
New Mechanisms to Prevent Heart Failure with Preserved Ejection Fraction Using Glucagon-like Peptide-1 Receptor Agonism (GLP-1 RA) in Metabolic Syndrome and in Type 2 Diabetes: A Review

Abstract
:1. Obesity and Heart Failure with Preserved Ejection Fraction (HFpEF)
2. Consequences of Obesity on the Heart and on Heart Failure with Preserved Ejection Fraction
2.1. Obesity, Low-Grade Systemic Inflammation, Cardiac Damage, and HFpEF
2.2. Visceral Adipose Tissue and HFpEF
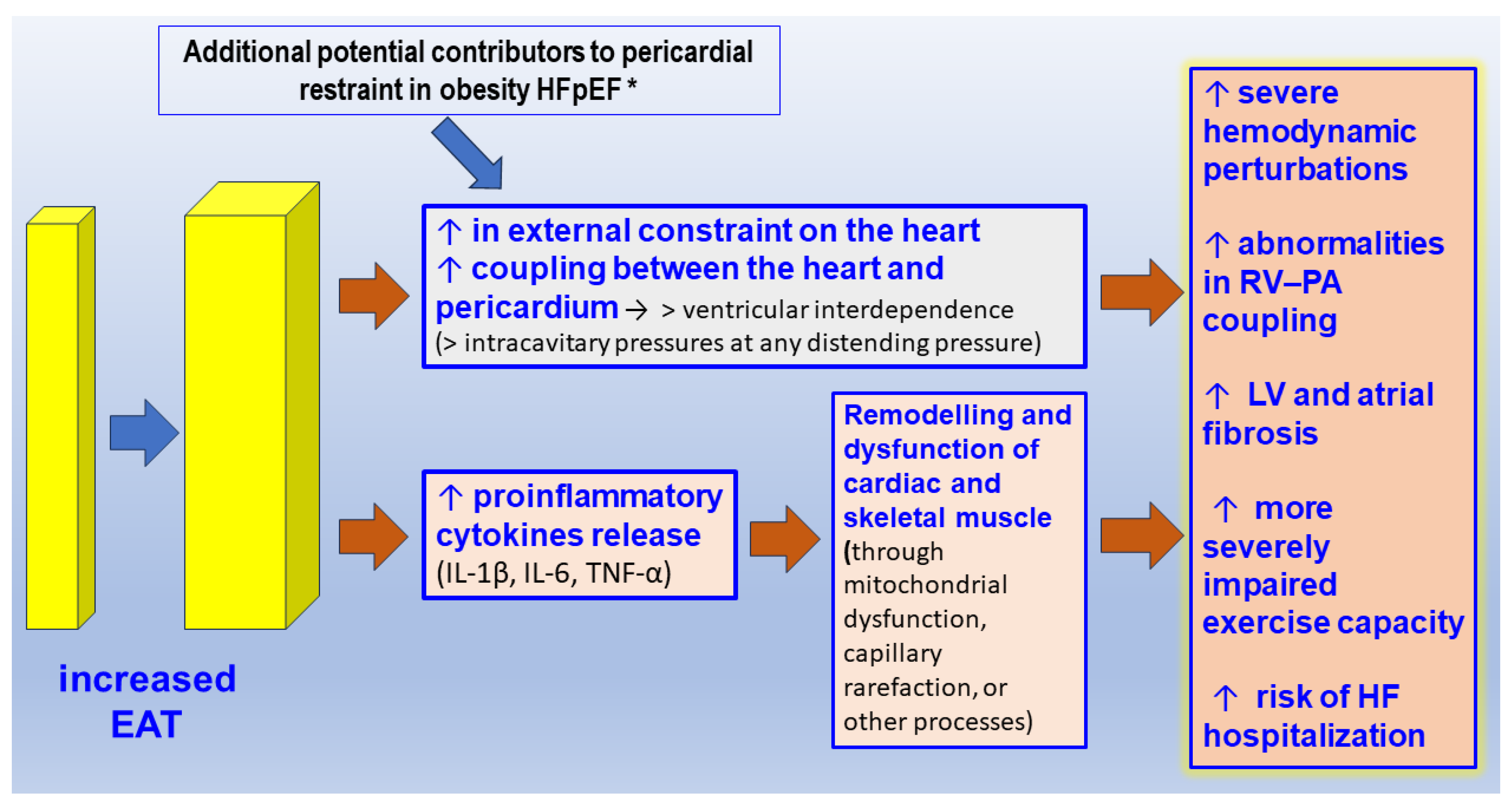
2.3. The Pericardial/Epicardial Adipose Tissue and HFpEF
2.4. The Relationship between Type 2 Diabetes, Obesity, and HFpEF
2.5. Current Effective Therapeutic Interventions in Patients with the Obese–HFpEF Phenotype
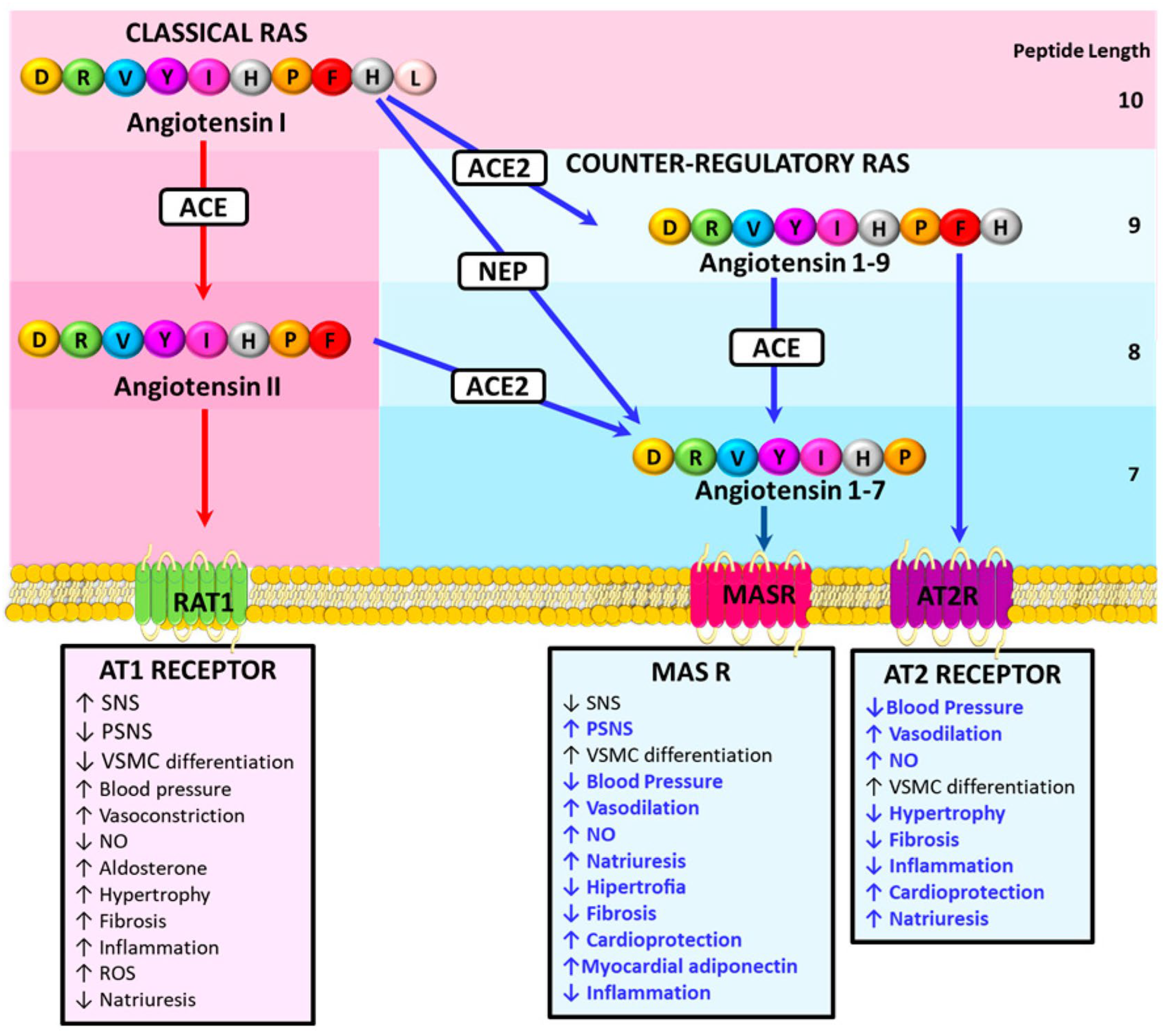
3. The Counter-Regulatory Renin Angiotensin System and Prevention of Cardiovascular Damage
4. The Glucagon-like Peptide 1 (GLP-1) Axis: A Novel Anti Diabetic, Anti-Obesity, and Anti Myocardial Remodeling Path
4.1. Effects of Glucagon-like Peptide 1 (GLP-1) upon Glucose Regulation, Obesity, and Diabetes
4.2. Effects of GLP-1R Agonism on the RAS, Cardiac Remodeling Pathways, and HFpEF Mechanisms
4.3. Current Evidence on Glucagon-like Peptide-1 Receptor Agonists (GLP-1 RAs) for Cardiovascular Prevention and Clinical Innovations
Author Contributions
Funding
Conflicts of Interest
References
- Dhore-Patil, A.; Thannoun, T.; Samson, R.; Le Jemtel, T.H. Diabetes Mellitus and Heart Failure with Preserved Ejection Fraction: Role of Obesity. Front. Physiol. 2022, 12, 785879. [Google Scholar] [CrossRef]
- Volpe, M.; Gallo, G. Cardiometabolic phenotype of heart failure with preserved ejection fraction as a target of sodium-glucose co-transporter 2 inhibitors and glucagon-like peptide receptor agonists. Cardiovasc. Res. 1994, 117, 1992–1994. [Google Scholar] [CrossRef]
- Stoicescu, L.; Crişan, D.; Morgovan, C.; Avram, L.; Ghibu, S. Heart Failure with Preserved Ejection Fraction: The Pathophysiological Mechanisms behind the Clinical Phenotypes and the Therapeutic Approach. Int. J. Mol. Sci. 2024, 25, 794. [Google Scholar] [CrossRef]
- Samson, R.; Jaiswal, A.; Ennezat, P.V.; Cassidy, M.; Le Jemtel, T.H. Clinical Phenotypes in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2016, 5, e002477. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J. CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-Preserved Trial. Lancet 2003, 362, 777–781. [Google Scholar]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322, Erratum in Circulation 2016, 133, e417. [Google Scholar] [CrossRef]
- Altara, R.; Giordano, M.; Nordén, E.S.; Cataliotti, A.; Kurdi, M.; Bajestani, S.N.; Booz, G.W. Targeting Obesity and Diabetes to Treat Heart Failure with Preserved Ejection Fraction. Front. Endocrinol. 2017, 8, 160. [Google Scholar] [CrossRef]
- WHO. The Global Health Observatory. Available online: https://www.who.int/data/gho (accessed on 27 February 2024).
- Noubiap, J.J.; Nansseu, J.R.; Lontchi-Yimagou, E.; Nkeck, J.R.; Nyaga, U.F.; Ngouo, A.T.; Tounouga, D.N.; Tianyi, F.L.; Foka, A.J.; Ndoadoumgue, A.L.; et al. Geographic distribution of metabolic syndrome and its components in the general adult population: A meta-analysis of global data from 28 million individuals. Diabetes Res. Clin. Pract. 2022, 188, 109924. [Google Scholar] [CrossRef] [PubMed]
- Ostchega, Y.; Fryar, C.D.; Nwankwo, T.; Nguyen, D.T. Hypertension Prevalence among Adults Aged 18 and Over: United States, 2017–2018; NCHS Data Brief, no 364; National Center for Health Statistics: Hyattsville, MD, USA, 2020. Available online: https://www.cdc.gov/nchs/products/databriefs/db364.htm (accessed on 15 March 2024).
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef]
- Hirode, G.; Wong, R.J. Trends in the Prevalence of Metabolic Syndrome in the United States, 2011–2016. JAMA 2020, 323, 2526–2528. [Google Scholar] [CrossRef]
- Prausmüller, S.; Weidenhammer, A.; Heitzinger, G.; Spinka, G.; Goliasch, G.; Arfsten, H.; Abdel Mawgoud, R.; Gabler, C.; Strunk, G.; Hengstenberg, C.; et al. Obesity in heart failure with preserved ejection fraction with and without diabetes: Risk factor or innocent bystander? Eur. J. Prev. Cardiol. 2023, 30, 1247–1254. [Google Scholar] [CrossRef]
- Burger, P.M.; Koudstaal, S.; Dorresteijn, J.A.N.; Savarese, G.; van der Meer, M.G.; de Borst, G.J.; Mosterd, A.; Visseren, F.L.J. UCC-SMART study group. Metabolic syndrome and risk of incident heart failure in non-diabetic patients with established cardiovascular disease. Int. J. Cardiol. 2023, 379, 66–375. [Google Scholar]
- Teramoto, K.; Teng, T.K.; Chandramouli, C.; Tromp, J.; Sakata, Y.; Lam, C.S. Epidemiology and Clinical Features of Heart Failure with Preserved Ejection Fraction. Card. Fail. Rev. 2022, 8, e27. [Google Scholar] [CrossRef] [PubMed]
- Kosiborod, M.N.; Abildstrøm, S.Z.; Borlaug, B.A.; Butler, J.; Rasmussen, S.; Davies, M.; Hovingh, G.K.; Kitzman, D.W.; Lindegaard, M.L.; Møller, D.V.; et al. Semaglutide in Patients with Heart Failure with Preserved Ejection Fraction and Obesity. N. Engl. J. Med. 2023, 389, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Lasbleiz, A.; Gaborit, B.; Soghomonian, A.; Bartoli, A.; Ancel, P.; Jacquier, A.; Dutour, A. COVID-19 and Obesity: Role of Ectopic Visceral and Epicardial Adipose Tissues in Myocardial Injury. Front. Endocrinol. 2021, 12, 726967. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Park, S.M. Epicardial Adipose Tissue and Heart Failure, Friend or Foe? Diabetes Metab. J. 2024. ahead of print. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef]
