
Characterization of Human B Cell Hematological Malignancies Using Protein-Based Approaches

 , , , and
, , , and
Abstract
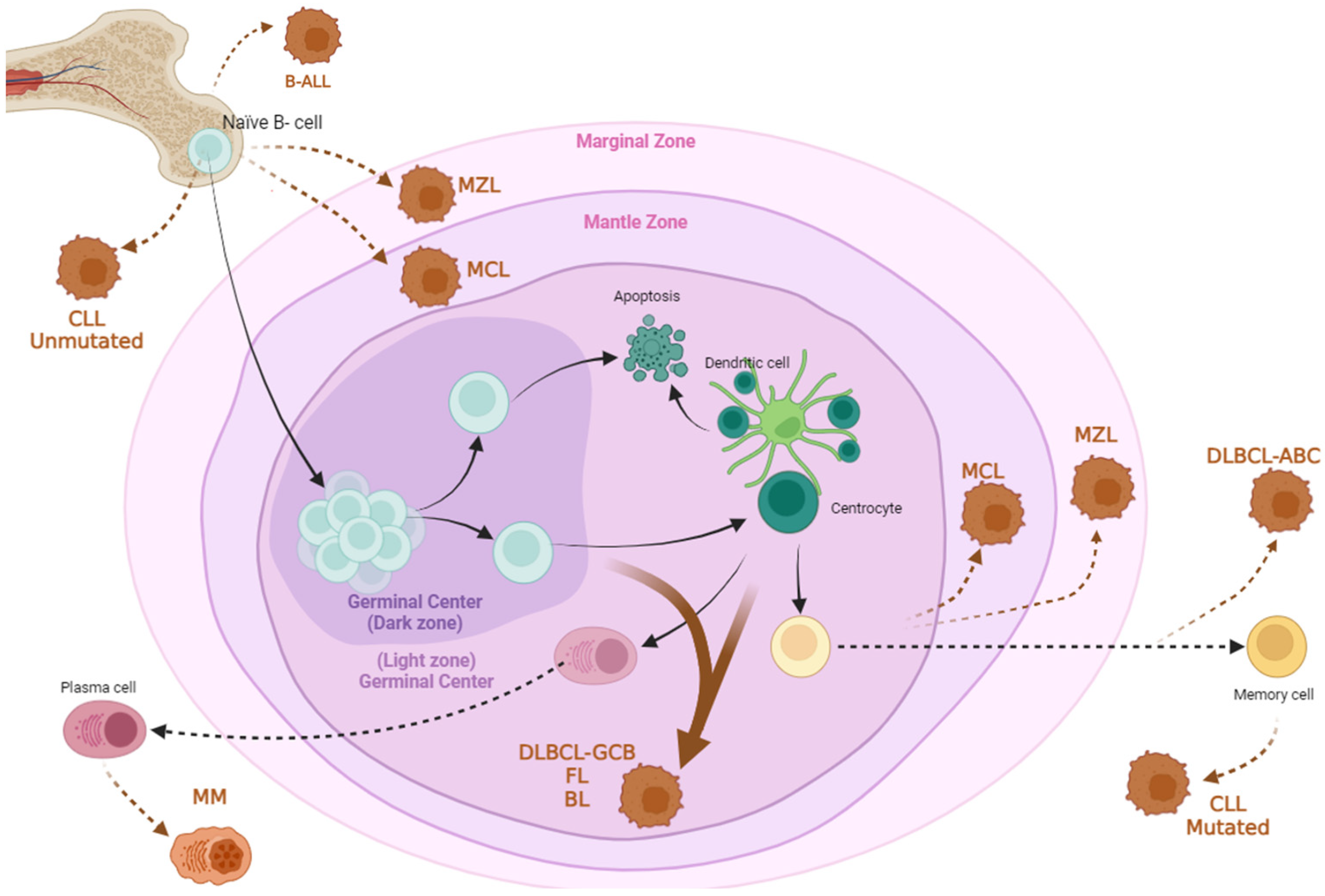
:1. Introduction to B Cell Hematological Disorders
1.1. Leukemias
1.1.1. Chronic Lymphocytic Leukemia (CLL)
1.1.2. Acute Lymphoblastic Leukemia (B-ALL)
1.2. B Cell Lymphomas
1.2.1. Diffuse Large B Cell Lymphoma (DLBCL)
1.2.2. Follicular Lymphoma (FL)

{kind=link}
{kind=link}
{kind=link}
| Leukemia | Lymphoma | Myeloma | ||||||
|---|---|---|---|---|---|---|---|---|
| B-ALL [36,37,38,39,40,41] | CLL [41,42,43,44] | DLBCL [41,44,45,46,47] | FL [41,42,45] | MCL [41,42,44,45] | MZL [41,44,48] | BL [41,45] | MM [41,42,49] | |
| CD3 | − | − | − | − | − | − | − | − |
| CD4 | − | − | − | − | − | − | − | − |
| CD5 | − | + | −/+ | − | + | − | − | − |
| CD7 | − | − | − | − | − | − | − | − |
| CD8 | − | − | − | − | − | − | − | − |
| CD9 | + | |||||||
| CD10 | +/− | − | −/+ | +/− | − | − | + | − |
| CD11c | +/− | −/+ | −/+ | − | + | − | − | |
| CD13 | +/− | |||||||
| CD19 | + | + | + | + | + | + | + | − |
| CD20 | + | low | + | + | + | + | + | dim+ |
| CD21 | − | |||||||
| CD22 | + | − | + | + | + | + | + | |
| CD23 | − | + | − | − | − | − | −/+ | +/− |
| CD24 | + | |||||||
| CD25 | +/− | − | − | − | −/+ | − | − | |
| CD27 | + | + | + | + | + | −/+ | −/dim+ | |
| CD28 | + | |||||||
| CD30 | − | −/+ | − | − | − | |||
| CD33 | +/− | + | ||||||
| CD34 | + | − | − | |||||
| CD38 | + | +/− | −/+ | + | + | +/− | + | + |
| CD43 | + | −/+ | − | + | −/+ | + | +/− | |
| CD44 | low/− | |||||||
| CD45 | + | + | + | + | + | + | + | −/+ |
| CD54 | dim+ | |||||||
| CD56 | − | − | + | |||||
| CD58 | + | |||||||
| CD66c | −/+ | |||||||
| CD73 | −/+ | |||||||
| CD79a | + | + | + | + | + | + | −/dim+ | |
| CD79b | −/low | −/+ | +/− | + | +/− | +/− | ||
| CD81 | + | low/+ | + | + | + | + | + | −/dim+ |
| CD103 | − | − | − | − | − | − | ||
| CD117 | − | + | ||||||
| CD123 | + | |||||||
| CD138 | − | − | − | + | ||||
| CD185 | + | + | + | + | + | + | ||
| CD200 | +++ | −/+ | − | − | − | − | +/++ | |
| CD304 | −/+ | |||||||
| CD305 | − | − | − | − | − | − | ||
| CD307 | ++ | |||||||
| BCL-2 | + | +/− | ++ | + | − | +/− | ||
| BCL-6 | − | −/+ | + | − | + | |||
| CCND1 | + | |||||||
| HLA-DR | + | + | + | + | + | + | + | |
| FCM7 | + | |||||||
| Igκ/Igλ | dim/low | + | + | + | + | + | + | |
| IgM | −/+ | − | + | −/+ | + | −/+ | + | +/− |
| Ki67 | + | + | ||||||
| MPO | − | |||||||
| NG2 | − | |||||||
| PAX5 | + | + | ||||||
| TdT | +/− | − | − | |||||
1.2.3. Mantle Cell Lymphoma (MCL)
1.2.4. Marginal Zone Lymphoma (MZL)
1.2.5. Burkitt Lymphoma (BL)
1.3. Multiple Myeloma (MM)
2. Protein-Based Technologies to Study B Cell Malignancies
2.1. Mass Spectrometry (MS)
2.2. Flow Cytometry (FCM)
2.3. Mass Cytometry (CyTOF)
2.4. Other Proteomics Techniques
2.5. Integration of Proteomics with Other-Omics Approaches
3. Proteomics Studies for the Understanding of B Cell Hematological Malignancies
3.1. Proteomics Studies on Chronic Lymphocytic Leukemia
3.2. Proteomics Studies on Acute Lymphoblastic Leukemia (B-ALL)
3.3. Proteomics Studies on Diffuse Large B Cell Lymphoma (DLBCL)
3.4. Proteomics Studies on Follicular Lymphoma (FL)
3.5. Proteomics Studies on Mantle Cell Lymphoma (MCL)
3.6. Proteomics Studies on Marginal Zone Lymphoma (MZL)
3.7. Proteomics Studies on Burkitt Lymphoma (BL)
3.8. Proteomics Studies on Multiple Myeloma (MM)
3.9. Case Studies Using Proteomics
4. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Al-Sawaf, O. Chronic Lymphocytic Leukemia: 2022 Update on Diagnostic and Therapeutic Procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic Lymphocytic Leukaemia: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Kreuzer, K.; Soosapilla, A.; Spacek, M.; Stehlikova, O.; Gambell, P.; McIver-Brown, N.; Villamor, N.; Psarra, K.; Arroz, M.; et al. Reproducible Diagnosis of Chronic Lymphocytic Leukemia by Flow Cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation Project. Cytom. B Clin. Cytom. 2018, 94, 121–128. [Google Scholar] [CrossRef]
- Fischer, K.; Bahlo, J.; Fink, A.M.; Goede, V.; Herling, C.D.; Cramer, P.; Langerbeins, P.; von Tresckow, J.; Engelke, A.; Maurer, C.; et al. Long-Term Remissions after FCR Chemoimmunotherapy in Previously Untreated Patients with CLL: Updated Results of the CLL8 Trial. Blood 2016, 127, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Fink, A.-M.; Bahlo, J.; Busch, R.; Kovacs, G.; Maurer, C.; Lange, E.; Köppler, H.; Kiehl, M.; Sökler, M.; et al. First-Line Chemoimmunotherapy with Bendamustine and Rituximab versus Fludarabine, Cyclophosphamide, and Rituximab in Patients with Advanced Chronic Lymphocytic Leukaemia (CLL10): An International, Open-Label, Randomised, Phase 3, Non-Inferiority Trial. Lancet Oncol. 2016, 17, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Wierda, W.G.; Kipps, T.J.; Mayer, J.; Stilgenbauer, S.; Williams, C.D.; Hellmann, A.; Robak, T.; Furman, R.R.; Hillmen, P.; Trneny, M.; et al. Ofatumumab As Single-Agent CD20 Immunotherapy in Fludarabine-Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2010, 28, 1749–1755. [Google Scholar] [CrossRef]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Pui, C.-H.; Robison, L.L.; Look, A.T. Acute Lymphoblastic Leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Riva, G.; Nasillo, V.; Ottomano, A.M.; Bergonzini, G.; Paolini, A.; Forghieri, F.; Lusenti, B.; Barozzi, P.; Lagreca, I.; Fiorcari, S.; et al. Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges. Cancers 2021, 13, 4582. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome–Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-Rearranged Subtypes in Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Hoelzer, D.; Bassan, R.; Boissel, N.; Roddie, C.; Ribera, J.M.; Jerkeman, M. ESMO Clinical Practice Guideline Interim Update on the Use of Targeted Therapy in Acute Lymphoblastic Leukaemia. Ann. Oncol. 2024, 35, 15–28. [Google Scholar] [CrossRef]
- Leoni, V.; Biondi, A. Tyrosine Kinase Inhibitors in BCR-ABL Positive Acute Lymphoblastic Leukemia. Haematologica 2015, 100, 295–299. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Pourhassan, H.; Agrawal, V.; Pullarkat, V.; Aldoss, I. Positioning Blinatumomab in the Frontline of Adult B-Cell Acute Lymphoblastic Leukemia Treatment. Front. Oncol. 2023, 13, 1237031. [Google Scholar] [CrossRef]
- Chung, C. Driving toward Precision Medicine for B Cell Lymphomas: Targeting the Molecular Pathogenesis at the Gene Level. J. Oncol. Pharm. Pract. 2020, 26, 943–966. [Google Scholar] [CrossRef]
- Singh, R.; Dubey, A.; Rathore, A.; Kapoor, R.; Sharma, D.; Singh, N.; Maggo, S. Diffuse Large B-Cell Lymphoma-Review. J. Med. Sci. 2018, 38, 137. [Google Scholar] [CrossRef]
- Wang, S.S. Epidemiology and Etiology of Diffuse Large B-Cell Lymphoma. Semin. Hematol. 2023, 60, 255–266. [Google Scholar] [CrossRef]
