
Advancing Adverse Drug Reaction Prediction with Deep Chemical Language Model for Drug Safety Evaluation


Abstract
1. Introduction
2. Results
2.1. Model Performance on Three Curated Datasets
2.2. Attention Analysis for Drugs with a High Risk of DIQT
2.3. Attention Analysis for Antiepileptic Drugs with a High Risk of DIT
2.4. Attention Analysis for Statins
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Data Preparation
4.3. Model Construction
4.4. Evaluation of Model Performance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, Y.; Hu, Y.; Liu, X.; Yin, Z.; Chen, X.-W.; Liu, M. Improving drug safety: From adverse drug reaction knowledge discovery to clinical implementation. Methods 2016, 110, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Poudel, D.R.; Acharya, P.; Ghimire, S.; Dhital, R.; Bharati, R. Burden of hospitalizations related to adverse drug events in the USA: A retrospective analysis from large inpatient database. Pharmacoepidemiol. Drug Saf. 2017, 26, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Hohenegger, M. Drug induced rhabdomyolysis. Curr. Opin. Pharmacol. 2012, 12, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Liu, Z.; Tong, W.; Laaksonen, R.; Bai, J.P.F. Drug-Induced Rhabdomyolysis: From Systems Pharmacology Analysis to Biochemical Flux. Chem. Res. Toxicol. 2014, 27, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Stahl, K.; Rastelli, E.; Schoser, B. A systematic review on the definition of rhabdomyolysis. J. Neurol. 2019, 267, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.R.; Stein, C.M.; Ray, W.A. Postmarketing surveillance for drug safety: Surely we can do better. Clin. Pharmacol. Ther. 2004, 75, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Alomar, M.; Tawfiq, A.M.; Hassan, N.; Palaian, S. Post marketing surveillance of suspected adverse drug reactions through spontaneous reporting: Current status, challenges and the future. Ther. Adv. Drug Saf. 2020, 11, 2042098620938595. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, P.A.-O. Interpretation of Quantitative Structure-Activity Relationship Models: Past, Present, and Future. J. Chem. Inf. Model. 2017, 57, 2618–2639. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Wang, R.; Xue, Y.; Li, Z.R.; Yang, S.Y.; Wei, Y.Q.; Chen, Y.Z. Advances in machine learning prediction of toxicological properties and adverse drug reactions of pharmaceutical agents. Curr. Drug Saf. 2008, 3, 100–114. [Google Scholar] [CrossRef]
- Patlewicz, G.; Fitzpatrick, J.M. Current and Future Perspectives on the Development, Evaluation, and Application of in Silico Approaches for Predicting Toxicity. Chem. Res. Toxicol. 2016, 29, 438–451. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, F.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico Prediction of Drug Induced Liver Toxicity Using Substructure Pattern Recognition Method. Mol. Inform. 2016, 35, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Duan, Y.; Chen, C.; Li, S.; Li, M.; Chen, L.; Wen, Z. Structure-Activity Relationship (SAR) Model for Predicting Teratogenic Risk of Antiseizure Medications in Pregnancy by Using Support Vector Machine. Front. Pharmacol. 