Wang Z, Aquib A, Gadiyar V, Rodrigues AA, Pulica R, Desind S, Davra V, Calianese DC, et al. Regulation of Mertk Surface Expression via ADAM17 and γ-Secretase Proteolytic Processing. International Journal of Molecular Sciences. 2024; 25(8):4404. https://doi.org/10.3390/ijms25084404
Chicago/Turabian StyleLahey, Kevin C., Christopher Varsanyi, Ziren Wang, Ahmed Aquib, Varsha Gadiyar, Alcina A. Rodrigues, Rachael Pulica, Samuel Desind, Viralkumar Davra, David C. Calianese, and et al. 2024. "Regulation of Mertk Surface Expression via ADAM17 and γ-Secretase Proteolytic Processing" International Journal of Molecular Sciences 25, no. 8: 4404. https://doi.org/10.3390/ijms25084404
APA StyleLahey, K. C., Varsanyi, C., Wang, Z., Aquib, A., Gadiyar, V., Rodrigues, A. A., Pulica, R., Desind, S., Davra, V., Calianese, D. C., Liu, D., Cho, J.-H., Kotenko, S. V., De Lorenzo, M. S., & Birge, R. B. (2024). Regulation of Mertk Surface Expression via ADAM17 and γ-Secretase Proteolytic Processing. International Journal of Molecular Sciences, 25(8), 4404. https://doi.org/10.3390/ijms25084404