- Smith, A.; Howell, D.; Patmore, R.; Jack, A.; Roman, E. Incidence of Haematological Malignancy by Sub-Type: A Report from the Haematological Malignancy Research Network. Br. J. Cancer 2011, 105, 1684–1692. [Google Scholar] [CrossRef]
- Liu, Y.; Barta, S.K. Diffuse Large B-Cell Lymphoma: 2019 Update on Diagnosis, Risk Stratification, and Treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef]
- Frontzek, F.; Lenz, G. Novel Insights into the Pathogenesis of Molecular Subtypes of Diffuse Large B-Cell Lymphoma and Their Clinical Implications. Expert. Rev. Clin. Pharmacol. 2019, 12, 1059–1067. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- McPhail, E.D.; Maurer, M.J.; Macon, W.R.; Feldman, A.L.; Kurtin, P.J.; Ketterling, R.P.; Vaidya, R.; Cerhan, J.R.; Ansell, S.M.; Porrata, L.F.; et al. Inferior Survival in High-Grade B-Cell Lymphoma with MYC and BCL2 and/or BCL6 Rearrangements Is Not Associated with MYC/IG Gene Rearrangements. Haematologica 2018, 103, 1899–1907. [Google Scholar] [CrossRef]
- Susanibar-Adaniya, S.; Barta, S.K. 2021 Update on Diffuse Large B Cell Lymphoma: A Review of Current Data and Potential Applications on Risk Stratification and Management. Am. J. Hematol. 2021, 96, 617–629. [Google Scholar] [CrossRef]
- Freedman, A.; Jacobsen, E. Follicular Lymphoma: 2020 Update on Diagnosis and Management. Am. J. Hematol. 2020, 95, 316–327. [Google Scholar] [CrossRef]
- Luminari, S.; Bellei, M.; Biasoli, I.; Federico, M. Follicular Lymphoma. Rev. Bras. Hematol. Hemoter. 2011, 34, 54–59. [Google Scholar] [CrossRef]
- Sungalee, S.; Mamessier, E.; Morgado, E.; Grégoire, E.; Brohawn, P.Z.; Morehouse, C.A.; Jouve, N.; Monvoisin, C.; Menard, C.; Debroas, G.; et al. Germinal Center Reentries of BCL2-Overexpressing B Cells Drive Follicular Lymphoma Progression. J. Clin. Investig. 2014, 124, 5337–5351. [Google Scholar] [CrossRef]
- Dreyling, M.; Ghielmini, M.; Rule, S.; Salles, G.; Ladetto, M.; Tonino, S.H.; Herfarth, K.; Seymour, J.F.; Jerkeman, M.; ESMO Guidelines Committee. Newly Diagnosed and Relapsed Follicular Lymphoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 298–308. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Jacobsen, E. Follicular Lymphoma: 2023 Update on Diagnosis and Management. Am. J. Hematol. 2022, 97, 1638–1651. [Google Scholar] [CrossRef]
- Olaniyi, J. Flow Cytometric Immunophenotyping of Hematological Malignancies: The Way Forward in Nigeria. Pathol. Lab. Med. Int. 2011, 3, 17–24. [Google Scholar] [CrossRef]
- Boris, E.; Theron, A.; Montagnon, V.; Rouquier, N.; Almeras, M.; Moreaux, J.; Bret, C. Immunophenotypic Portrait of leukemia-associated-phenotype Markers in B Acute Lymphoblastic Leukemia. Cytom. B Clin. Cytom. 2024, 106, 45–57. [Google Scholar] [CrossRef]
- Sędek, Ł.; Theunissen, P.; Sobral da Costa, E.; van der Sluijs-Gelling, A.; Mejstrikova, E.; Gaipa, G.; Sonsala, A.; Twardoch, M.; Oliveira, E.; Novakova, M.; et al. Differential Expression of CD73, CD86 and CD304 in Normal vs. Leukemic B-Cell Precursors and Their Utility as Stable Minimal Residual Disease Markers in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia. J. Immunol. Methods 2019, 475, 112429. [Google Scholar] [CrossRef]
- Ahmadi, A.; Poorfathollah, A.-A.; Aghaiipour, M.; Rezaei, M.; Nikoo-ghoftar, M.; Abdi, M.; Gharib, A.; Amini, A. Diagnostic Value of CD117 in Differential Diagnosis of Acute Leukemias. Tumor Biol. 2014, 35, 6763–6768. [Google Scholar] [CrossRef]
- Jiang, Z.; Wu, D.; Lin, S.; Li, P. CD34 and CD38 Are Prognostic Biomarkers for Acute B Lymphoblastic Leukemia. Biomark. Res. 2016, 4, 23. [Google Scholar] [CrossRef]
- Böttcher, S.; Engelmann, R.; Grigore, G.; Fernandez, P.; Caetano, J.; Flores-Montero, J.; van der Velden, V.H.J.; Novakova, M.; Philippé, J.; Ritgen, M.; et al. Expert-Independent Classification of Mature B-Cell Neoplasms Using Standardized Flow Cytometry: A Multicentric Study. Blood Adv. 2022, 6, 976–992. [Google Scholar] [CrossRef] [PubMed]
- Paulus, A.; Chitta, K.S.; Wallace, P.K.; Advani, P.P.; Akhtar, S.; Kuranz-Blake, M.; Ailawadhi, S.; Chanan-Khan, A.A. Immunophenotyping of Waldenströms Macroglobulinemia Cell Lines Reveals Distinct Patterns of Surface Antigen Expression: Potential Biological and Therapeutic Implications. PLoS ONE 2015, 10, e0122338. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Geyer, S.M.; Bone, N.D.; Tschumper, R.C.; Witzig, T.E.; Nowakowski, G.S.; Zent, C.S.; Call, T.G.; LaPlant, B.; Dewald, G.W.; et al. CD49d Expression Is an Independent Predictor of Overall Survival in Patients with Chronic Lymphocytic Leukaemia: A Prognostic Parameter with Therapeutic Potential. Br. J. Haematol. 2008, 140, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Zalcberg, I.; D’Andrea, M.G.; Monteiro, L.; Pimenta, G.; Xisto, B. Multidisciplinary Diagnostics of Chronic Lymphocytic Leukemia: European Research Initiative on CLL—ERIC Recommendations. Hematol. Transfus. Cell Ther. 2020, 42, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, P. Primary Gastrointestinal Lymphoma. World J. Gastroenterol. 2011, 17, 697. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Jung, J.S.; Kim, S.H. Clinicopathologic Significance of CD11c, CD16 and FOXP3 Expression in Diffuse Large B-Cell Lymphoma (DLBCL) Patients Receiving Rituximab, Cyclophosphamide, Anthracycline, Vincristine, and Prednisolone (R-CHOP) Combination Chemotherapy. Blood 2012, 120, 5112. [Google Scholar] [CrossRef]
- Koff, J.L.; Cashman, K.S.; Warren, V.; Smith, K.; Flowers, C.R.; Sanz, I. Circulating B Cell Subsets from Untreated Diffuse Large B Cell Lymphoma (DLBCL) Patients Resemble Those of Patients with Autoimmune Disease. Blood 2018, 132, 4221. [Google Scholar] [CrossRef]
- Dong, H.Y.; Shahsafaei, A.; Dorfman, D.M. CD148 and CD27 Are Expressed in B Cell Lymphomas Derived from Both Memory and Naïve B Cells. Leuk. Lymphoma 2002, 43, 1855–1858. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; de Tute, R.; Paiva, B.; Perez, J.J.; Böttcher, S.; Wind, H.; Sanoja, L.; Puig, N.; Lecrevisse, Q.; Vidriales, M.B.; et al. Immunophenotype of Normal vs. Myeloma Plasma Cells: Toward Antibody Panel Specifications for MRD Detection in Multiple Myeloma. Cytom. B Clin. Cytom. 2016, 90, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US Lymphoid Malignancy Statistics by World Health Organization Subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef]