- Chen, H.; Liu, L.; Li, M.; Zhu, D.; Tian, G. Epicardial Adipose Tissue-Derived Leptin Promotes Myocardial Injury in Metabolic Syndrome Rats Through PKC/NADPH Oxidase/ROS Pathway. J. Am. Heart Assoc. 2023, 15, e029415. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xiao, C.; Sun, L.; Zhang, F. Assessment of left atrial function in patients with metabolic syndrome by four-dimensional automatic left atrial quantification. Diabetes Res. Clin. Pract. 2024, 207, 111080. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.P.; Hsu, H.L.; Hung, W.C.; Yu, T.H.; Chen, Y.H.; Chiu, C.A.; Lu, L.F.; Chung, F.M.; Shin, S.J.; Lee, Y.J. Increased epicardial adipose tissue (EAT) volume in type 2 diabetes mellitus and association with metabolic syndrome and severity of coronary atherosclerosis. Clin. Endocrinol. 2009, 70, 876–882. [Google Scholar] [CrossRef]
- Li, Y.; Liu, B.; Li, Y.; Jing, X.; Deng, S.; Yan, Y.; She, Q. Epicardial fat tissue in patients with diabetes mellitus: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 3. [Google Scholar] [CrossRef]
- Lan, Y.; Ma, Q.; Luo, G.; Yang, H.; Li, Y.; Zhang, Q. Epicardial adipose tissue in patients with chronic obstructive pulmonary disease: Systematic review with meta analysis and trial sequential analysis. BMC Pulm. Med. 2023, 23, 241. [Google Scholar] [CrossRef]
- Song, G.; Sun, F.; Wu, D.; Bi, W. Association of epicardial adipose tissues with obstructive sleep apnea and its severity: A meta-analysis study. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1115–1120. [Google Scholar] [CrossRef]
- Conte, M.; Petraglia, L.; Poggio, P.; Valerio, V.; Cabaro, S.; Campana, P.; Comentale, G.; Attena, E.; Russo, V.; Pilato, E.; et al. Inflammation and Cardiovascular Diseases in the Elderly: The Role of Epicardial Adipose Tissue. Front. Med. 2022, 9, 844266. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Maeda, M.; Oba, K.; Maimaituxun, G.; Arasaki, O.; Yagi, S.; Kusunose, K.; Soeki, T.; Yamada, H.; Fukuda, D.; et al. Sex differences in the association between epicardial adipose tissue volume and left atrial volume index. BMC Cardiovasc. Disord. 2024, 24, 46. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat. Rev. Endocrinol. 2015, 11, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wang, X.; Song, G. Association of epicardial adipose tissue with the severity and adverse clinical outcomes of COVID-19: A meta-analysis. Int. J. Infect. Dis. 2022, 120, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Drugs That Ameliorate Epicardial Adipose Tissue Inflammation May Have Discordant Effects in Heart Failure With a Preserved Ejection Fraction as Compared with a Reduced Ejection Fraction. J. Card. Fail. 2019, 25, 986–1003. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef] [PubMed]
- Vilariño-García, T.; Polonio-González, M.L.; Pérez-Pérez, A.; Ribalta, J.; Arrieta, F.; Aguilar, M.; Obaya, J.C.; Gimeno-Orna, J.A.; Iglesias, P.; Navarro, J.; et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 2338. [Google Scholar] [CrossRef]
- Packer, M.; Kitzman, D.W. Obesity-Related Heart Failure With a Preserved Ejection Fraction: The Mechanistic Rationale for Combining Inhibitors of Aldosterone, Neprilysin, and Sodium-Glucose Cotransporter-2. JACC Heart Fail. 2018, 6, 633–639. [Google Scholar] [CrossRef]
- Shibata, R.; Ouchi, N.; Murohara, T. Adiponectin and cardiovascular disease. Circ. J. 2009, 73, 608–614. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, H.; Yin, C.; Lan, X.; Wu, L.; Du, X.; Griffiths, H.R.; Gao, D. Adipokines, Hepatokines and Myokines: Focus on Their Role and Molecular Mechanisms in Adipose Tissue Inflammation. Front. Endocrinol. 2022, 13, 873699. [Google Scholar] [CrossRef]
- Kumada, M.; Kihara, S.; Ouchi, N.; Kobayashi, H.; Okamoto, Y.; Ohashi, K.; Maeda, K.; Nagaretani, H.; Kishida, K.; Maeda, N.; et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 2004, 109, 2046–2049. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ouchi, N.; Kihara, S.; Walsh, K.; Kumada, M.; Abe, Y.; Funahashi, T.; Matsuzawa, Y. Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin. Circ. Res. 2004, 94, e27–e31. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kobayashi, H.; Kihara, S.; Kumada, M.; Sato, K.; Inoue, T.; Funahashi, T.; Walsh, K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J. Biol. Chem. 2004, 279, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Ali Ahmad, M.; Karavetian, M.; Moubareck, C.A.; Wazz, G.; Mahdy, T.; Venema, K. The Association between Peptide Hormones with Obesity and Insulin Resistance Markers in Lean and Obese Individuals in the United Arab Emirates. Nutrients 2022, 14, 1271. [Google Scholar] [CrossRef] [PubMed]
- Sapunar, J.; Fonseca, L.; Molina, V.; Ortiz, E.; Barra, M.I.; Reimer, C.; Charles, M.; Schneider, C.; Ortiz, M.; Brito, R.; et al. Adenovirus 36 seropositivity is related to obesity risk, glycemic control, and leptin levels in Chilean subjects. Int. J. Obes. 2020, 44, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Slouha, E.; Elkersh, E.M.; Shay, A.; Ghosh, S.; Mahmood, A.; Gorantla, V.R. Significance of Hormone Alteration Following Bariatric Surgery. Cureus 2023, 15, e49053. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zeng, J.; Zou, F.; Zhang, X.; Qian, Z.; Wang, Y.; Hou, X.; Zou, J. Causal effect of central obesity on left ventricular structure and function in preserved EF population: A Mendelian randomization study. Front. Cardiovasc. Med. 2023, 9, 1103011. [Google Scholar] [CrossRef] [PubMed]
- Turkbey, E.B.; McClelland, R.L.; Kronmal, R.A.; Burke, G.L.; Bild, D.E.; Tracy, R.P.; Arai, A.E.; Lima, J.A.; Bluemke, D.A. The impact of obesity on the left ventricle: The Multi-Ethnic Study of Atherosclerosis (MESA). JACC Cardiovasc. Imaging 2010, 3, 266–274. [Google Scholar] [CrossRef]
- Hughes, D.; Aminian, A.; Tu, C.; Okushi, Y.; Saijo, Y.; Wilson, R.; Chan, N.; Kumar, A.; Grimm, R.A.; Griffin, B.P.; et al. Impact of Bariatric Surgery on Left Ventricular Structure and Function. J. Am. Heart Assoc. 2024, 13, e031505. [Google Scholar] [CrossRef]