2022, 13, 747935. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Li, S.; He, Y.; Lin, J.; Li, M.; Wen, Z. Unraveling Structural Alerts in Marketed Drugs for Improving Adverse Outcome Pathway Framework of Drug-Induced QT Prolongation. Int. J. Mol. Sci. 2023, 24, 6771. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W.; Cai, C.Z.; Xue, Y.; Chen, Y.Z. Prediction of Torsade-Causing Potential of Drugs by Support Vector Machine Approach No funding was used to assist in conducting the study and the authors do not have any conflicts of interest directly relevant to the contents of the manuscript. Toxicol. Sci. 2004, 79, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Zhu XW, L.S. In Silico Prediction of Drug-Induced Liver Injury Based on Adverse Drug Reaction Report. Toxicol. Sci. 2017, 158, 391–400. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, S.; Zhao, Y.; Guo, M.; Liu, Y.; Li, M.; Wen, Z. Quantitative Structure–Activity Relationship (QSAR) Model for the Severity Prediction of Drug-Induced Rhabdomyolysis by Using Random Forest. Chem. Res. Toxicol. 2021, 34, 514–521. [Google Scholar] [CrossRef]
- Jamal, S.; Ali, W.; Nagpal, P.; Grover, S.; Grover, A. Computational models for the prediction of adverse cardiovascular drug reactions. J. Transl. Med. 2019, 17, 171. [Google Scholar] [CrossRef]
- Hong, H.; Thakkar, S.; Chen, M.; Tong, W. Development of Decision Forest Models for Prediction of Drug-Induced Liver Injury in Humans Using A Large Set of FDA-approved Drugs. Sci. Rep. 2017, 7, 17311. [Google Scholar] [CrossRef]
- Chen, M.; Hong, H.; Fang, H.; Kelly, R.; Zhou, G.; Borlak, J.; Tong, W. Quantitative Structure-Activity Relationship Models for Predicting Drug-Induced Liver Injury Based on FDA-Approved Drug Labeling Annotation and Using a Large Collection of Drugs. Toxicol. Sci. 2013, 136, 242–249. [Google Scholar] [CrossRef]
- Minerali, E.; Foil, D.H.; Zorn, K.M.; Lane, T.R.; Ekins, S. Comparing Machine Learning Algorithms for Predicting Drug-Induced Liver Injury (DILI). Mol. Pharm. 2020, 17, 2628–2637. [Google Scholar] [CrossRef]
- Frid, A.A.; Matthews, E.J. Prediction of drug-related cardiac adverse effects in humans-B: Use of QSAR programs for early detection of drug-induced cardiac toxicities. Regul. Toxicol. Pharmacol. 2010, 56, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, Z.; Guo, M.; Li, M.; Wen, Z. Drug-induced QT Prolongation Atlas (DIQTA) for enhancing cardiotoxicity management. Drug Discov. Today 2022, 27, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Liang, Y.; Hao, Y.; Delavan, B.; Huang, R.; Mikailov, M.; Tong, W.; Li, M.; Liu, Z. Drug-Induced Rhabdomyolysis Atlas (DIRA) for idiosyncratic adverse drug reaction management. Drug Discov. Today 2019, 24, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Chen, Y.-P.P. Prediction of drug adverse events using deep learning in pharmaceutical discovery. Brief. Bioinform. 2021, 22, 1884–1901. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, F.; Chen, M. Computational models for predicting liver toxicity in the deep learning era. Front. Toxicol. 2024, 5, 1340860. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.-A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Winter, R.; Noé, F.; Clevert, D.-A. Neuraldecipher–reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structures. Chem. Sci. 2020, 11, 10378–10389. [Google Scholar] [CrossRef] [PubMed]