- Lynch, D.T.; Koya, S.; Acharya, U.; Kumar, A. Mantle Cell Lymphoma; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Jain, P.; Wang, M.L. Mantle Cell Lymphoma in 2022-A Comprehensive Update on Molecular Pathogenesis, Risk Stratification, Clinical Approach, and Current and Novel Treatments. Am. J. Hematol. 2022, 97, 638–656. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, V.S.; Lokireddy, P.; Parihar, M.; Prakash, P.S.; Menon, H. Mantle Cell Lymphoma: A Clinical Review of the Changing Treatment Paradigms with the Advent of Novel Therapies, and an Insight into Indian Data. Cancer Rep. 2022, 5, e1590. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Natkunam, Y.; Allen, J.R.; Warnke, R.A. Selective Immunophenotyping for Diagnosis of B-Cell Neoplasms: Immunohistochemistry and Flow Cytometry Strategies and Results. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Dreyling, M.; Campo, E.; Hermine, O.; Jerkeman, M.; Le Gouill, S.; Rule, S.; Shpilberg, O.; Walewski, J.; Ladetto, M. Newly Diagnosed and Relapsed Mantle Cell Lymphoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2017, 28, iv62–iv71. [Google Scholar] [CrossRef] [PubMed]
- Cerhan, J.R.; Habermann, T.M. Epidemiology of Marginal Zone Lymphoma. Ann. Lymphoma 2021, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Goede, V. Marginal Zone Lymphoma in Elderly and Geriatric Patients. Best Pract. Res. Clin. Haematol. 2017, 30, 158–165. [Google Scholar] [CrossRef]
- Bertoni, F.; Rossi, D.; Zucca, E. Recent Advances in Understanding the Biology of Marginal Zone Lymphoma. F1000Reserch 2018, 7, 406. [Google Scholar] [CrossRef] [PubMed]
- Zucca, E.; Arcaini, L.; Buske, C.; Johnson, P.W.; Ponzoni, M.; Raderer, M.; Ricardi, U.; Salar, A.; Stamatopoulos, K.; Thieblemont, C.; et al. Marginal Zone Lymphomas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, 31, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Zucca, E.; Rossi, D.; Bertoni, F. Marginal Zone Lymphomas. Hematol. Oncol. 2023, 41, 88–91. [Google Scholar] [CrossRef]
- Linch, D.C. Burkitt Lymphoma in Adults. Br. J. Haematol. 2012, 156, 693–703. [Google Scholar] [CrossRef]
- López, C.; Burkhardt, B.; Chan, J.K.C.; Leoncini, L.; Mbulaiteye, S.M.; Ogwang, M.D.; Orem, J.; Rochford, R.; Roschewski, M.; Siebert, R. Burkitt Lymphoma. Nat. Rev. Dis. Prim. 2022, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Kalisz, K.; Alessandrino, F.; Beck, R.; Smith, D.; Kikano, E.; Ramaiya, N.H.; Tirumani, S.H. An Update on Burkitt Lymphoma: A Review of Pathogenesis and Multimodality Imaging Assessment of Disease Presentation, Treatment Response, and Recurrence. Insights Imaging 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Lynch, D.T. Burkitt Lymphoma; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Kersten, M.J.; Spanjaart, A.M.; Thieblemont, C. CD19-Directed CAR T-Cell Therapy in B-Cell NHL. Curr. Opin. Oncol. 2020, 32, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Cid Ruzafa, J.; Merinopoulou, E.; Baggaley, R.F.; Leighton, P.; Werther, W.; Felici, D.; Cox, A. Patient Population with Multiple Myeloma and Transitions across Different Lines of Therapy in the USA: An Epidemiologic Model. Pharmacoepidemiol. Drug Saf. 2016, 25, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D. Multiple Myeloma Epidemiology and Survival: A Unique Malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 Patients with Newly Diagnosed Multiple Myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef]
- Arroz, M.; Came, N.; Lin, P.; Chen, W.; Yuan, C.; Lagoo, A.; Monreal, M.; de Tute, R.; Vergilio, J.; Rawstron, A.C.; et al. Consensus Guidelines on Plasma Cell Myeloma Minimal Residual Disease Analysis and Reporting. Cytom. B Clin. Cytom. 2016, 90, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A. Update on the Treatment of Multiple Myeloma. Oncologist 2001, 6, 119–124. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: Every Year a New Standard? Hematol. Oncol. 2019, 37 (Suppl. S1), 62–65. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2020 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef]
- Kumar, S.K. Recycling Therapies for Myeloma: The Need for Prospective Trials. Cancer 2019, 125, 2920–2922. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2018 Update on Diagnosis, Risk-stratification, and Management. Am. J. Hematol. 2018, 93, 1091–1110. [Google Scholar] [CrossRef]
- Harousseau, J.L. How to Select among Available Options for the Treatment of Multiple Myeloma. Ann. Oncol. 2012, 23, x334–x338. [Google Scholar] [CrossRef]
- Mikhael, J.; Ismaila, N.; Cheung, M.C.; Costello, C.; Dhodapkar, M.V.; Kumar, S.; Lacy, M.; Lipe, B.; Little, R.F.; Nikonova, A.; et al. Treatment of Multiple Myeloma: ASCO and CCO Joint Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1228–1263. [Google Scholar] [CrossRef]
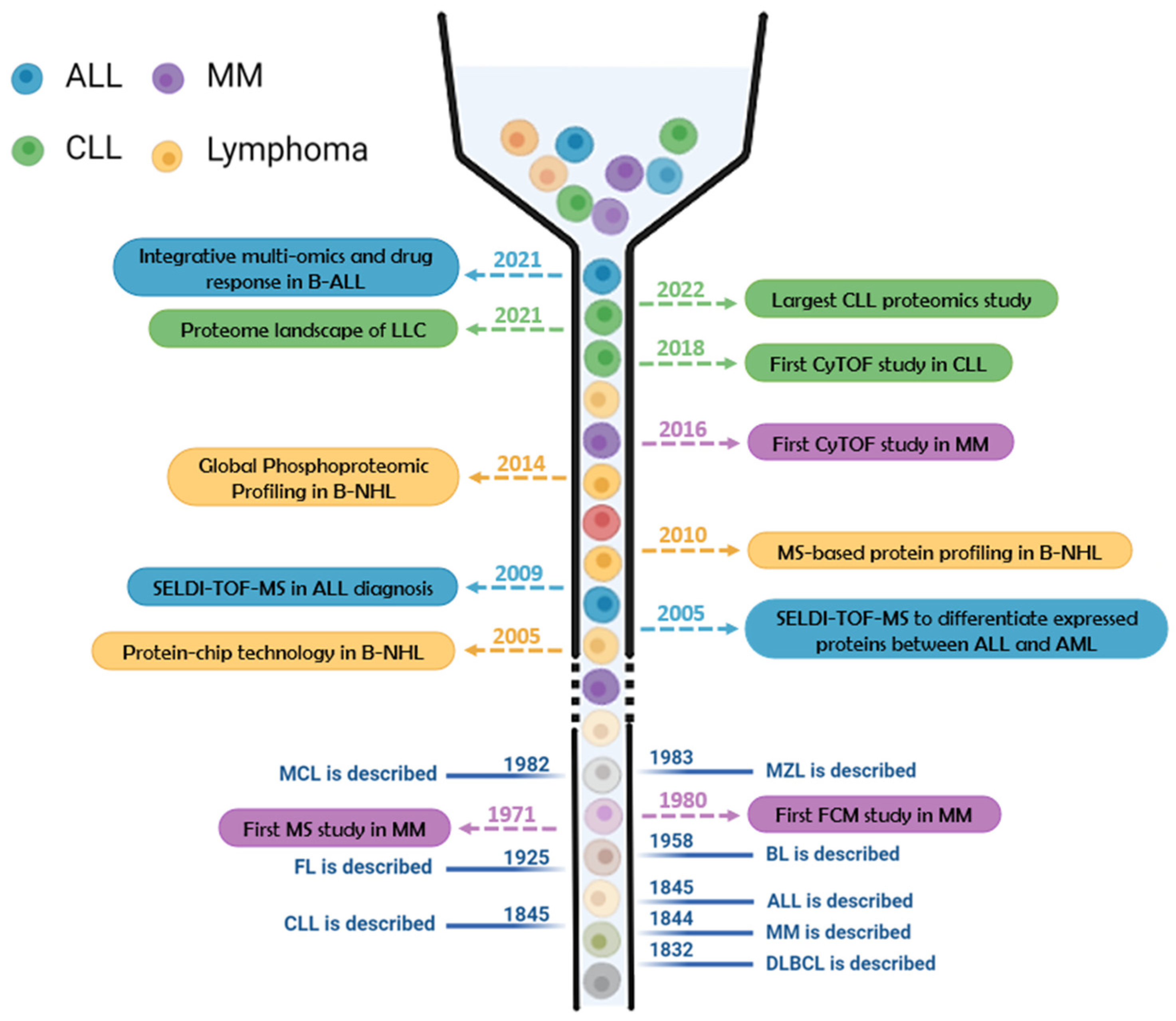
- Meier-Abt, F.; Lu, J.; Cannizzaro, E.; Pohly, M.F.; Kummer, S.; Pfammatter, S.; Kunz, L.; Collins, B.C.; Nadeu, F.; Lee, K.S.; et al. The Protein Landscape of Chronic Lymphocytic Leukemia. Blood 2021, 138, 2514–2525. [Google Scholar] [CrossRef]
- Herbst, S.A.; Vesterlund, M.; Helmboldt, A.J.; Jafari, R.; Siavelis, I.; Stahl, M.; Schitter, E.C.; Liebers, N.; Brinkmann, B.J.; Czernilofsky, F.; et al. Proteogenomics Refines the Molecular Classification of Chronic Lymphocytic Leukemia. Nat. Commun. 2022, 13, 6226. [Google Scholar] [CrossRef]
- Braoudaki, M.; Lambrou, G.I.; Vougas, K.; Karamolegou, K.; Tsangaris, G.T.; Tzortzatou-Stathopoulou, F. Protein Biomarkers Distinguish between High- and Low-Risk Pediatric Acute Lymphoblastic Leukemia in a Tissue Specific Manner. J. Hematol. Oncol. 2013, 6, 52. [Google Scholar] [CrossRef]
- Churchman, M.L.; Low, J.; Qu, C.; Paietta, E.M.; Kasper, L.H.; Chang, Y.; Payne-Turner, D.; Althoff, M.J.; Song, G.; Chen, S.-C.; et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 343–356. [Google Scholar] [CrossRef]
- Cavalcante, M.d.S.; Torres-Romero, J.C.; Lobo, M.D.P.; Moreno, F.B.M.B.; Bezerra, L.P.; Lima, D.S.; Matos, J.C.; Moreira, R.d.A.; Monteiro-Moreira, A.C.d.O. A Panel of Glycoproteins as Candidate Biomarkers for Early Diagnosis and Treatment Evaluation of B-Cell Acute Lymphoblastic Leukemia. Biomark. Res. 2016, 4, 1. [Google Scholar] [CrossRef]
- Dehghan-Nayeri, N.; Rezaei-Tavirani, M.; Omrani, M.D.; Gharehbaghian, A.; Goudarzi Pour, K.; Eshghi, P. Identification of Potential Predictive Markers of Dexamethasone Resistance in Childhood Acute Lymphoblastic Leukemia. J. Cell Commun. Signal 2017, 11, 137–145. [Google Scholar] [CrossRef]
- Guzmán-Ortiz, A.L.; Aparicio-Ozores, G.; Valle-Rios, R.; Medina-Contreras, O.; Patiño-López, G.; Quezada, H. Proteomic Changes in a Childhood Acute Lymphoblastic Leukemia Cell Line during the Adaptation to Vincristine. Bol. Med. Hosp. Infant. Mex. 2017, 74, 181–192. [Google Scholar] [CrossRef]
- Leo, I.R.; Aswad, L.; Stahl, M.; Kunold, E.; Post, F.; Erkers, T.; Struyf, N.; Mermelekas, G.; Joshi, R.N.; Gracia-Villacampa, E.; et al. Integrative Multi-Omics and Drug Response Profiling of Childhood Acute Lymphoblastic Leukemia Cell Lines. Nat. Commun. 2022, 13, 1691. [Google Scholar] [CrossRef]
- Yu, R.; Yang, S.; Liu, Y.; Zhu, Z. Identification and Validation of Serum Autoantibodies in Children with B-Cell Acute Lymphoblastic Leukemia by Serological Proteome Analysis. Proteome Sci. 2022, 20, 3. [Google Scholar] [CrossRef]