- Castagneto-Gissey, L.; Angelini, G.; Mingrone, G.; Cavarretta, E.; Tenori, L.; Licari, C.; Luchinat, C.; Tiepner, A.L.; Basso, N.; Bornstein, S.R.; et al. The early reduction of left ventricular mass after sleeve gastrectomy depends on the fall of branched-chain amino acid circulating levels. EBioMedicine 2022, 76, 103864. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, N.; Chen, J.Y.; Aggarwal, R.; Fadel, M.G.; Fehervari, M.; Ashrafian, H. The effects of bariatric surgery on cardiac function: A systematic review and meta-analysis. Int. J. Obes. 2024, 48, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Hsuan, C.F.; Huang, C.K.; Lin, J.W.; Lin, L.C.; Lee, T.L.; Tai, C.M.; Yin, W.H.; Tseng, W.K.; Hsu, K.L.; Wu, C.C. The effect of surgical weight reduction on left ventricular structure and function in severe obesity. Obesity 2010, 18, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Grymyr, L.M.D.; Nadirpour, S.; Gerdts, E.; Nedrebø, B.G.; Hjertaas, J.J.; Matre, K.; Cramariuc, D. One-year impact of bariatric surgery on left ventricular mechanics: Results from the prospective FatWest study. Eur. Heart J. Open 2021, 1, oeab024. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Patel, K.V.; Vaduganathan, M.; Sarma, S.; Haykowsky, M.J.; Berry, J.D.; Lavie, C.J. Physical Activity, Fitness, and Obesity in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2018, 6, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Savji, N.; Meijers, W.C.; Bartz, T.M.; Bhambhani, V.; Cushman, M.; Nayor, M.; Kizer, J.R.; Sarma, A.; Blaha, M.J.; Gansevoort, R.T.; et al. The Association of Obesity and Cardiometabolic Traits with Incident HFpEF and HFrEF. JACC Heart Fail. 2018, 6, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Cordola Hsu, A.R.; Xie, B.; Peterson, D.V.; LaMonte, M.J.; Garcia, L.; Eaton, C.B.; Going, S.B.; Phillips, L.S.; Manson, J.E.; Anton-Culver, H.; et al. Metabolically Healthy/Unhealthy Overweight/Obesity Associations With Incident Heart Failure in Postmenopausal Women: The Women’s Health Initiative. Circ. Heart Fail. 2021, 14, e007297. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Ayers, C.R.; Rohatgi, A.K.; Turer, A.T.; Berry, J.D.; Das, S.R.; Vega, G.L.; Khera, A.; McGuire, D.K.; Grundy, S.M.; et al. Associations of visceral and abdominal subcutaneous adipose tissue with markers of cardiac and metabolic risk in obese adults. Obesity 2013, 21, E439–E447. [Google Scholar] [CrossRef] [PubMed]
- Haykowsky, M.J.; Nicklas, B.J.; Brubaker, P.H.; Hundley, W.G.; Brinkley, T.E.; Upadhya, B.; Becton, J.T.; Nelson, M.D.; Chen, H.; Kitzman, D.W. Regional Adipose Distribution and its Relationship to Exercise Intolerance in Older Obese Patients Who Have Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2018, 6, 640–649. [Google Scholar] [CrossRef]
- Rao, V.N.; Zhao, D.; Allison, M.A.; Guallar, E.; Sharma, K.; Criqui, M.H.; Cushman, M.; Blumenthal, R.S.; Michos, E.D. Adiposity and Incident Heart Failure and its Subtypes: MESA (Multi-Ethnic Study of Atherosclerosis). JACC Heart Fail. 2018, 6, 999–1007. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Kajio, H. Abdominal Obesity Is Associated with an Increased Risk of All-Cause Mortality in Patients with HFpEF. J. Am. Coll. Cardiol. 2017, 70, 2739–2749, Erratum in J. Am. Coll. Cardiol. 2018, 71, 1059. [Google Scholar] [CrossRef]
- Haass, M.; Kitzman, D.W.; Anand, I.S.; Miller, A.; Zile, M.R.; Massie, B.M.; Carson, P.E. Body mass index and adverse cardiovascular outcomes in heart failure patients with preserved ejection fraction: Results from the Irbesartan in Heart Failure with Preserved Ejection Fraction (I-PRESERVE) trial. Circ. Heart Fail. 2011, 4, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.N.V.; Lewis, G.D.; Shah, S.J.; Obokata, M.; Abou-Ezzedine, O.F.; Fudim, M.; Sun, J.L.; Chakraborty, H.; McNulty, S.; LeWinter, M.M.; et al. Characterization of the Obese Phenotype of Heart Failure with Preserved Ejection Fraction: A RELAX Trial Ancillary Study. Mayo Clin. Proc. 2019, 94, 1199–1209. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Jackson, A.M.; Lam, C.S.P.; Redfield, M.M.; Anand, I.S.; Ge, J.; Lefkowitz, M.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; et al. Effects of Sacubitril-Valsartan Versus Valsartan in Women Compared with Men with Heart Failure and Preserved Ejection Fraction: Insights From PARAGON-HF. Circulation 2020, 141, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.; Harty, B.J.; Claggett, B.; Fleg, J.L.; McKinlay, S.M.; Anand, I.S.; Lewis, E.F.; Joseph, J.; Desai, A.S.; Sweitzer, N.K.; et al. Comparison of Outcomes in Patients with Diabetes Mellitus Treated with Versus Without Insulin + Heart Failure with Preserved Left Ventricular Ejection Fraction (from the TOPCAT Study). Am. J. Cardiol. 2019, 123, 611–617. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Faulkner, J.L.; Bruder-Nascimento, T.; Belin de Chantemèle, E.J. The regulation of aldosterone secretion by leptin: Implications in obesity-related cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 63–69. [Google Scholar] [CrossRef]
- Wang, T.J. The natriuretic peptides and fat metabolism. N. Engl. J. Med. 2012, 367, 377–378. [Google Scholar] [CrossRef]
- Vecchiola, A.; García, K.; González-Gómez, L.M.; Tapia-Castillo, A.; Artigas, R.; Baudrand, R.; Kalergis, A.M.; Carvajal, C.A.; Fardella, C.E. Plasminogen Activator Inhibitor-1 and Adiponectin Are Associated with Metabolic Syndrome Components. Am. J. Hypertens. 2022, 35, 311–318. [Google Scholar] [CrossRef]
- Berezin, A.E.; Berezin, A.A.; Lichtenauer, M. Emerging Role of Adipocyte Dysfunction in Inducing Heart Failure Among Obese Patients with Prediabetes and Known Diabetes Mellitus. Front. Cardiovasc. Med. 2020, 7, 583175. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Le Jemtel, T.H.; Samson, R.; Milligan, G.; Jaiswal, A.; Oparil, S. Visceral Adipose Tissue Accumulation and Residual Cardiovascular Risk. Curr. Hypertens. Rep. 2018, 20, 77. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- McLaughlin, T.; Ackerman, S.E.; Shen, L.; Engleman, E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Investig. 2017, 127, 5–13. [Google Scholar] [CrossRef]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef] [PubMed]