- Clevert, D.-A.; Le, T.; Winter, R.; Montanari, F. Img2Mol–accurate SMILES recognition from molecular graphical depictions. Chem. Sci. 2021, 12, 14174–14181. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Wu, C.-K.; Yi, J.-C.; Zeng, X.-X.; Yang, C.-Q.; Lu, A.-P.; Hou, T.-J.; Cao, D.-S. Pushing the Boundaries of Molecular Property Prediction for Drug Discovery with Multitask Learning BERT Enhanced by SMILES Enumeration. Research 2022, 2022, 4. [Google Scholar] [CrossRef]
- Li, X.; Fourches, D. SMILES Pair Encoding: A Data-Driven Substructure Tokenization Algorithm for Deep Learning. J. Chem. Inf. Model. 2021, 61, 1560–1569. [Google Scholar] [CrossRef]
- Wu, C.-K.; Wu, C.-K.; Zhang, X.-C.; Zhang, X.-C.; Yang, Z.-J.; Yang, Z.-J.; Lu, A.-P.; Lu, A.-P.; Hou, T.-J.; Hou, T.-J.; et al. Learning to SMILES: BAN-based strategies to improve latent representation learning from molecules. Brief. Bioinform. 2021, 22, bbab327. [Google Scholar] [CrossRef] [PubMed]
- Ucak, U.V.; Ashyrmamatov, I.; Lee, J. Improving the quality of chemical language model outcomes with atom-in-SMILES tokenization. J. Cheminformatics 2023, 15, 55. [Google Scholar] [CrossRef]
- Ross, J.; Belgodere, B.; Chenthamarakshan, V.; Padhi, I.; Mroueh, Y.; Das, P. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 2022, 4, 1256–1264. [Google Scholar] [CrossRef]
- Colatsky, T.; Fermini, B.; Gintant, G.; Pierson, J.B.; Sager, P.; Sekino, Y.; Strauss, D.G.; Stockbridge, N. The Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative—Update on progress. J. Pharmacol. Toxicol. Methods 2016, 81, 15–20. [Google Scholar] [CrossRef]
- Sushko, I.; Salmina, E.; Potemkin, V.A.; Poda, G.; Tetko, I.V. ToxAlerts: A Web Server of Structural Alerts for Toxic Chemicals and Compounds with Potential Adverse Reactions. J. Chem. Inf. Model. 2012, 52, 2310–2316. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.; Dalvie, D.; O’Donnell, J.; Taylor, T.; Sahakian, D. On the Diversity of Oxidative Bioactivation Reactions on Nitrogen- Containing Xenobiotics. Curr. Drug Metab. 2002, 3, 379–424. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S. Designing around Structural Alerts in Drug Discovery. J. Med. Chem. 2019, 63, 6276–6302. [Google Scholar] [CrossRef]
- Islam, I.; Skibo, E.B.; Dorr, R.T.; Alberts, D.S. Structure-activity studies of antitumor agents based on pyrrolo[1,2-a]benzimidazoles: New reductive alkylating DNA cleaving agents. J. Med. Chem. 1991, 34, 2954–2961. [Google Scholar] [CrossRef]