- Bram Ednersson, S.; Stern, M.; Fagman, H.; Enblad, G.; Mellqvist, U.-H.; Nilsson-Ehle, H.; Hasselblom, S.; Andersson, P.-O. Quantitative Proteomics in Diffuse Large B-Cell Lymphoma Patients Reveal Novel Overexpressed Proteins and Potentially Druggable Targets in the ABC Subtype. Blood 2019, 134, 3967. [Google Scholar] [CrossRef]
- van der Meeren, L.E.; Kluiver, J.; Rutgers, B.; Alsagoor, Y.; Kluin, P.M.; van den Berg, A.; Visser, L. A Super-SILAC Based Proteomics Analysis of Diffuse Large B-Cell Lymphoma-NOS Patient Samples to Identify New Proteins That Discriminate GCB and Non-GCB Lymphomas. PLoS ONE 2019, 14, e0223260. [Google Scholar] [CrossRef]
- Bram Ednersson, S.; Stern, M.; Fagman, H.; Nilsson-Ehle, H.; Hasselblom, S.; Thorsell, A.; Andersson, P.-O. Proteomic Analysis in Diffuse Large B-Cell Lymphoma Identifies Dysregulated Tumor Microenvironment Proteins in Non-GCB/ABC Subtype Patients. Leuk. Lymphoma 2021, 62, 2360–2373. [Google Scholar] [CrossRef]
- Lin, Z.; Crockett, D.K.; Jenson, S.D.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Quantitative Proteomic and Transcriptional Analysis of the Response to the P38 Mitogen-Activated Protein Kinase Inhibitor SB203580 in Transformed Follicular Lymphoma Cells. Mol. Cell Proteom. 2004, 3, 820–833. [Google Scholar] [CrossRef]
- Miles, R.R.; Crockett, D.K.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Analysis of BCL6-Interacting Proteins by Tandem Mass Spectrometry. Mol. Cell Proteom. 2005, 4, 1898–1909. [Google Scholar] [CrossRef]
- Crauste, C.; Lefebvre, I.; Hovaneissian, M.; Puy, J.Y.; Roy, B.; Peyrottes, S.; Cohen, S.; Guitton, J.; Dumontet, C.; Perigaud, C. Development of a Sensitive and Selective LC/MS/MS Method for the Simultaneous Determination of Intracellular 1-Beta-D-Arabinofuranosylcytosine Triphosphate (AraCTP), Cytidine Triphosphate (CTP) and Deoxycytidine Triphosphate (DCTP) in a Human Follicular Lymphoma Cell Line. J. Chromatogr. B 2009, 877, 1417–1425. [Google Scholar] [CrossRef]
- Everton, K.L.; Abbott, D.R.; Crockett, D.K.; Elenitoba-Johnson, K.S.J.; Lim, M.S. Quantitative Proteomic Analysis of Follicular Lymphoma Cells in Response to Rituximab. J. Chromatogr. B 2009, 877, 1335–1343. [Google Scholar] [CrossRef]
- Duś-Szachniewicz, K.; Rymkiewicz, G.; Agrawal, A.K.; Kołodziej, P.; Wiśniewski, J.R. Large-Scale Proteomic Analysis of Follicular Lymphoma Reveals Extensive Remodeling of Cell Adhesion Pathway and Identifies Hub Proteins Related to the Lymphomagenesis. Cancers 2021, 13, 630. [Google Scholar] [CrossRef]
- Enemark, M.B.H.; Wolter, K.; Campbell, A.J.; Andersen, M.D.; Sørensen, E.F.; Hybel, T.E.; Madsen, C.; Lauridsen, K.L.; Plesner, T.L.; Hamilton-Dutoit, S.J.; et al. Proteomics Identifies Apoptotic Markers as Predictors of Histological Transformation in Patients with Follicular Lymphoma. Blood Adv. 2023, 7, 7418–7432. [Google Scholar] [CrossRef]
- Weinkauf, M.; Christopeit, M.; Hiddemann, W.; Dreyling, M. Proteome- and Microarray-Based Expression Analysis of Lymphoma Cell Lines Identifies a P53-Centered Cluster of Differentially Expressed Proteins in Mantle Cell and Follicular Lymphoma. Electrophoresis 2007, 28, 4416–4426. [Google Scholar] [CrossRef]
- Stranneheim, H.; Orre, L.M.; Lehtiö, J.; Flygare, J. A Comparison between Protein Profiles of B Cell Subpopulations and Mantle Cell Lymphoma Cells. Proteome Sci. 2009, 7, 43. [Google Scholar] [CrossRef]
- Miguet, L.; Béchade, G.; Fornecker, L.; Zink, E.; Felden, C.; Gervais, C.; Herbrecht, R.; Van Dorsselaer, A.; Mauvieux, L.; Sanglier-Cianferani, S. Proteomic Analysis of Malignant B-Cell Derived Microparticles Reveals CD148 as a Potentially Useful Antigenic Biomarker for Mantle Cell Lymphoma Diagnosis. J. Proteome Res. 2009, 8, 3346–3354. [Google Scholar] [CrossRef]
- Rolland, D.; Bouamrani, A.; Houlgatte, R.; Barbarat, A.; Ramus, C.; Arlotto, M.; Ballester, B.; Berger, F.; Felman, P.; Callet-Bauchu, E.; et al. Identification of Proteomic Signatures of Mantle Cell Lymphoma, Small Lymphocytic Lymphoma, and Marginal Zone Lymphoma Biopsies by Surface Enhanced Laser Desorption/Ionization-Time of Flight Mass Spectrometry. Leuk. Lymphoma 2011, 52, 648–658. [Google Scholar] [CrossRef]
- Streckfus, C.F.; Romaguera, J.; Guajardo-Streckfus, C. The Use of Salivary Protein Secretions as an in Vivo Model to Study Mantel Cell Lymphoma Progression and Treatment. Cancer Investig. 2013, 31, 494–499. [Google Scholar] [CrossRef]
- Lorkova, L.; Scigelova, M.; Arrey, T.N.; Vit, O.; Pospisilova, J.; Doktorova, E.; Klanova, M.; Alam, M.; Vockova, P.; Maswabi, B.; et al. Detailed Functional and Proteomic Characterization of Fludarabine Resistance in Mantle Cell Lymphoma Cells. PLoS ONE 2015, 10, e0135314. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen Presentation Profiling Reveals Recognition of Lymphoma Immunoglobulin Neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef]
- Psatha, K.; Kollipara, L.; Drakos, E.; Deligianni, E.; Brintakis, K.; Patsouris, E.; Sickmann, A.; Rassidakis, G.Z.; Aivaliotis, M. Interruption of P53-MDM2 Interaction by Nutlin-3a in Human Lymphoma Cell Models Initiates a Cell-Dependent Global Effect on Transcriptome and Proteome Level. Cancers 2023, 15, 3903. [Google Scholar] [CrossRef]
- Zhu, D.; Bhatt, S.; Lu, X.; Guo, F.; Veelken, H.; Hsu, D.K.; Liu, F.-T.; Alvarez Cubela, S.; Kunkalla, K.; Vega, F.; et al. Chlamydophila Psittaci-Negative Ocular Adnexal Marginal Zone Lymphomas Express Self Polyreactive B-Cell Receptors. Leukemia 2015, 29, 1587–1599. [Google Scholar] [CrossRef]
- Cui, L.; Elzakra, N.; Xu, S.; Xiao, G.G.; Yang, Y.; Hu, S. Investigation of Three Potential Autoantibodies in Sjogren’s Syndrome and Associated MALT Lymphoma. Oncotarget 2017, 8, 30039–30049. [Google Scholar] [CrossRef]
- Panea, R.I.; Love, C.L.; Shingleton, J.R.; Reddy, A.; Bailey, J.A.; Moormann, A.M.; Otieno, J.A.; Ong’echa, J.M.; Oduor, C.I.; Schroeder, K.M.S.; et al. The Whole-Genome Landscape of Burkitt Lymphoma Subtypes. Blood 2019, 134, 1598–1607. [Google Scholar] [CrossRef]
- Weng, Q.; Lan, X.; Wang, Y.; Fan, C.; Xu, R.-A.; Zhang, P. Effect of Sophocarpine on the Pharmacokinetics of Umbralisib in Rat Plasma Using a Novel UPLC-MS/MS Method. Front. Pharmacol. 2022, 13, 749095. [Google Scholar] [CrossRef]
- El-Mallawany, N.K.; Day, N.; Ayello, J.; Van de Ven, C.; Conlon, K.; Fermin, D.; Basrur, V.; Elenitoba-Johnson, K.; Lim, M.; Cairo, M.S. Differential Proteomic Analysis of Endemic and Sporadic Epstein-Barr Virus-Positive and Negative Burkitt Lymphoma. Eur. J. Cancer 2015, 51, 92–100. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Larson, D.R.; Rajkumar, S.V.; Kyle, R.A.; Kumar, S.K.; Kourelis, T.; Arendt, B.; Willrcih, M.; Dasari, S.; Murray, D. N-Glycosylation of Monoclonal Light Chains on Routine MASS-FIX Testing Is a Risk Factor for MGUS Progression. Leukemia 2020, 34, 2749–2753. [Google Scholar] [CrossRef]