- Abad-Jiménez, Z.; López-Domènech, S.; Díaz-Rúa, R.; Iannantuoni, F.; Gómez-Abril, S.Á.; Periañez-Gómez, D.; Morillas, C.; Víctor, V.M.; Rocha, M. Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin. Antioxidants 2020, 9, 892. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, J.; Zeng, Y.; Chen, K.; Wang, C.; Yang, S.; Sun, N.; Chen, H.; Duan, K.; Zeng, G. Pyroptosis: A pro-inflammatory type of cell death in cardiovascular disease. Clin. Chim. Acta 2020, 510, 62–72. [Google Scholar] [CrossRef]
- Dhorepatil, A.; Ball, S.; Ghosh, R.K.; Kondapaneni, M.; Lavie, C.J. Canakinumab: Promises and Future in Cardiometabolic Diseases and Malignancy. Am. J. Med. 2019, 132, 312–324. [Google Scholar] [CrossRef]
- Ghoneim, S.; Shah, A.; Dhorepatil, A.; Butt, M.U.; Waghray, N. The Risk of Cerebrovascular Accidents in Inflammatory Bowel Disease in the United States: A Population-Based National Study. Clin. Exp. Gastroenterol. 2020, 13, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, S.; Dhorepatil, A.; Shah, A.R.; Ram, G.; Ahmad, S.; Kim, C.; Asaad, I. Non-alcoholic steatohepatitis and the risk of myocardial infarction: A population-based national study. World J. Hepatol. 2020, 12, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J. Unfolding Discoveries in Heart Failure. N. Engl. J. Med. 2020, 382, 679–682. [Google Scholar] [CrossRef]
- González, A.; López, B.; Ravassa, S.; San José, G.; Latasa, I.; Butler, J.; Díez, J. Myocardial Interstitial Fibrosis in Hypertensive Heart Disease: From Mechanisms to Clinical Management. Hypertension 2024, 81, 218–228. [Google Scholar] [CrossRef]
- Zhang, Y.; Van Laer, A.O.; Baicu, C.F.; Neff, L.S.; Hoffman, S.; Katz, M.R.; Zeigler, S.M.; Zile, M.R.; Bradshaw, A.D. Phenotypic characterization of primary cardiac fibroblasts from patients with HFpEF. PLoS ONE 2022, 17, e0262479. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, J.X. Sirtuin 3, Endothelial Metabolic Reprogramming, and Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. 2019, 74, 315–323. [Google Scholar] [CrossRef]
- Lebovitz, H.E.; Banerji, M.A. Point: Visceral adiposity is causally related to insulin resistance. Diabetes Care 2005, 28, 2322–2325. [Google Scholar] [CrossRef]
- Rawshani, A.; Eliasson, B.; Rawshani, A.; Henninger, J.; Mardinoglu, A.; Carlsson, Å.; Sohlin, M.; Ljungberg, M.; Hammarstedt, A.; Rosengren, A.; et al. Adipose tissue morphology, imaging and metabolomics predicting cardiometabolic risk and family history of type 2 diabetes in non-obese men. Sci. Rep. 2020, 10, 9973. [Google Scholar] [CrossRef]
- Rao, V.N.; Bush, C.G.; Mongraw-Chaffin, M.; Hall, M.E.; Clark, D., 3rd; Fudim, M.; Correa, A.; Hammill, B.G.; O’Brien, E.; Min, Y.I.; et al. Regional Adiposity and Risk of Heart Failure and Mortality: The Jackson Heart Study. J. Am. Heart Assoc. 2021, 10, e020920. [Google Scholar] [CrossRef]
- Abbasi, S.A.; Hundley, W.G.; Bluemke, D.A.; Jerosch-Herold, M.; Blankstein, R.; Petersen, S.E.; Rider, O.J.; Lima, J.A.; Allison, M.A.; Murthy, V.L.; et al. Visceral adiposity and left ventricular remodeling: The Multi-Ethnic Study of Atherosclerosis. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 667–676. [Google Scholar] [CrossRef]
- Sorimachi, H.; Obokata, M.; Takahashi, N.; Reddy, Y.N.V.; Jain, C.C.; Verbrugge, F.H.; Koepp, K.E.; Khosla, S.; Jensen, M.D.; Borlaug, B.A. Pathophysiologic importance of visceral adipose tissue in women with heart failure and preserved ejection fraction. Eur. Heart J. 2021, 42, 1595–1605. [Google Scholar] [CrossRef]
- Yafei, S.; Elsewy, F.; Youssef, E.; Ayman, M.; Elshafei, M.; Abayazeed, R. Echocardiographic association of epicardial fat with carotid intima-media thickness in patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2019, 16, 378–384. [Google Scholar] [CrossRef]
- Marchington, J.M.; Mattacks, C.A.; Pond, C.M. Adipose tissue in the mammalian heart and pericardium: Structure, foetal development and biochemical properties. Comp. Biochem. Physiol. B 1989, 94, 225–232. [Google Scholar] [CrossRef]
- Malavazos, A.E.; Iacobellis, G.; Dozio, E.; Basilico, S.; Di Vincenzo, A.; Dubini, C.; Menicanti, L.; Vianello, E.; Meregalli, C.; Ruocco, C.; et al. Human epicardial adipose tissue expresses glucose-dependent insulinotropic polypeptide, glucagon, and glucagon-like peptide-1 receptors as potential targets of pleiotropic therapies. Eur. J. Prev. Cardiol. 2023, 30, 680–693. [Google Scholar] [CrossRef]
- Jakubowska, A.; Roux, C.W.L.; Viljoen, A. The Road towards Triple Agonists: Glucagon-Like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide and Glucagon Receptor—An Update. Endocrinol. Metab. 2024, 39, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Kalogeropoulos, A.; Georgiopoulou, V.; de Rekeneire, N.; Rodondi, N.; Smith, A.L.; Hoffmann, U.; Kanaya, A.; Newman, A.B.; Kritchevsky, S.B.; et al. Serum resistin concentrations and risk of new onset heart failure in older persons: The health, aging, and body composition (Health ABC) study. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1144–11449. [Google Scholar] [CrossRef] [PubMed]
- Brankovic, M.; Akkerhuis, K.M.; van Boven, N.; Anroedh, S.; Constantinescu, A.; Caliskan, K.; Manintveld, O.; Cornel, J.H.; Baart, S.; Rizopoulos, D.; et al. Patient-specific evolution of renal function in chronic heart failure patients dynamically predicts clinical outcome in the Bio-SHiFT study. Kidney Int. 2018, 93, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Acquarone, E.; Monacelli, F.; Borghi, R.; Nencioni, A.; Odetti, P. Resistin: A reappraisal. Mech. Ageing Dev. 2019, 178, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, S.W.; Lee, J.S.; Lee, S.K.; Abbott, R.; Lee, K.Y.; Lim, H.E.; Sung, K.C.; Cho, G.Y.; Koh, K.K.; et al. Association of pericardial adipose tissue with left ventricular structure and function: A region-specific effect? Cardiovasc. Diabetol. 2021, 20, 26. [Google Scholar] [CrossRef]