- Tomson, T.; Battino, D.; Bonizzoni, E.; Craig, J.; Lindhout, D.; Sabers, A.; Perucca, E.; Vajda, F. Dose-dependent risk of malformations with antiepileptic drugs: An analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011, 10, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Díaz, S.; Smith, C.R.; Shen, A.; Mittendorf, R.; Hauser, W.A.; Yerby, M.; Holmes, L.B.; For the North American AED Pregnancy Registry; Cragan, J.; Hauser, A.; et al. Comparative safety of antiepileptic drugs during pregnancy. Neurology 2012, 78, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Kennedy, F.; Russell, A.; Smithson, W.H.; Parsons, L.; Morrison, P.J.; Liggan, B.; Irwin, B.; Delanty, N.; Hunt, S.J.; et al. Malformation risks of antiepileptic drug monotherapies in pregnancy: Updated results from the UK and Ireland Epilepsy and Pregnancy Registers. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Tomson, T.; Battino, D.; Bonizzoni, E.; Craig, J.; Lindhout, D.; Perucca, E.; Sabers, A.; Thomas, S.V.; Vajda, F.; Faravelli, F.; et al. Comparative risk of major congenital malformations with eight different antiepileptic drugs: A prospective cohort study of the EURAP registry. Lancet Neurol. 2018, 17, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Gardner, I.; Obach, R.S.; Shaffer, C.L.; Callegari, E.; Henne, K.R.; Mutlib, A.E.; Dalvie, D.K.; Lee, J.S.; Nakai, Y.; et al. A comprehensive listing of bioactivation pathways of organic functional groups.pdf. Curr. Drug Metab. 2005, 6, 161–225. [Google Scholar] [CrossRef]
- Nendza, M.; Wenzel, A.; Müller, M.; Lewin, G.; Simetska, N.; Stock, F.; Arning, J. Screening for potential endocrine disruptors in fish: Evidence from structural alerts and in vitro and in vivo toxicological assays. Environ. Sci. Eur. 2016, 28, 26. [Google Scholar] [CrossRef] [PubMed]
- Stepan, A.F.; Walker, D.P.; Bauman, J.; Price, D.A.; Baillie, T.A.; Kalgutkar, A.S.; Aleo, M.D. Structural Alert/Reactive Metabolite Concept as Applied in Medicinal Chemistry to Mitigate the Risk of Idiosyncratic Drug Toxicity: A Perspective Based on the Critical Examination of Trends in the Top 200 Drugs Marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. [Google Scholar] [CrossRef]
- Bitzur, R.; Cohen, H.; Kamari, Y.; Harats, D. Intolerance to Statins: Mechanisms and Management. Diabetes Care 2013, 36, S325–S330. [Google Scholar] [CrossRef]
- Needham, M.; Mastaglia, F.L. Statin myotoxicity: A review of genetic susceptibility factors. Neuromuscul. Disord. 2014, 24, 4–15. [Google Scholar] [CrossRef]
- Liebler, D.C.; Guengerich, F.P. Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 2005, 4, 410–420. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C. Structure alerts for carcinogenicity, and the Salmonella assay system: A novel insight through the chemical relational databases technology. Mutat. Res./Rev. Mutat. Res. 2008, 659, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| DIQTA | DITD | DIRA | |
|---|---|---|---|
| Accuracy | 0.859 ± 0.029 | 0.750 ± 0.037 | 0.866 ± 0.034 |
| Recall rate | 0.942 ± 0.027 | 0.862 ± 0.160 | 0.974 ± 0.027 |
| Precision | 0.845 ± 0.029 | 0.771 ± 0.010 | 0.872 ± 0.029 |
| MCC | 0.702 ± 0.062 | 0.503 ± 0.047 | 0.535 ± 0.095 |