- Ferguson, I.D.; Patiño-Escobar, B.; Tuomivaara, S.T.; Lin, Y.-H.T.; Nix, M.A.; Leung, K.K.; Kasap, C.; Ramos, E.; Nieves Vasquez, W.; Talbot, A.; et al. The Surfaceome of Multiple Myeloma Cells Suggests Potential Immunotherapeutic Strategies and Protein Markers of Drug Resistance. Nat. Commun. 2022, 13, 4121. [Google Scholar] [CrossRef]
- Di Meo, F.; Iyer, A.; Akama, K.; Cheng, R.; Yu, C.; Cesarano, A.; Kurihara, N.; Tenshin, H.; Aljoufi, A.; Marino, S.; et al. A Target Discovery Pipeline Identified ILT3 as a Target for Immunotherapy of Multiple Myeloma. Cell Rep. Med. 2023, 4, 101110. [Google Scholar] [CrossRef]
- van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; van der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow Antibody Panels for Standardized N-Dimensional Flow Cytometric Immunophenotyping of Normal, Reactive and Malignant Leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Grigore, G.; Fluxá, R.; Hernández, J.; Fernandez, P.; Almeida, J.; Muñoz, N.; Böttcher, S.; Sedek, L.; van der Velden, V.; et al. EuroFlow Lymphoid Screening Tube (LST) Data Base for Automated Identification of Blood Lymphocyte Subsets. J. Immunol. Methods 2019, 475, 112662. [Google Scholar] [CrossRef]
- Criado, I.; Blanco, E.; Rodríguez-Caballero, A.; Alcoceba, M.; Contreras, T.; Gutiérrez, M.L.; Romero, A.; Fernández-Navarro, P.; González, M.; Solano, F.; et al. Residual Normal B-Cell Profiles in Monoclonal B-Cell Lymphocytosis versus Chronic Lymphocytic Leukemia. Leukemia 2018, 32, 2701–2705. [Google Scholar] [CrossRef]
- Abaza, H.M.; Alfeky, M.A.A.; Eissa, D.S.; Abdel Fattah, M.F.; Annaka, L.M.; Ebeid, F.S. Neuropilin-1/CD304 Expression by Flow Cytometry in Pediatric Precursor B-Acute Lymphoblastic Leukemia: A Minimal Residual Disease and Potential Prognostic Marker. J. Pediatr. Hematol. Oncol. 2018, 40, 200–207. [Google Scholar] [CrossRef]
- Rusak, M.; Bołkun, Ł.; Chociej-Stypułkowska, J.; Pawlus, J.; Kłoczko, J.; Dąbrowska, M. Flow-Cytometry-Based Evaluation of Peripheral Blood Lymphocytes in Prognostication of Newly Diagnosed DLBCL Patients. Blood Cells Mol. Dis. 2016, 59, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Le Gallou, S.; Lhomme, F.; Irish, J.M.; Mingam, A.; Pangault, C.; Monvoisin, C.; Ferrant, J.; Azzaoui, I.; Rossille, D.; Bouabdallah, K.; et al. Nonclassical Monocytes Are Prone to Migrate Into Tumor in Diffuse Large B-Cell Lymphoma. Front. Immunol. 2021, 12, 755623. [Google Scholar] [CrossRef]
- Azoulay, D.; Cohen, H.I.; Dementiev, E.; Eshel, E.; Akria, L.; Shaoul, E.; Horowitz, N. Flow Cytometry Aneuploidy and Cell Cycle Indexing as a Possible Tool for Differentiating between CD10+ Diffuse Large B-Cell Lymphoma and Follicular Lymphoma. Cytom. B Clin. Cytom. 2020, 98, 449–453. [Google Scholar] [CrossRef]
- Schniederjan, S.D.; Li, S.; Saxe, D.F.; Lechowicz, M.J.; Lee, K.L.; Terry, P.D.; Mann, K.P. A Novel Flow Cytometric Antibody Panel for Distinguishing Burkitt Lymphoma from CD10+ Diffuse Large B-Cell Lymphoma. Am. J. Clin. Pathol. 2010, 133, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Kase, S.; Ishijima, K.; Uraki, T.; Suimon, Y.; Suzuki, Y.; Kase, M.; Ishida, S. Usefulness of Flow Cytometry in Diagnosis of IgG4-Related Ophthalmic Disease and Extranodal Marginal Zone B-Cell Lymphoma of the Ocular Adnexa. Anticancer Res. 2017, 37, 5001–5004. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 Expression and Complement Inhibitors Affect Response and Resistance to Daratumumab Therapy in Myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.C.; Zweegman, S.; van Meerloo, J.; Musters, R.J.P.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.-T.; Goicoechea, I.; Alameda, D.; San José-Eneriz, E.; Merino, J.; Rodríguez-Otero, P.; et al. Immunogenomic Identification and Characterization of Granulocytic Myeloid-Derived Suppressor Cells in Multiple Myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Baeta, H.; Henriques, A.F.A.; Ejtehadifar, M.; Tranfield, E.M.; Sousa, A.L.; Farinho, A.; Silva, B.C.; Cabeçadas, J.; Gameiro, P.; et al. Proteomic Landscape of Extracellular Vesicles for Diffuse Large B-Cell Lymphoma Subtyping. Int. J. Mol. Sci. 2021, 22, 11004. [Google Scholar] [CrossRef]
- Hosen, N.; Yoshihara, S.; Takamatsu, H.; Ri, M.; Nagata, Y.; Kosugi, H.; Shimomura, Y.; Hanamura, I.; Fuji, S.; Minauchi, K.; et al. Expression of Activated Integrin Β7 in Multiple Myeloma Patients. Int. J. Hematol. 2021, 114, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Yang, Y.; Zhou, J.; Xu, L.; Wu, L.; Huang, P.; Feng, C.; Ke, P.; He, A.; Li, G.; et al. Combinatory Strategy Using Nanoscale Proteomics and Machine Learning for T Cell Subtyping in Peripheral Blood of Single Multiple Myeloma Patients. Anal. Chim. Acta 2021, 1173, 338672. [Google Scholar] [CrossRef]
- Wierz, M.; Janji, B.; Berchem, G.; Moussay, E.; Paggetti, J. High-Dimensional Mass Cytometry Analysis Revealed Microenvironment Complexity in Chronic Lymphocytic Leukemia. Oncoimmunology 2018, 7, e1465167. [Google Scholar] [CrossRef]
- Andrieu, T.; Mondière, P.; Jouve, P.-E.; Dussurgey, S.; Malassigné, V.; Servanton, H.; Baseggio, L.; Davi, F.; Michallet, A.-S.; Defrance, T. Mass Cytometry Analysis Reveals Attrition of Naïve and Anergized Self-Reactive Non-Malignant B Cells in Chronic Lymphocytic Leukemia Patients. Front. Oncol. 2022, 12, 1020740. [Google Scholar] [CrossRef]
- Nissen, M.D.; Kusakabe, M.; Wang, X.; Simkin, G.; Gracias, D.; Tyshchenko, K.; Hill, A.; Meskas, J.; Hung, S.; Chavez, E.A.; et al. Single Cell Phenotypic Profiling of 27 DLBCL Cases Reveals Marked Intertumoral and Intratumoral Heterogeneity. Cytom. A 2020, 97, 620–629. [Google Scholar] [CrossRef]
- Ferrant, J.; Le Gallou, S.; Padonou, F.; Léonard, S.; Papa, I.; Pangault, C.; Barthel, A.; Launay, V.; Manson, G.; Hoareau, B.; et al. Single-Cell Profiling Identifies Clinically Relevant Interactions between Tumor Associated Macrophages and Blood Endothelial Cells in Diffuse Large B Cell Lymphoma. bioRxiv 2022, preprint. [Google Scholar] [CrossRef]
- Shi, Y.; Ding, W.; Gu, W.; Shen, Y.; Li, H.; Zheng, Z.; Zheng, X.; Liu, Y.; Ling, Y. Single-Cell Phenotypic Profiling to Identify a Set of Immune Cell Protein Biomarkers for Relapsed and Refractory Diffuse Large B Cell Lymphoma: A Single-Center Study. J. Leukoc. Biol. 2022, 112, 1633–1648. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Wu, G.; Xu, Y.; Zhuang, W.; Lu, J.; Han, S.; Zhuang, Y.; Dong, X.; Yang, H. Peripheral Immune Cell Profiling of Double-Hit Lymphoma by Mass Cytometry. BMC Cancer 2023, 23, 184. [Google Scholar] [CrossRef]
- Hansmann, L.; Blum, L.; Ju, C.-H.; Liedtke, M.; Robinson, W.H.; Davis, M.M. Mass Cytometry Analysis Shows That a Novel Memory Phenotype B Cell Is Expanded in Multiple Myeloma. Cancer Immunol. Res. 2015, 3, 650–660. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Villasboas, J.C.; Jessen, E.; Dasari, S.; Dispenzieri, A.; Jevremovic, D.; Kumar, S. Mass Cytometry Dissects T Cell Heterogeneity in the Immune Tumor Microenvironment of Common Dysproteinemias at Diagnosis and after First Line Therapies. Blood Cancer J. 2019, 9, 72. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, Y.; Tu, C.; Zhang, H.; Vanderkerken, K.; Menu, E.; Liu, J. Identification of the Immune Checkpoint Signature of Multiple Myeloma Using Mass Cytometry-Based Single-Cell Analysis. Clin. Transl. Immunol. 2020, 9, e01132. [Google Scholar] [CrossRef] [PubMed]