- Ng, A.C.T.; Strudwick, M.; van der Geest, R.J.; Ng, A.C.C.; Gillinder, L.; Goo, S.Y.; Cowin, G.; Delgado, V.; Wang, W.Y.S.; Bax, J.J. Impact of Epicardial Adipose Tissue, Left Ventricular Myocardial Fat Content, and Interstitial Fibrosis on Myocardial Contractile Function. Circ. Cardiovasc. Imaging 2018, 11, e007372. [Google Scholar] [CrossRef]
- Oduah, M.T.; Sundaram, V.; Reddy, Y.N. Epicardial Fat in Heart Failure with Preserved Ejection Fraction: Bad Actor or Just Lying Around? Card. Fail. Rev. 2023, 9, e06. [Google Scholar] [CrossRef] [PubMed]
- Zamani, S.K.; Sarma, S.; MacNamara, J.P.; Hynan, L.S.; Haykowsky, M.J.; Hearon, C.M.; Wakeham, D.; Brazile, T.; Levine, B.D.; Zaha, V.G.; et al. Excess Pericardial Fat Is Related to Adverse Cardio-Mechanical Interaction in Heart Failure with Preserved Ejection Fraction. Circulation 2023, 148, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Backhaus, S.J.; Lange, T.; Evertz, R.; Kutty, S.; Kowallick, J.T.; Hasenfuß, G.; Schuster, A. Impact of epicardial adipose tissue on cardiac function and morphology in patients with diastolic dysfunction. ESC Heart Fail. 2024. ahead of print. [Google Scholar] [CrossRef]
- Henry, J.A.; Abdesselam, I.; Deal, O.; Lewis, A.J.; Rayner, J.; Bernard, M.; Dutour, A.; Gaborit, B.; Kober, F.; Soghomonian, A.; et al. Changes in epicardial and visceral adipose tissue depots following bariatric surgery and their effect on cardiac geometry. Front. Endocrinol. 2023, 14, 1092777. [Google Scholar] [CrossRef] [PubMed]
- Crum, Y.; Hoendermis, E.S.; van Veldhuisen, D.J.; van Woerden, G.; Lobeek, M.; Dickinson, M.G.; Meems, L.M.G.; Voors, A.A.; Rienstra, M.; Gorter, T.M. Epicardial adipose tissue and pericardial constraint in heart failure with preserved ejection fraction. ESC Heart Fail. 2024. ahead of print. [Google Scholar] [CrossRef]
- Pugliese, N.R.; Paneni, F.; Mazzola, M.; De Biase, N.; Del Punta, L.; Gargani, L.; Mengozzi, A.; Virdis, A.; Nesti, L.; Taddei, S.; et al. Impact of epicardial adipose tissue on cardiovascular haemodynamics, metabolic profile, and prognosis in heart failure. Eur. J. Heart Fail. 2021, 23, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Kenchaiah, S.; Ding, J.; Carr, J.J.; Allison, M.A.; Budoff, M.J.; Tracy, R.P.; Burke, G.L.; McClelland, R.L.; Arai, A.E.; Bluemke, D.A. Pericardial Fat and the Risk of Heart Failure. J. Am. Coll. Cardiol. 2021, 77, 2638–2652. [Google Scholar] [CrossRef]
- He, S.; Zhu, H.; Zhang, J.; Yang, X.; Zhao, L. Genome-wide screening for circRNAs in epicardial adipose tissue of heart failure patients with preserved ejection fraction. Am. J. Transl. Res. 2023, 15, 4610–4619. [Google Scholar]
- Lv, J.; Liu, Y.; Yan, Y.; Sun, D.; Fan, L.; Guo, Y.; Fernandez, C.; Bazzano, L.; He, J.; Li, S.; et al. Relationship Between Left Ventricular Hypertrophy and Diabetes Is Likely Bidirectional: A Temporality Analysis. J. Am. Heart Assoc. 2023, 12, e028219. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.M.; Liu, S.C.; Leung, C.H.; Lee, Y.T.; Chien, K.L. High left ventricular mass associated with increased risk of incident diabetes. Sci. Rep. 2024, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Avagimyan, A.; Fogacci, F.; Pogosova, N.; Kakrurskiy, L.; Kogan, E.; Urazova, O.; Kobalava, Z.; Mikhaleva, L.; Vandysheva, R.; Zarina, G.; et al. Diabetic Cardiomyopathy: 2023 Update by the International Multidisciplinary Board of Experts. Curr. Probl. Cardiol. 2024, 49 Pt A, 102052. [Google Scholar] [CrossRef]
- Vig, H.; Ravinandan, A.P.; Vishwas, H.N.; Tyagi, S.; Rathore, S.; Wal, A.; Wal, P. An Insight into the Pathogenesis of Diabetic Cardiomyopathy Along with the Novel Potential Therapeutic Approaches. Curr. Diabetes Rev. 2024, 20, e020523216416. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B.; Sun, Q.; Lopaschuk, G.D. Concurrent diabetes and heart failure: Interplay and novel therapeutic approaches. Cardiovasc. Res. 2022, 118, 686–715. [Google Scholar] [CrossRef] [PubMed]
- Abudureyimu, M.; Luo, X.; Wang, X.; Sowers, J.R.; Wang, W.; Ge, J.; Ren, J.; Zhang, Y. Heart failure with preserved ejection fraction (HFpEF) in type 2 diabetes mellitus: From pathophysiology to therapeutics. J. Mol. Cell Biol. 2022, 14, mjac028. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Catrinoiu, D.; Chandramouli, C.; Cosentino, F.; Dombrowsky, A.C.; Itzhak, B.; Lalic, N.M.; Prattichizzo, F.; Schnell, O.; Seferović, P.M.; et al. Heart failure in type 2 diabetes: Current perspectives on screening, diagnosis and management. Cardiovasc. Diabetol. 2021, 20, 218. [Google Scholar] [CrossRef] [PubMed]
- Gallego, M.; Zayas-Arrabal, J.; Alquiza, A.; Apellaniz, B.; Casis, O. Electrical Features of the Diabetic Myocardium. Arrhythmic and Cardiovascular Safety Considerations in Diabetes. Front. Pharmacol. 2021, 12, 687256. [Google Scholar] [CrossRef]
- Frisk, M.; Le, C.; Shen, X.; Røe, Å.T.; Hou, Y.; Manfra, O.; Silva, G.J.J.; van Hout, I.; Norden, E.S.; Aronsen, J.M.; et al. Etiology-Dependent Impairment of Diastolic Cardiomyocyte Calcium Homeostasis in Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2021, 77, 405–419. [Google Scholar] [CrossRef]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef]
- Lejeune, S.; Roy, C.; Slimani, A.; Pasquet, A.; Vancraeynest, D.; Vanoverschelde, J.L.; Gerber, B.L.; Beauloye, C.; Pouleur, A.C. Diabetic phenotype and prognosis of patients with heart failure and preserved ejection fraction in a real life cohort. Cardiovasc. Diabetol. 2021, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, N.R.; De Biase, N.; Conte, L.; Gargani, L.; Mazzola, M.; Fabiani, I.; Natali, A.; Dini, F.L.; Frumento, P.; Rosada, J.; et al. Cardiac Reserve and Exercise Capacity: Insights from Combined Cardiopulmonary and Exercise Echocardiography Stress Testing. J. Am. Soc. Echocardiogr. 2021, 34, 38–50. [Google Scholar] [CrossRef]