| BACC | 0.835 ± 0.033 | 0.719 ± 0.058 | 0.703 ± 0.042 |
| F1 score | 0.891 ± 0.022 | 0.800 ± 0.052 | 0.920 ± 0.023 |
| AUROC | 0.829 ± 0.060 | 0.702 ± 0.048 | 0.703 ± 0.042 |
| AUPRC | 0.822 ± 0.079 | 0.742 ± 0.101 | 0.832 ± 0.062 |
| Specificity | 0.747 ± 0.036 | 0.576 ± 0.260 | 0.432 ± 0.088 |
| Generic/ Proper Name(s) | Canonical SMILES | Structures Extracted by Attention Map | SAs* |
|---|---|---|---|
| Quinidine gluconate | C=CC1CN2CCC1CC2C(O)c1ccnc2ccc(OC)cc12 |  |  |
| Vandetanib | COc1cc2c(Nc3ccc(Br)cc3F)ncnc2cc1OCC1CCN(C)CC1 |  | 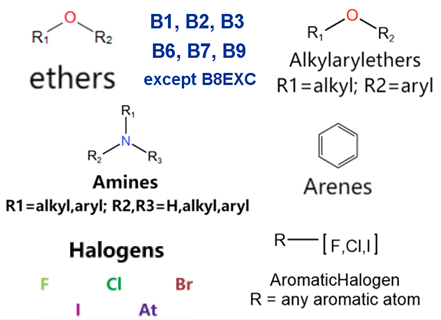 |
| Ibutilide fumarate | CCCCCCCN(CC)CCCC(O)c1ccc(NS(C)(=O)=O)cc1 |  |  |
| Dofetilide | CN(CCOc1ccc(NS(C)(=O)=O)cc1)CCc1ccc(NS(C)(=O)=O)cc1 |  | 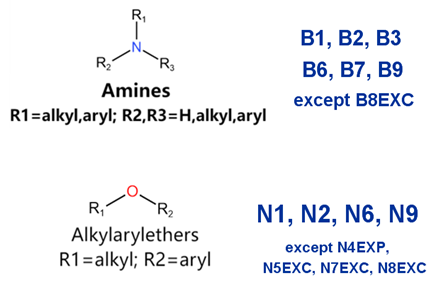 |
| Disopyramide phosphate | CC(C)N(CCC(C(N)=O)(c1ccccc1)c1ccccn1)C(C)C | 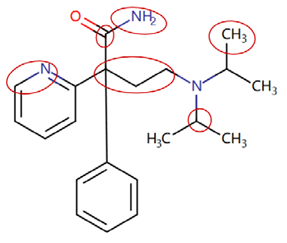 | 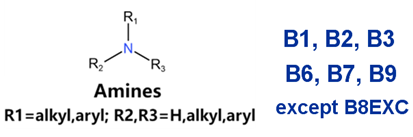 |
| Generic/ Proper Name(s) | Canonical SMILES | Structures Extracted by Attention Map | SAs* |
|---|---|---|---|
| Phenytoin | C1=CC=C(C=C1)C2(C(=O)NC(=O)N2)C3=CC=CC=C3 |  |  |
| Valproate | CCCC(CCC)C(=O)O |  | 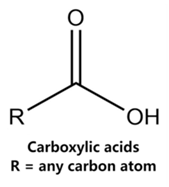 |
| Generic/ Proper Name(s) | Canonical SMILES | Structures Extracted by Attention Map | SAs* |
|---|---|---|---|
| Fluvastatin sodium | CC(C)n1c(C=CC(O)CC(O)CC(=O)O)c(-c2ccc(F)cc2)c2ccccc21 |  |  |
| Lovastatin | CCC(C)C(=O)OC1CC(C)C=C2C=CC(C)C(CCC3CC(O)CC(=O)O3)C21 | 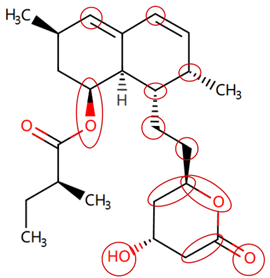 |  |
| Pravastatin sodium | CCC(C)C(=O)OC1CC(O)C=C2C=CC(C)C(CCC(O)CC(O)CC(=O)O)C21 |  |  |
| Atorvastatin calcium | CC(C)c1c(C(=O)Nc2ccccc2)c(-c2ccccc2)c(-c2ccc(F)cc2)n1CCC(O)CC(O)CC(=O)O |  |  |
| Rosuvastatin calcium | CC(C)c1nc(N(C)S(C)(=O)=O)nc(-c2ccc(F)cc2)c1C=CC(O)CC(O)CC(=O)O |  |  |
| Pitavastatin calcium | O=C(O)CC(O)CC(O)C=Cc1c(C2CC2)nc2ccccc2c1-c1ccc(F)cc1 |  |  |
| Simvastatin | CCC(C)(C)C(=O)OC1CC(C)C=C2C=CC(C)C(CCC3CC(O)CC(=O)O3)C21 | 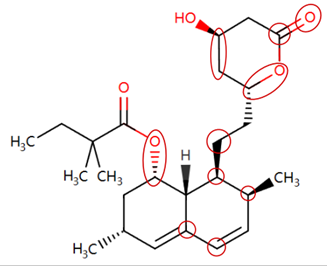 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; He, Y.; Ru, C.; Long, W.; Li, M.; Wen, Z. Advancing Adverse Drug Reaction Prediction with Deep Chemical Language Model for Drug Safety Evaluation. Int. J. Mol. Sci. 2024, 25, 4516. https://doi.org/10.3390/ijms25084516
Lin J, He Y, Ru C, Long W, Li M, Wen Z. Advancing Adverse Drug Reaction Prediction with Deep Chemical Language Model for Drug Safety Evaluation. International Journal of Molecular Sciences. 2024; 25(8):4516. https://doi.org/10.3390/ijms25084516
Chicago/Turabian StyleLin, Jinzhu, Yujie He, Chengxiang Ru, Wulin Long, Menglong Li, and Zhining Wen. 2024. "Advancing Adverse Drug Reaction Prediction with Deep Chemical Language Model for Drug Safety Evaluation" International Journal of Molecular Sciences 25, no. 8: 4516. https://doi.org/10.3390/ijms25084516
APA StyleLin, J., He, Y., Ru, C., Long, W., Li, M., & Wen, Z. (2024). Advancing Adverse Drug Reaction Prediction with Deep Chemical Language Model for Drug Safety Evaluation. International Journal of Molecular Sciences, 25(8), 4516. https://doi.org/10.3390/ijms25084516