- Seymour, F.; Cavenagh, J.D.; Mathews, J.; Gribben, J.G. NK Cells CD56bright and CD56dim Subset Cytokine Loss and Exhaustion Is Associated with Impaired Survival in Myeloma. Blood Adv. 2022, 6, 5152–5159. [Google Scholar] [CrossRef] [PubMed]
- Jakubikova, J.; Cholujova, D.; Beke, G.; Hideshima, T.; Klucar, L.; Leiba, M.; Jamroziak, K.; Richardson, P.G.; Kastritis, E.; Dorfman, D.M.; et al. Heterogeneity of B Cell Lymphopoiesis in Patients with Premalignant and Active Myeloma. JCI Insight 2023, 8, e159924. [Google Scholar] [CrossRef]
- Wong, S.; Hamidi, H.; Costa, L.J.; Bekri, S.; Neparidze, N.; Vij, R.; Nielsen, T.G.; Raval, A.; Sareen, R.; Wassner-Fritsch, E.; et al. Multi-Omic Analysis of the Tumor Microenvironment Shows Clinical Correlations in Ph1 Study of Atezolizumab +/− SoC in MM. Front. Immunol. 2023, 14, 1085893. [Google Scholar] [CrossRef]
- Baughn, L.B.; Jessen, E.; Sharma, N.; Tang, H.; Smadbeck, J.B.; Long, M.D.; Pearce, K.; Smith, M.; Dasari, S.; Sachs, Z.; et al. Mass Cytometry Reveals Unique Phenotypic Patterns Associated with Subclonal Diversity and Outcomes in Multiple Myeloma. Blood Cancer J. 2023, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Griffen, T.L.; Hoff, F.W.; Qiu, Y.; Lillard, J.W.; Ferrajoli, A.; Thompson, P.; Toro, E.; Ruiz, K.; Burger, J.; Wierda, W.; et al. Proteomic Profiling Based Classification of CLL Provides Prognostication for Modern Therapy and Identifies Novel Therapeutic Targets. Blood Cancer J. 2022, 12, 43. [Google Scholar] [CrossRef]
- Gulmann, C.; Espina, V.; Petricoin, E.; Longo, D.L.; Santi, M.; Knutsen, T.; Raffeld, M.; Jaffe, E.S.; Liotta, L.A.; Feldman, A.L. Proteomic Analysis of Apoptotic Pathways Reveals Prognostic Factors in Follicular Lymphoma. Clin. Cancer Res. 2005, 11, 5847–5855. [Google Scholar] [CrossRef]
- Lokhande, L.; Kuci Emruli, V.; Eskelund, C.W.; Kolstad, A.; Hutchings, M.; Räty, R.; Niemann, C.U.; Grønbaek, K.; Jerkeman, M.; Ek, S. Serum Proteome Modulations upon Treatment Provides Biological Insight on Response to Treatment in Relapsed Mantle Cell Lymphoma. Cancer Rep. 2022, 5, e1524. [Google Scholar] [CrossRef]
- Boyd, R.S.; Jukes-Jones, R.; Walewska, R.; Brown, D.; Dyer, M.J.S.; Cain, K. Protein Profiling of Plasma Membranes Defines Aberrant Signaling Pathways in Mantle Cell Lymphoma. Mol. Cell Proteom. 2009, 8, 1501–1515. [Google Scholar] [CrossRef]
- Jiang, Y.; Rex, D.A.B.; Schuster, D.; Neely, B.A.; Rosano, G.L.; Volkmar, N.; Momenzadeh, A.; Peters-Clarke, T.M.; Egbert, S.B.; Kreimer, S.; et al. Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry. arXiv 2023, arXiv:2311.07791. [Google Scholar]
- van Bergen, W.; Heck, A.J.R.; Baggelaar, M.P. Recent Advancements in Mass Spectrometry-Based Tools to Investigate Newly Synthesized Proteins. Curr. Opin. Chem. Biol. 2022, 66, 102074. [Google Scholar] [CrossRef] [PubMed]
- Tamara, S.; den Boer, M.A.; Heck, A.J.R. High-Resolution Native Mass Spectrometry. Chem. Rev. 2022, 122, 7269–7326. [Google Scholar] [CrossRef]
- Müller, W.H.; Verdin, A.; De Pauw, E.; Malherbe, C.; Eppe, G. Surface-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging: A Review. Mass. Spectrom. Rev. 2022, 41, 373–420. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-H.; Wang, C.-C.; Chen, C.W.; Liu, B.-H.; Lin, S.H.; Lee, Y.T.; Wang, Y.-S. Analysis of Initial Reactions of MALDI Based on Chemical Properties of Matrixes and Excitation Condition. J. Phys. Chem. B 2012, 116, 9635–9643. [Google Scholar] [CrossRef]
- Boesl, U. Time-of-Flight Mass Spectrometry: Introduction to the Basics. Mass. Spectrom. Rev. 2017, 36, 86–109. [Google Scholar] [CrossRef] [PubMed]
- Pareige, C.; Lefebvre-Ulrikson, W.; Vurpillot, F.; Sauvage, X. Time-of-Flight Mass Spectrometry and Composition Measurements. In Atom Probe Tomography; Elsevier: Amsterdam, The Netherlands, 2016; pp. 123–154. [Google Scholar]
- Deslignière, E.; Rolland, A.; Ebberink, E.H.T.M.; Yin, V.; Heck, A.J.R. Orbitrap-Based Mass and Charge Analysis of Single Molecules. Acc. Chem. Res. 2023, 56, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A.; Makarov, A. Orbitrap Mass Spectrometry. Anal. Chem. 2013, 85, 5288–5296. [Google Scholar] [CrossRef]
- Glish, G.L.; Burinsky, D.J. Hybrid Mass Spectrometers for Tandem Mass Spectrometry. J. Am. Soc. Mass. Spectrom. 2008, 19, 161–172. [Google Scholar] [CrossRef]
- Loos, G.; Van Schepdael, A.; Cabooter, D. Quantitative Mass Spectrometry Methods for Pharmaceutical Analysis. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150366. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Griffin, T.J.; Donohoe, S. Isotope-Coded Affinity Tagging of Proteins. CSH Protoc. 2007, 2007, pdb.prot4728. [Google Scholar] [CrossRef] [PubMed]
- Vélez-Bermúdez, I.C.; Wen, T.-N.; Lan, P.; Schmidt, W. Isobaric Tag for Relative and Absolute Quantitation (ITRAQ)-Based Protein Profiling in Plants. Methods Mol. Biol. 2016, 1450, 213–221. [Google Scholar] [CrossRef]
- Chen, X.; Wei, S.; Ji, Y.; Guo, X.; Yang, F. Quantitative Proteomics Using SILAC: Principles, Applications, and Developments. Proteomics 2015, 15, 3175–3192. [Google Scholar] [CrossRef] [PubMed]
- Zecha, J.; Satpathy, S.; Kanashova, T.; Avanessian, S.C.; Kane, M.H.; Clauser, K.R.; Mertins, P.; Carr, S.A.; Kuster, B. TMT Labeling for the Masses: A Robust and Cost-Efficient, In-Solution Labeling Approach. Mol. Cell Proteom. 2019, 18, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Kaeslin, J.; Zenobi, R. Resolving Isobaric Interferences in Direct Infusion Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2022, 36, e9266. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Gigliobianco, M.R.; Magnoni, F.; Censi, R.; Di Martino, P. Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review. Molecules 2020, 25, 3047. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, K.M. Flow Cytometry: An Overview. Curr. Protoc. Immunol. 2018, 120, 5.1.1–5.1.11. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P. The Evolution of Spectral Flow Cytometry. Cytom. Part A 2022, 101, 812–817. [Google Scholar] [CrossRef]
- Cossarizza, A.; Chang, H.; Radbruch, A.; Akdis, M.; Andrä, I.; Annunziato, F.; Bacher, P.; Barnaba, V.; Battistini, L.; Bauer, W.M.; et al. Guidelines for the Use of Flow Cytometry and Cell Sorting in Immunological Studies*. Eur. J. Immunol. 2017, 47, 1584–1797. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Crowell, H.L.; Zanotelli, V.R.T.; Engler, S.; Robinson, M.D.; Bodenmiller, B. Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst. 2018, 6, 612–620.e5. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Guo, T. Immunometabolism the CyTOF Way. Immunity 2021, 54, 610–613. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, B.; Xue, L. Applications of CyTOF in Brain Immune Component Studies. Engineering 2022, 16, 187–197. [Google Scholar] [CrossRef]