- Espino-Gonzalez, E.; Tickle, P.G.; Benson, A.P.; Kissane, R.W.P.; Askew, G.N.; Egginton, S.; Bowen, T.S. Abnormal skeletal muscle blood flow, contractile mechanics and fibre morphology in a rat model of obese-HFpEF. J. Physiol. 2021, 599, 981–1001. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, B.; Haykowsky, M.J.; Eggebeen, J.; Kitzman, D.W. Sarcopenic obesity and the pathogenesis of exercise intolerance in heart failure with preserved ejection fraction. Curr. Heart Fail. Rep. 2015, 12, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Levelt, E.; Rodgers, C.T.; Clarke, W.T.; Mahmod, M.; Ariga, R.; Francis, J.M.; Liu, A.; Wijesurendra, R.S.; Dass, S.; Sabharwal, N.; et al. Cardiac energetics, oxygenation, and perfusion during increased workload in patients with type 2 diabetes mellitus. Eur. Heart J. 2016, 37, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.; Pugliese, N.R.; Sciuto, P.; Natali, A. Type 2 diabetes and reduced exercise tolerance: A review of the literature through an integrated physiology approach. Cardiovasc. Diabetol. 2020, 19, 134. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, C.; Kozawa, J.; Hosakawa, Y.; Yoneda, S.; Kimura, T.; Fujita, Y.; Fukui, K.; Iwahashi, H.; Shimomura, I. Pancreatic fat is related to the longitudinal decrease in the increment of C-peptide in glucagon stimulation test in type 2 diabetes patients. J. Diabetes Investig. 2020, 11, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Launbo, N.; Zobel, E.H.; von Scholten, B.J.; Faerch, K.; Jørgensen, P.G.; Christensen, R.H. Targeting epicardial adipose tissue with exercise, diet, bariatric surgery or pharmaceutical interventions: A systematic review and meta-analysis. Obes. Rev. 2021, 22, e13136. [Google Scholar] [CrossRef]
- Anagnostopoulos, I.; Kousta, M.; Kossyvakis, C.; Paraskevaidis, N.T.; Vrachatis, D.; Deftereos, S.; Giannopoulos, G. Epicardial Adipose Tissue and Atrial Fibrillation Recurrence following Catheter Ablation: A Systematic Review and Meta-Analysis. J. Clin. Med. 2023, 12, 6369. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Mohseni, M.; Bianco, S.D.; Banga, P.K. Liraglutide causes large and rapid epicardial fat reduction. Obesity 2017, 25, 311–316. [Google Scholar] [CrossRef]
- Cohen, J.B.; Schrauben, S.J.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Yarde, M.; Wang, Z.; Bhattacharya, P.T.; Chirinos, D.A.; et al. Clinical Phenogroups in Heart Failure with Preserved Ejection Fraction: Detailed Phenotypes, Prognosis, and Response to Spironolactone. JACC Heart Fail. 2020, 8, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Elkholey, K.; Papadimitriou, L.; Butler, J.; Thadani, U.; Stavrakis, S. Effect of Obesity on Response to Spironolactone in Patients with Heart Failure with Preserved Ejection Fraction. Am. J. Cardiol. 2021, 146, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Qin, L.; Wang, L.; Guo, Y.; Zhang, X.; Yu, J. Effectiveness and Safety of Sacubitril/Valsartan in Heart Failure with Preserved Ejection Fraction: A Systematic Review and Meta-Analysis. Altern. Ther. Health Med. 2023. ahead of print. [Google Scholar] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction: A Review. JAMA 2023, 329, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Keidar, S.; Strizevsky, A.; Raz, A.; Gamliel-Lazarovich, A. ACE2 activity is increased in monocyte-derived macrophages from prehypertensive subjects. Nephrol. Dial. Transplant. 2007, 22, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Tang, W.H.; Chen, S.Y.; Van Lente, F.; Francis, G.S.; Sen, S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: Insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J. Am. Coll. Cardiol. 2008, 52, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Valderas, P.; Moya, J.; Gabrielli, L.; Godoy, I.; Córdova, S.; Nab, P.M.; García, L.; Farías, L.; Jalil, J.E. Rho kinase cascade activation in circulating leukocytes in patients with diabetes mellitus type 2. Cardiovasc. Diabetol. 2020, 19, 56. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Moya, J.; Jalil, J.E.; Lavandero, S.; Kalergis, A.M.; Molina, C.; Gabrielli, L.; Godoy, I.; Córdova, S.; Castro, P.; et al. Rho-kinase pathway activation and apoptosis in circulating leucocytes in patients with heart failure with reduced ejection fraction. J. Cell Mol. Med. 2020, 24, 1413–1427. [Google Scholar] [CrossRef]
- Antlanger, M.; Domenig, O.; Kaltenecker, C.C.; Kovarik, J.J.; Rathkolb, V.; Müller, M.M.; Schwaiger, E.; Hecking, M.; Poglitsch, M.; Säemann, M.D.; et al. Combined sodium glucose co-transporter-2 inhibitor and angiotensin-converting enzyme inhibition upregulates the renin-angiotensin system in chronic kidney disease with type 2 diabetes: Results of a randomized, double-blind, placebo-controlled exploratory trial. Diabetes Obes. Metab. 2022, 24, 816–826. [Google Scholar]
- El-Sayed Moustafa, J.S.; Jackson, A.U.; Brotman, S.M.; Guan, L.; Villicaña, S.; Roberts, A.L.; Zito, A.; Bonnycastle, L.; Erdos, M.R.; Narisu, N.; et al. ACE2 expression in adipose tissue is associated with cardio-metabolic risk factors and cell type composition-implications for COVID-19. Int. J. Obes. 2022, 46, 1478–1486. [Google Scholar] [CrossRef]
- Fernandes, F.B.; Fernandes, A.B.; Febba, A.C.S.; Leite, A.P.O.; Leite, C.A.; Vitalle, M.S.S.; Jung, F.F.; Casarini, D.E. Association of Ang-(1-7) and des-Arg9BK as new biomarkers of obesity and cardiometabolic risk factors in adolescents. Hypertens. Res. 2021, 44, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The Ascending GLP-1 Road From Clinical Safety to Reduction of Cardiovascular Complications. Diabetes 2018, 67, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Le glucagon-like peptide-1 (GLP-1), nouvelle cible dans le traitement du diabète de type 2 [Glucagon-like peptide-1 (GLP-1), new target for the treatment of type 2 diabetes. Rev. Med. Liege 2007, 62, 217–221. [Google Scholar]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Noyan-Ashraf, M.H.; Momen, M.A.; Ban, K.; Sadi, A.M.; Zhou, Y.Q.; Riazi, A.M.; Baggio, L.L.; Henkelman, R.M.; Husain, M.; Drucker, D.J. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 2009, 58, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, V.; Sbraccia, P. GLP-1 receptor independent pathways: Emerging beneficial effects of GLP-1 breakdown products. Eat. Weight Disord. 2017, 22, 231–240. [Google Scholar] [CrossRef]
- Pan, X.; Xu, S.; Li, J.; Tong, N. The Effects of DPP-4 Inhibitors, GLP-1RAs, and SGLT-2/1 Inhibitors on Heart Failure Outcomes in Diabetic Patients with and Without Heart Failure History: Insights From CVOTs and Drug Mechanism. Front. Endocrinol. 2020, 11, 599355. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Krauthamer, M.; Bjalme-Evans, M. Wegovy (semaglutide): A new weight loss drug for chronic weight management. J. Investig. Med. 2022, 70, 5–13. [Google Scholar] [CrossRef]
- Miñambres, I.; Pérez, A. Is there a justification for classifying GLP-1 receptor agonists as basal and prandial? Diabetol. Metab. Syndr. 2017, 9, 6. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Abdalla, M.A.; Sathyapalan, T.; Banach, M.; Jamialahmadi, T.; Sahebkar, A. The Effects of Glucagon-Like Peptide-1 Receptor Agonists and Dipeptydilpeptidase-4 Inhibitors on Blood Pressure and Cardiovascular Complications in Diabetes. J. Diabetes Res. 2021, 2021, 6518221. [Google Scholar] [CrossRef]
- Le, Y.; Zheng, Z.; Xue, J.; Cheng, M.; Guan, M.; Xue, Y. Effects of exendin-4 on the intrarenal renin-angiotensin system and interstitial fibrosis in unilateral ureteral obstruction mice: Exendin-4 and unilateral ureteral obstruction. J. Renin Angiotensin Aldosterone Syst. 2016, 17, 1470320316677918. [Google Scholar] [CrossRef]
- Skov, J.; Dejgaard, A.; Frøkiær, J.; Holst, J.J.; Jonassen, T.; Rittig, S.; Christiansen, J.S. Glucagon-like peptide-1 (GLP-1): Effect on kidney hemodynamics and renin-angiotensin-aldosterone system in healthy men. J Clin Endocrinol Metab. 2013, 98, E664–E671. [Google Scholar] [CrossRef]
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor Mediates Cardiovascular Protection by Liraglutide in Mice with Experimental Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef]
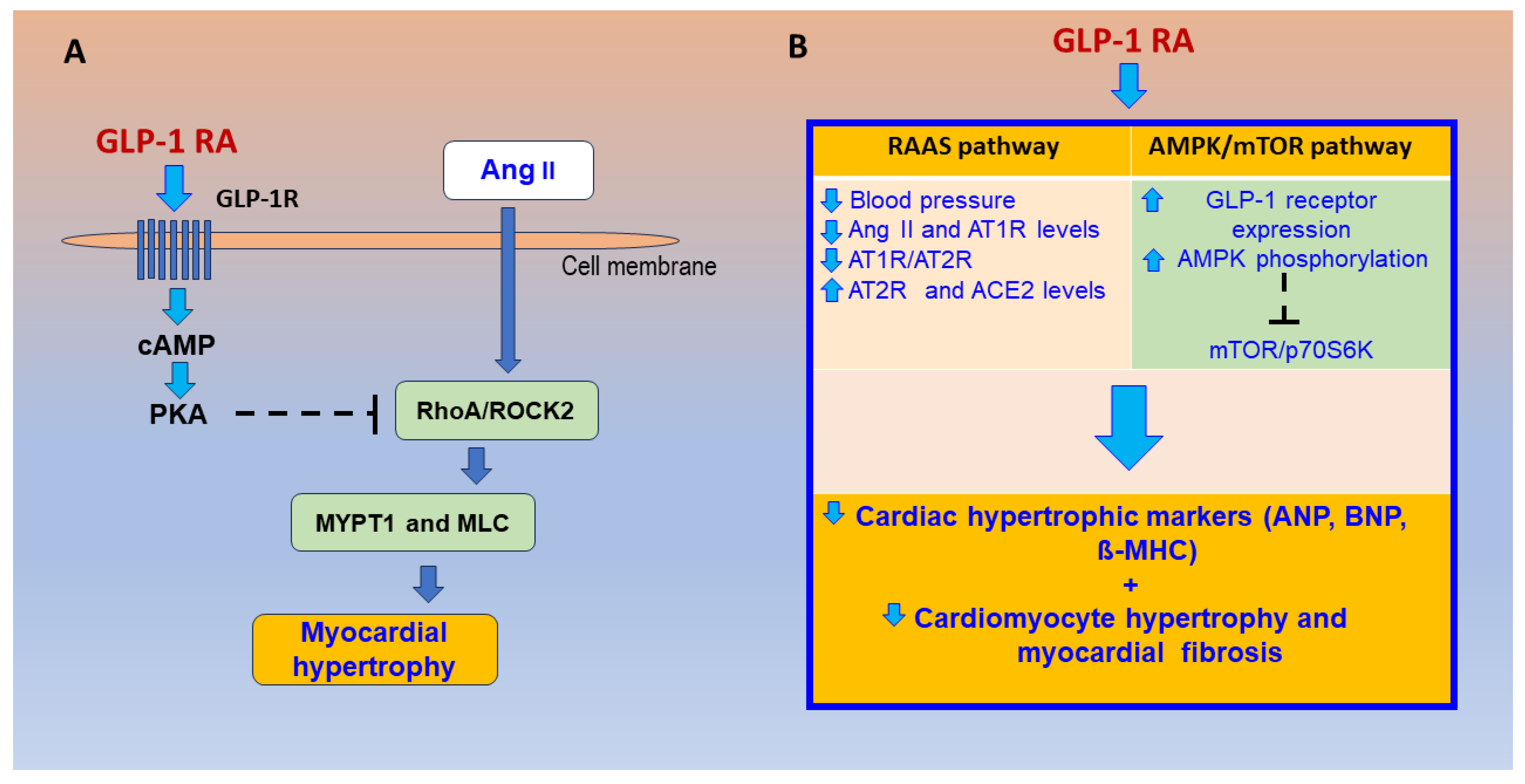
- Fan, S.; Xiong, Q.; Zhang, X.; Zhang, L.; Shi, Y. Glucagon-like peptide 1 reverses myocardial hypertrophy through cAMP/PKA/RhoA/ROCK2 signaling. Acta Biochim. Biophys. Sin. 2020, 52, 612–619. [Google Scholar] [CrossRef]
- Petersen, N.; Frimurer, T.M.; Terndrup Pedersen, M.; Egerod, K.L.; Wewer Albrechtsen, N.J.; Holst, J.J.; Grapin-Botton, A.; Jensen, K.B.; Schwartz, T.W. Inhibiting RHOA Signaling in Mice Increases Glucose Tolerance and Numbers of Enteroendocrine and Other Secretory Cells in the Intestine. Gastroenterology 2018, 155, 1164–1176.e2. [Google Scholar]
- Wu, L.; Wang, K.; Wang, W.; Wen, Z.; Wang, P.; Liu, L.; Wang, D.W. Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARα pathway. Aging Cell 2018, 17, e12763. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, S.; Xiong, Q.; Niu, Y.; Zhang, X.; Qin, J.; Shi, Y.; Zhang, L. Glucagon-like peptide-1 attenuates cardiac hypertrophy via the AngII/AT1R/ACE2 and AMPK/mTOR/p70S6K pathways. Acta Biochim. Biophys. Sin. 2021, 53, 1189–1197. [Google Scholar] [CrossRef]
- de Marañón, A.M.; Díaz-Pozo, P.; Canet, F.; Díaz-Morales, N.; Abad-Jiménez, Z.; López-Domènech, S.; Vezza, T.; Apostolova, N.; Morillas, C.; Rocha, M.; et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 2022, 53, 102342. [Google Scholar] [CrossRef] [PubMed]
- Baek, C.H.; Kim, H.; Moon, S.Y.; Yang, W.S. Liraglutide, a glucagon-like peptide-1 receptor agonist, induces ADAM10-dependent ectodomain shedding of RAGE via AMPK activation in human aortic endothelial cells. Life Sci. 2022, 292, 120331. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chen, J.H.; Lin, L.J.; Liao, J.K. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J. Am. Coll. Cardiol. 2007, 49, 1619–1624. [Google Scholar] [CrossRef]
- Leguina-Ruzzi, A.; Pereira, J.; Pereira-Flores, K.; Valderas, J.P.; Mezzano, D.; Velarde, V.; Sáez, C.G. Increased RhoA/Rho-Kinase Activity and Markers of Endothelial Dysfunction in Young Adult Subjects with Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Jalil, J.E.; Altamirano, R.; de León, A.; Moya, J.; Lonis, A.; Gabrielli, L.; Nab, P.M.; Córdova, S.; Paredes, A.; et al. Reverse Remodeling in Human Heart Failure after Cardiac Resynchronization Therapy Is Associated with Reduced RHO-Kinase Activation. Front. Pharmacol. 2021, 12, 565724. [Google Scholar] [CrossRef]
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.W.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, T.; Ozeki, A.; Asai, K.; Saka, M.; Hobo, A.; Furuta, S. Liraglutide Improves Glycemic and Blood Pressure Control and Ameliorates Progression of Left Ventricular Hypertrophy in Patients with Type 2 Diabetes Mellitus on Peritoneal Dialysis. Ther. Apher. Dial. 2015, 19, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, T.; Ozeki, A.; Ishikawa, H.; Furuta, S. Long Term Effects of Liraglutide in Japanese Patients with type 2 Diabetes Among the Subgroups with Different Renal Functions: Results of 2-Year Prospective Study. Drug Res. 2017, 67, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, M.; Li, T.; Dong, N.; Yi, L.; Zhang, Q.; Mi, M. GLP-1 regulates exercise endurance and skeletal muscle remodeling via GLP-1R/AMPK pathway. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119300. [Google Scholar] [CrossRef] [PubMed]