- Mahmood, T.; Yang, P.-C. Western Blot: Technique, Theory, and Trouble Shooting. N. Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Lourido, L.; Diez, P.; Dasilva, N.; Gonzalez-Gonzalez, M.; Ruiz-Romero, C.; Blanco, F.; Orfao, A.; LaBaer, J.; Fuentes, M. Protein Microarrays: Overview, Applications and Challenges. In Genomics and Proteomics for Clinical Discovery and Development; Springer: Berlin/Heidelberg, Germany, 2014; pp. 147–173. [Google Scholar]
- Abdelkader, Y.; Perez-Davalos, L.; LeDuc, R.; Zahedi, R.P.; Labouta, H.I. Omics Approaches for the Assessment of Biological Responses to Nanoparticles. Adv. Drug Deliv. Rev. 2023, 200, 114992. [Google Scholar] [CrossRef]
- Cui, M.; Cheng, C.; Zhang, L. High-Throughput Proteomics: A Methodological Mini-Review. Lab. Investig. 2022, 102, 1170–1181. [Google Scholar] [CrossRef]
- Gerritsen, J.S.; White, F.M. Phosphoproteomics: A Valuable Tool for Uncovering Molecular Signaling in Cancer Cells. Expert Rev. Proteom. 2021, 18, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Hwa, C.; Lee, G.-H.; Park, J.-M.; An, J.-Y. Integrative Multi-Omics Approaches in Cancer Research: From Biological Networks to Clinical Subtypes. Mol. Cells 2021, 44, 433–443. [Google Scholar] [CrossRef]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The Cellular Immune System in Myelomagenesis: NK Cells and T Cells in the Development of MM and Their Uses in Immunotherapies. Blood Cancer J. 2015, 5, e321. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cohen, A.D.; Kaushal, A.; Garfall, A.L.; Manalo, R.J.; Carr, A.R.; McCachren, S.S.; Stadtmauer, E.A.; Lacey, S.F.; Melenhorst, J.J.; et al. Changes in Bone Marrow Tumor and Immune Cells Correlate with Durability of Remissions Following BCMA CAR T Therapy in Myeloma. Blood Cancer Discov. 2022, 3, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Vescovini, R.; Marchica, V.; Storti, P.; Notarfranchi, L.; Dalla Palma, B.; Toscani, D.; Burroughs-Garcia, J.; Catarozzo, M.T.; Sammarelli, G.; et al. PD-L1/PD-1 Pattern of Expression Within the Bone Marrow Immune Microenvironment in Smoldering Myeloma and Active Multiple Myeloma Patients. Front. Immunol. 2020, 11, 613007. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.H.; Yee, M.; Song, L.; Canonico, M.M.; Verhoef, P.A.; Shimabukuro, K.A. Chronic Lymphocytic Leukemia With Leptomeningeal Involvement Presenting as an Acute Encephalopathy. Perm. J. 2022, 26, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, M.; Zhang, Z.; Jing, H.; Zou, Y.; Bu, H. Primary Meningeal Central Nervous System Lymphoma: A Case Report and Literature Review. Medicine 2022, 101, e32567. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, R.; Alvarez, B.; Masso, P.; Gonzalez, F.A.; Conejo, L.; Velasco, D.; Alonso, J.; Villarrubia, J.; Cava, F.; Llamas, P. The Utility of Multiparametric Seven-color Flow Cytometry in the Detection of Double Hit Lymphoma in Ascitic Fluid Samples. Cytom. B Clin. Cytom. 2016, 90, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Nabe, Y.; Kikuchi, S.; Kamihara, Y.; Wada, A.; Murakami, J.; Sato, T. Early Complete Response of Primary Bone Marrow B-cell Lymphoma Treated with Rituximab-based CHOP Therapy, Assessed by Flow Cytometry and Immunogloblin Heavy Chain Rearrangement. Clin. Case Rep. 2021, 9, e04657. [Google Scholar] [CrossRef] [PubMed]
- Baldini, C.; Giusti, L.; Ciregia, F.; Da Valle, Y.; Giacomelli, C.; Donadio, E.; Ferro, F.; Galimberti, S.; Donati, V.; Bazzichi, L.; et al. Correspondence between Salivary Proteomic Pattern and Clinical Course in Primary Sjögren Syndrome and Non-Hodgkin’s Lymphoma: A Case Report. J. Transl. Med. 2011, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Rodríguez, J.C.; Blanco-Bustos, M.P.; Cuervo-Maldonado, S.I.; Gómez-Rincón, J.C.; Reyes, Á. Geotricosis: Fungemia En Paciente Con Leucemia Linfoblástica Aguda. Biomédica 2023, 43, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Tachita, T.; Fujinami, H.; Oshima, Y.; Sasaki, H.; Marumo, Y.; Narita, T.; Ito, A.; Ri, M.; Kusumoto, S.; et al. Exophiala dermatitidis Fungemia Diagnosed Using Time-of-Flight Mass Spectrometry during Chemotherapy for Malignant Lymphoma and Successful Treatment with Voriconazole. Intern. Med. 2019, 58, 2219–2224. [Google Scholar] [CrossRef]
- Salvador, F.; Porte, L.; Durán, L.; Marcotti, A.; Pérez, J.; Thompson, L.; Noriega, L.M.; Lois, V.; Weitzel, T. Breakthrough Bacteremia Due to Clostridium Tertium in a Patient with Neutropenic Fever, and Identification by MALDI-TOF Mass Spectrometry. Int. J. Infect. Dis. 2013, 17, e1062–e1063. [Google Scholar] [CrossRef]
- Kim, T.; Yu, H.-J.; Kang, O.; Kim, C.-K.; Jung, C.; Huh, K.; Kang, C.-I.; Huh, H.; Lee, N. Fatal Prototheca Zopfii Algaemia in a Patient with Acute Lymphoblastic Leukemia: A Case Report. Clin. Lab. 2022, 68, 829–832. [Google Scholar] [CrossRef] [PubMed]
- van der Pan, K.; Khatri, I.; de Jager, A.L.; Louis, A.; Kassem, S.; Naber, B.A.E.; de Laat, I.F.; Hameetman, M.; Comans, S.E.T.; Orfao, A.; et al. Performance of Spectral Flow Cytometry and Mass Cytometry for the Study of Innate Myeloid Cell Populations. Front. Immunol. 2023, 14, 1191992. [Google Scholar] [CrossRef] [PubMed]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
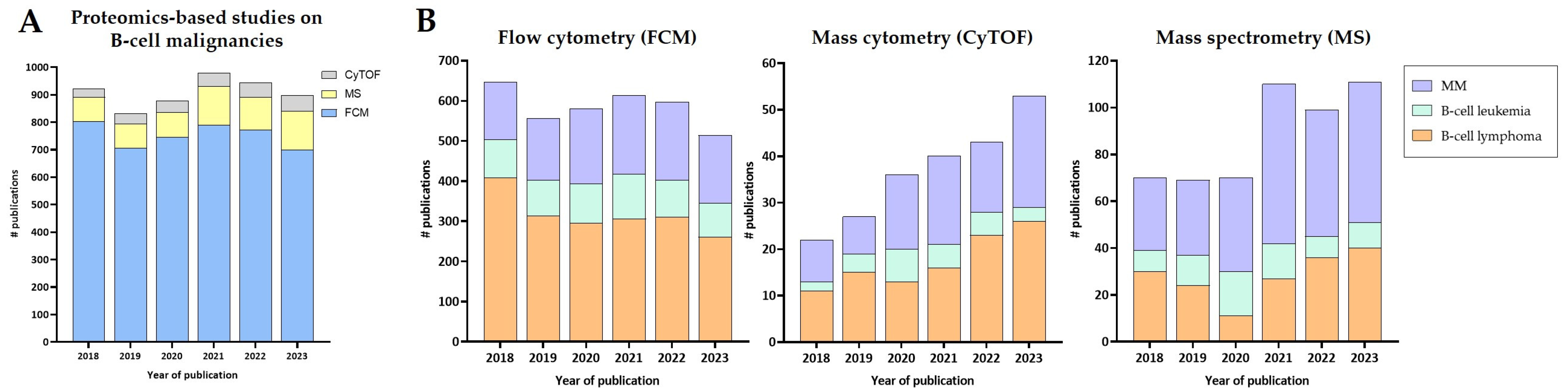
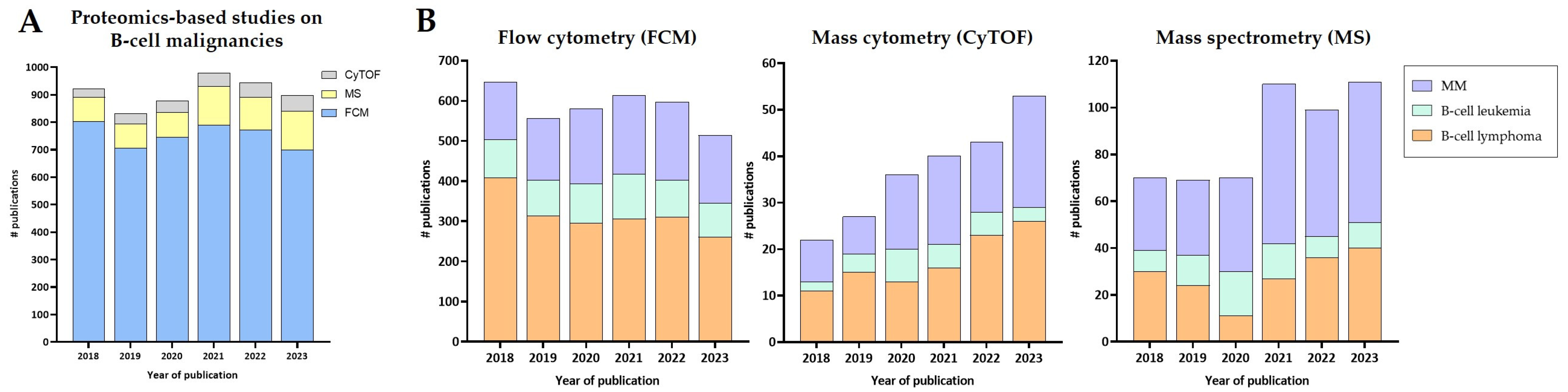
| Technology | Disease | Application | Reference |
|---|---|---|---|
| Mass Spectrometry | CLL | Correlation of genomic alterations with protein level variations | [77] |
| Identification of a novel CLL subgroup associated with unfavorable clinical outcome | [78] | ||
| B-ALL | Discovery of candidate biomarkers related to diagnosis, prognosis, and targeted therapy | [79] | |
| Definition of the role of IKZF1 alterations in ALL pathogenesis | [80] | ||
| Development of a panel of candidate biomarkers for early diagnosis and treatment evaluation | [81] | ||
| Identification of potential predictive markers of dexamethasone resistance in childhood ALL | [82] | ||
| Study of the mechanisms involved in tolerance to vincristine | [83] | ||
| Connection of the molecular phenotypes with drug responses and identification of therapeutic candidates for high-risk subtypes | [84] | ||
| Screening of serum autoantibodies for early detection of B-ALL in children | [85] | ||
| DLBCL | Characterization of pathways enriched in the different DLBCL subtypes | [86] | |
| Identification of proteins that discriminate between GCB and non-GCB lymphomas | [87] | ||
| Finding of proteins differentially expressed between GCB and non-GCB subtypes in extracellular vesicles | [88] | ||