- Kosiborod, M.N.; Verma, S.; Borlaug, B.A.; Butler, J.; Davies, M.J.; Jon Jensen, T.; Rasmussen, S.; Erlang Marstrand, P.; Petrie, M.C.; Shah, S.J.; et al. Effects of Semaglutide on Symptoms, Function, and Quality of Life in Patients with Heart Failure with Preserved Ejection Fraction and Obesity: A Prespecified Analysis of the STEP-HFpEF Trial. Circulation 2024, 149, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Abildstrøm, S.Z.; Borlaug, B.A.; Davies, M.J.; Kitzman, D.W.; Petrie, M.C.; Shah, S.J.; Verma, S.; Abhayaratna, W.P.; Chopra, V.; et al. Semaglutide in Patients with Obesity and Heart Failure Across Mildly Reduced or Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2023, 82, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, Q.; Yang, X.; Chen, X. Protective effect of glucagon-like peptide-1 agents on reperfusion injury for acute myocardial infarction: A meta-analysis of randomized controlled trials. Ann. Med. 2017, 49, 552–561. [Google Scholar] [CrossRef]
- Khan, M.S.; Fonarow, G.C.; McGuire, D.K.; Hernandez, A.F.; Vaduganathan, M.; Rosenstock, J.; Handelsman, Y.; Verma, S.; Anker, S.D.; McMurray, J.J.V.; et al. Glucagon-Like Peptide 1 Receptor Agonists and Heart Failure: The Need for Further Evidence Generation and Practice Guidelines Optimization. Circulation 2020, 142, 1205–1218. [Google Scholar] [CrossRef]
- Marx, N.; Husain, M.; Lehrke, M.; Verma, S.; Sattar, N. GLP-1 Receptor Agonists for the Reduction of Atherosclerotic Cardiovascular Risk in Patients with Type 2 Diabetes. Circulation 2022, 146, 1882–1894. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785, Erratum in Lancet Diabetes Endocrinol. 2020, 8, e2. [Google Scholar] [CrossRef]
- Avogaro, A.; Azzolina, D.; Gregori, D.; De Kreutzenberg, S.; Fadini, G.P.; Mannucci, E. The effect of GLP-1 receptor agonists on N-terminal pro-brain natriuretic peptide. A scoping review and metanalysis. Int. J. Cardiol. 2022, 357, 123–127. [Google Scholar] [CrossRef]
- Gomes, D.A.; Presume, J.; de Araújo Gonçalves, P.; Almeida, M.S.; Mendes, M.; Ferreira, J. Association Between the Magnitude of Glycemic Control and Body Weight Loss with GLP-1 Receptor Agonists and Risk of Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-analyses of Randomized Diabetes Cardiovascular Outcomes Trials. Cardiovasc. Drugs Ther. 2024. ahead of print. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; Kushner, R.F.; et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 389, 2221–2232. [Google Scholar] [CrossRef]
- Cho, Y.K.; La Lee, Y.; Jung, C.H. The Cardiovascular Effect of Tirzepatide: A Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Polypeptide Dual Agonist. J. Lipid Atheroscler. 2023, 12, 213–222. [Google Scholar] [CrossRef]
- Lv, X.; Wang, H.; Chen, C.; Zhao, Y.; Li, K.; Wang, Y.; Wang, L.; Fu, S.; Liu, J. The Effect of Tirzepatide on Weight, Lipid Metabolism and Blood Pressure in Overweight/Obese Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Diabetes Metab. Syndr. Obes. 2024, 17, 701–714. [Google Scholar] [CrossRef]
- Kaur, M.; Misra, S. A review of an investigational drug retatrutide, a novel triple agonist agent for the treatment of obesity. Eur. J. Clin. Pharmacol. 2024, 80, 669–676. [Google Scholar] [CrossRef]
- Rosenstock, J.; Frias, J.; Jastreboff, A.M.; Du, Y.; Lou, J.; Gurbuz, S.; Thomas, M.K.; Hartman, M.L.; Haupt, A.; Milicevic, Z.; et al. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: A randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet 2023, 402, 529–544. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Kaplan, L.M.; Frías, J.P.; Wu, Q.; Du, Y.; Gurbuz, S.; Coskun, T.; Haupt, A.; Milicevic, Z.; Hartman, M.L.; et al. Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial. N. Engl. J. Med. 2023, 389, 514–526. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Comorbidities and Concomitant Conditions in HFpEF | ||
|---|---|---|

| Modifiable | Non-Modifiable |
| Hypertension Obesity Diabetes Dyslipidemia Metabolic syndrome Renal dysfunction Atrial fibrillation Sleep apnea Pulmonary Hypertension Amyloidosis | Advanced age Female sex | |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalil, J.E.; Gabrielli, L.; Ocaranza, M.P.; MacNab, P.; Fernández, R.; Grassi, B.; Jofré, P.; Verdejo, H.; Acevedo, M.; Cordova, S.; et al. New Mechanisms to Prevent Heart Failure with Preserved Ejection Fraction Using Glucagon-like Peptide-1 Receptor Agonism (GLP-1 RA) in Metabolic Syndrome and in Type 2 Diabetes: A Review. Int. J. Mol. Sci. 2024, 25, 4407. https://doi.org/10.3390/ijms25084407
Jalil JE, Gabrielli L, Ocaranza MP, MacNab P, Fernández R, Grassi B, Jofré P, Verdejo H, Acevedo M, Cordova S, et al. New Mechanisms to Prevent Heart Failure with Preserved Ejection Fraction Using Glucagon-like Peptide-1 Receptor Agonism (GLP-1 RA) in Metabolic Syndrome and in Type 2 Diabetes: A Review. International Journal of Molecular Sciences. 2024; 25(8):4407. https://doi.org/10.3390/ijms25084407
Chicago/Turabian StyleJalil, Jorge E., Luigi Gabrielli, María Paz Ocaranza, Paul MacNab, Rodrigo Fernández, Bruno Grassi, Paulina Jofré, Hugo Verdejo, Monica Acevedo, Samuel Cordova, and et al. 2024. "New Mechanisms to Prevent Heart Failure with Preserved Ejection Fraction Using Glucagon-like Peptide-1 Receptor Agonism (GLP-1 RA) in Metabolic Syndrome and in Type 2 Diabetes: A Review" International Journal of Molecular Sciences 25, no. 8: 4407. https://doi.org/10.3390/ijms25084407
APA StyleJalil, J. E., Gabrielli, L., Ocaranza, M. P., MacNab, P., Fernández, R., Grassi, B., Jofré, P., Verdejo, H., Acevedo, M., Cordova, S., Sanhueza, L., & Greig, D. (2024). New Mechanisms to Prevent Heart Failure with Preserved Ejection Fraction Using Glucagon-like Peptide-1 Receptor Agonism (GLP-1 RA) in Metabolic Syndrome and in Type 2 Diabetes: A Review. International Journal of Molecular Sciences, 25(8), 4407. https://doi.org/10.3390/ijms25084407