| FL | Identification of differentially expressed proteins after p38 MAPK inhibitor treatment | [89] | |
| Distinguishing of proteins that interact with BCL6 and modulate its activity in transcriptional regulation | [90] | ||
| Understanding nucleoside analogue resistance by quantification of intracellular araCTP, CTP, and dCTP | [91] | ||
| Characterization of rituximab action mechanism | [92] | ||
| Finding biomarkers for early disease detection and management | [93] | ||
| Discovery of predictive indicators of histological transformation | [94] | ||
| FL and MCL | Finding differentially expressed proteins between FL and MCL | [95] | |
| MCL | Identification of molecular signatures that differentiate MCL from B cells of the different compartments | [96] | |
| Identification of proteomic biomarkers to distinguish MCL | [97] | ||
| Searching for specific proteomic biomarkers overexpressed in MCL tumor biopsies | [98] | ||
| Identification of tyrosine-phosphorylated proteins | [99] | ||
| Provision of insights into the dynamics of the disease and response to treatment | [99] | ||
| Evaluation of resistance to antinucleoside drugs | [100] | ||
| Discovery of neo-antigen peptides that mediate the killing of autologous lymphoma cells by circulating CD4 T cells | [101] | ||
| Characterization of the action mechanism of the MDM2-antagonist nutlin-3a | [102] | ||
| MZL | Discovery of the mechanisms involved in the pathogenesis of ocular adnexa extranodal MZL | [103] | |
| Identification of biomarkers for the diagnosis of primary Sjögren’s syndrome/MALT and prediction of progression | [104] | ||
| Establishment of the role of ID3 in regulating cell proliferation | [105] | ||
| Study of the pharmacokinetics of umbralisib | [106] | ||
| BL | Analysis of differentially expressed proteins between endemic and sporadic BL variants and EBV+ and EBV− BL cell lines | [107] | |
| MM | Prediction of MGUS progression for an early diagnosis of MM | [108] | |
| Analysis of the tumor microenvironment to identify determinants of durable responses to BCMA CAR T therapy | [109] | ||
| Quantification of surface proteins to identify immunotherapy targets and biomarkers associated with resistance and response to treatment | [109] | ||
| Identification of cell surface targets for immune-based therapies | [110] | ||
| Flow Cytometry | CLL | Design of panels for rapid disease diagnosis and progression assessment | [111,112] |
| Comparison of residual normal B cell profiles between CLL and MBL | [113] | ||
| B-ALL | Evaluation of neuropilin-1/CD304 as minimal residual disease and prognostic marker | [114] | |
| DLBCL | Assessment of the absolute counts of B cells, T cells, and Treg cells for the prognostication of newly diagnosed DLBCL patients | [115] | |
| Evaluation of the monocytic population distribution as an independent prognostic factor | [116] | ||
| DLBCL & FL | Usage of aneuploidy and cell cycle indexing as tools for differentiating between CD10+ DLBCL and FL | [117] | |
| DLBCL & BL | Identification of cell markers to differentiate between BL and CD10+ DLBCL | [118] | |
| MZL | Distinguishing IgG4-related ophthalmic disease, idiopathic orbital inflammation, and extranodal MZL based on the expression of different markers | [119] | |
| MM | Prediction of response to daratumumab monotherapy based on baseline CD38 expression levels and CD38 reduction | [120,121,122] | |
| Identification of markers for optimal monitoring of granulocytic myeloid-derived suppressor cells | [123] | ||
| Study of the PD-L1/PD-1 immune profile in patients with smoldering and active MM | [124] | ||
| Identification of targets for CAR T cell therapy | [125] | ||
| Characterization of global proteomes of CD3+, CD4+, and CD8+ T cells and development of a strategy to classify T cell subtypes | [126] | ||
| Mass Cytometry | CLL | Analysis of the tumor microenvironment to find differences in protein expression between tumor and normal cells | [127] |
| Assessment of the healthy B cell pool of patients to find disease mechanisms | [128] | ||
| DLBCL | Evaluation of the intertumoral and intratumoral heterogeneity | [129] | |
| Identification of clinically relevant interactions between tumor-associated macrophages and blood endothelial cells | [130] | ||
| Finding proteins overexpressed in relapsed and refractory patients | [131] | ||
| Comparison of major immune subsets in DLBCL and double-hit lymphoma | [132] | ||
| MM | Identification of differences in immune cell compartments across various stages of MM and healthy individuals | [133] | |
| Description of the immune tumor microenvironment in patients with MGUS and MM at diagnosis and post-initial therapies | [134] | ||
| Analysis of the immune checkpoint signature and regulation | [135] | ||
| Characterization of NK cells in newly diagnosed cases | [136] | ||
| Understanding of the molecular and cellular complexities underlying disease heterogeneity and prognosis | [137] | ||
| Provision of insights into the mechanism of action of daratumumab and the anti-PD-L1 monoclonal antibody atezolizumab | [138] | ||
| Employment of protein profiling as a tool for prognosis and treatment stratification | [139] | ||
| Other Tools | |||
| RRPA | CLL | Prediction of survival outcomes based on the proteomic signature | [140] |
| Protein Microarrays | FL | Identification of antibodies that distinguish lymphoid follicles in FL and benign follicular hyperplasia | [141] |
| MCL | Monitoring of patient serum proteomes to identify treatment-modulated proteins linked to the presence of minimal residual disease | [142] | |
| Western Blot | MCL | Definition of the pathologic hallmark of MCL as a tool for the diagnosis | [143] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez, C.; Garrote-de-Barros, A.; López-Portugués, C.; Hernández-Sánchez, M.; Díez, P. Characterization of Human B Cell Hematological Malignancies Using Protein-Based Approaches. Int. J. Mol. Sci. 2024, 25, 4644. https://doi.org/10.3390/ijms25094644
Jiménez C, Garrote-de-Barros A, López-Portugués C, Hernández-Sánchez M, Díez P. Characterization of Human B Cell Hematological Malignancies Using Protein-Based Approaches. International Journal of Molecular Sciences. 2024; 25(9):4644. https://doi.org/10.3390/ijms25094644
Chicago/Turabian StyleJiménez, Cristina, Alba Garrote-de-Barros, Carlos López-Portugués, María Hernández-Sánchez, and Paula Díez. 2024. "Characterization of Human B Cell Hematological Malignancies Using Protein-Based Approaches" International Journal of Molecular Sciences 25, no. 9: 4644. https://doi.org/10.3390/ijms25094644
APA StyleJiménez, C., Garrote-de-Barros, A., López-Portugués, C., Hernández-Sánchez, M., & Díez, P. (2024). Characterization of Human B Cell Hematological Malignancies Using Protein-Based Approaches. International Journal of Molecular Sciences, 25(9), 4644. https://doi.org/10.3390/ijms25094644